
Abstract
Iron-sulfur clusters are essential for life and defects in their biosynthesis lead to human diseases. The mechanism of cluster assembly and delivery to cytosolic and nuclear client proteins via the cytosolic iron-sulfur cluster assembly (CIA) pathway is not well understood. Here we report cryo-EM structures of the HEAT-repeat protein Met18 from Saccharomyces cerevisiae, a key component of the CIA targeting complex (CTC) that identifies cytosolic and nuclear client proteins and delivers a mature iron-sulfur cluster. We find that in the absence of other CTC proteins, Met18 adopts tetrameric and hexameric states. Using mass photometry and negative stain EM, we show that upon the addition of Cia2, these higher order oligomeric states of Met18 disassemble. We also use pulldown assays to identify residues of critical importance for Cia2 binding and recognition of the Leu1 client, many of which are buried when Met18 oligomerizes. Our structures show conformations of Met18 that have not been previously observed in any Met18 homolog, lending support to the idea that a highly flexible Met18 may be key to how the CTC is able to deliver iron-sulfur clusters to client proteins of various sizes and shapes, i.e. Met18 conforms to the dimensions needed.
Similar content being viewed by others


Introduction
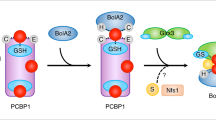
Iron-sulfur (Fe-S) clusters are versatile and important cofactors that allow numerous proteins to carry out their functions1,2,3. Fe-S clusters are involved in electron transport, gene expression regulation, and enzyme catalysis1,2. Although the importance of mitochondrial Fe-S proteins for primary metabolism is well-known, the prevalence of extramitochondrial Fe-S enzymes has been underappreciated. Cytosolic and nuclear Fe-S proteins are essential for life-sustaining processes such as: DNA replication and repair, transcription, translation, and nucleotide and amino acid biosynthesis [1-3]. Examples include DNA and RNA polymerases; primases; some helicases, such as the Rad3 component of the TFIIH complex required for transcription and nucleotide excision repair; Rli1, which is important for export of ribosomal subunits from the nucleus and promotion of translation initiation complex assembly; and Leu1, an isopropylmalate isomerase that catalyzes the second step in the leucine biosynthetic pathway1,2,4,5. These cytosolic and nuclear Fe-S cluster-containing proteins obtain their cofactors via the cytosolic iron-sulfur cluster assembly (CIA) pathway1,2,3 (Fig. 1). Due to the importance of the CIA pathway for maturation of essential Fe-S enzymes, CIA factors are highly conserved from yeast to humans. In the last step of the pathway, the CIA targeting complex (CTC) identifies apo-client proteins and delivers a mature iron-sulfur cluster1,2,3. The Saccharomyces cerevisiae CTC is comprised of the proteins Met18 (MMS19 in humans), Cia1 (CIAO1 in humans) and Cia2 (CIAO2B/CIA2B in humans). It is not well understood how the CTC recognizes its clients, now numbering > 30 eukaryotic proteins. A molecular level understanding of how the CTC controls flux through the CIA pathways is critical for elucidating how dysregulation of CIA impacts chromosomal integrity and contributes to carcinogenesis4,6,7,8.

A sulfur-containing metabolite is exported from the mitochondria through Atm1 and combined with iron to assemble into an iron-sulfur cluster via upstream factors in the CIA pathway. The CTC consisting of Met18-Cia2-Cia1 is believed to receive the mature iron-sulfur cluster from Nar1 and deliver this cluster to apo-clients required for a variety of biochemical processes. Red and yellow circles depict iron and sulfur, respectively. Model for CTC complex is based on the 12-Å cryo-EM map for the apo CTC-primase complex5.
Here, we focus on the CTC protein Met18 from S. cerevisiae (ScMet18), which was first discovered as a methionine auxotrophic9 and methyl methanesulfonate-sensitive (MMS) mutant10, and later shown to be involved in recognition of apo-client proteins in the cytosol and nucleus4,5,6. Met18 is a 118 kDa alpha solenoid protein comprised entirely of HEAT (Huntingtin, elongation factor 3 (EF3) repeats, protein phosphatase 1A, and the yeast kinase TOR1) repeats. Arrays of HEAT-repeats, each comprising two alpha helices, are well known to mediate protein-protein interactions and are often a part of large protein complexes11, consistent with Met18’s proposed role in client identification during the final step of the CIA pathway. Structural data on Met18 and on the CTC complex are limited, but recent structural work from Kassube and Thomä5 has provided key insights. These authors were able to determine a 3.6 Å resolution crystal structure of a complex between Cia2b-Cia1 from Drosophila melanogaster (DmCia2b-DmCia1) and Met18 from Mus musculus (MmMet18)5. This hybrid-species structure was a dimer of CTC units, mediated by homodimeric interactions between the two Met18 protomers as well as the two Cia2b proteins. Each CTC unit showed Met18 bound to Cia2b via Met18’s final HEAT-repeat (helices 45 and 46), Cia2b bound to Cia1, with no direct interactions between Met18 and Cia15, in agreement with previous biochemical studies12,13,14. Notably, lysine residues from helices 45 and 46 of Homo sapiens Met18 (K993, K1002, K1007, K1008, and K1013, numbering from human Met18) which were demonstrated to be important for the HsMet18-HsCia2b interaction via co-immunoprecipitation studies, are the target of ubiquitination15, suggesting that CTC complex formation may protect Met18 from degradation in eukaryotes; three (K1008, K1009, and K1014, numbering from S. cerevisiae Met18) of the five lysines are conserved between yeast and humans (Fig. S1).
To further examine how CTC interacts with its clients, Kassube and Thomä obtained low-resolution (8-12 Å) cryo-EM data on human CTC in the presence of client proteins DNA2 (CTC-DNA2) and the PriS-PriL primase heterodimer (CTC-primase)5. The arrangement of MmMet18-DmCia2b-DmCia1 observed in the hybrid-species crystal structure appeared to be conserved in the human CTC, however, the CTC dimer observed with the mouse and fly orthologs is not observed in the client-bound state. The primase heterodimer could be modeled into the cryo-EM maps, but DNA2 could not. Despite this limitation, these client-bound CTC maps supported the proposal that clients dock on the CTC via a bipartate interaction using the N-terminus of Met18 together with the conserved patch on the side of Cia15,14. Since Met18 was found to have a different superhelical curvature in all CTC structures, Kassube and Thomä proposed that this flexibility could allow for Met18 to recognize a variety of client proteins. Despite these important insights into the function of the CTC, the CTC-primase structure was not in a relevant state for Fe-S cluster transfer: no cluster was bound; and the primase [4Fe-4S] cluster binding site was 70 Å from the proposed cluster binding site on Cia2b12,16. Thus, a conformational change or additional factors are needed for Fe-S cluster transfer from the CTC to client proteins. The observed flexibility of Met18 in the different CTC structures5 suggested Met18 could flex to bring Cia2 closer to the client to afford Fe-S cluster transfer.
In this study, we use a combination of cryo-EM, negative stain EM, pulldown assays, and mass photometry to biophysically investigate S. cerevisiae Met18 (ScMet18). To interrogate the proposal that flexibility is key to Met18’s function5, we used two methods of cryo-grid preparation to determine structures of ScMet18 in isolation. We find that ScMet18 adopts a different conformation from those previously observed for the human and mouse homologs that allows it to form hexamers and tetramers in the absence of S. cerevisiae Cia2 (ScCia2). The addition of ScCia2 causes these higher order oligomeric states of ScMet18 to dissociate to form Met18-Cia2 complexes. Through pull-down assays, we identify residues at ScMet18’s N-terminus that are important for binding client protein S. cerevisiae Leu1 (ScLeu1) and at Met18’s C-terminus that are required to bind ScCia2. We compare these structures to the previously determined structures of Met18 in complex with other proteins5. Insights from these comparisons and from the biochemical data into Met18 regulation and function are described herein.
Results
3.3 Å resolution cryo-EM structure of ScMet18 in a hexameric state was obtained using Vitrobot plunged grids
To gain insight into the structure of ScMet18, a member of the CTC in the CIA pathway (Fig. 1), we purified recombinantly expressed SUMO-tagged ScMet18, cleaved the SUMO tag, and used untagged ScMet18 to prepare cryo-EM grids with a Vitrobot cryoplunger. Due to preferential orientation of ScMet18 particles on grids prepared using the Vitrobot, we collected 3 datasets on a Titan Krios (totaling 8031 micrographs) applying 0°, 25°, and 40° stage tilts to obtain additional particle orientations (Fig. S2 and Table 1). We processed the tilted data separately and obtained 2D class averages of particles. We then pooled the particles from 2D class averaging for 3D ab initio modeling. We first applied C1 symmetry and observed 2-fold and 3-fold symmetry in the map (Fig. S2). Therefore, D3 symmetry was applied throughout the rest of the data processing to generate Map 1 (Fig. S2). Map 1 is at 3.3 Å resolution (Fig S3a and Table 1). Due to air-water denaturation of particles and/or flexibility of the N-terminus, we applied masked focus classification to Map 1 during data processing (Fig. S2). Three classes were generated, and the particles and map from the class with the best density at the N-terminus (Class 3) was used for masked refinement to generate Map 2 (Fig. S2). Map 2 is at 3.6 Å resolution (Fig S3b and Table 1). We combined Map 1 and Map 2 to create a map for model building using WARP Frankenamp17 (Fig. S2), and used this combined map, along with Maps 1 and 2, to manually build residues 138–1029. The MmMet18 structure (PDB 6TC0) was docked into the combined map and used as a starting model for building residues 9–137 (Table 1).
To the best of our knowledge, our cryo-EM structure shows for the first time that ScMet18 can form an intertwined hexamer in the absence of other proteins (Fig. 2a and Supplemental Movie 1). Our final model contains six copies of Met18 with each protomer consisting of residues 9-1029 with two loops missing at residues 225–241 and 315–337 (Fig. 2 and Table 2). The hexamer is 154 Å long and has both 3-fold and 2-fold symmetry (Fig. 2a, b). For all chains, the N-terminus is solvent-exposed, and the C-terminus is buried (Fig. 2c). Each chain of the hexamer interacts with 4 other chains (i.e., chain A touches chains B, C, D, and F, but not chain E (Fig. S4a)). The hexamer is comprised of trimers and dimers where chains A, B, and C and chains D, E, and F can form trimers, and chains A and F, B and E, and C and D form dimers (Fig. S4a). Notably, the dimer from ScMet18 hexamer is not the same as the dimer observed in the crystal structure of MmMet18 in complex with DmCia2b-DmCia1(Fig. S4a, b). Numerous contacts are made between the ScMet18 dimers (Fig. S5). Aliphatic residues pack against each other at the center of the dimer interface, and this hydrophobic patch is flanked by salt bridges (Fig. S5).

a Views of the 2-fold and 3-fold axes of the hexameric form of ScMet18 with each chain colored differently. b Frankenmap combined cryo-EM Map 1 and Map 2 of the Vitrobot-plunged cryo-EM grids of ScMet18. c Chain A of the hexamer is shown in sandy brown in the Frankenmap combined map (transparent blue) without any other chains for clarity. The N-terminus is exposed, and the C-terminus is buried. d Protomer of the hexamer shown in rainbow in the same orientation as in c. e The curvature of Met18 is different in the ScMet18 hexamer compared to the MmMet18 structure in complex with Cia2-Cia1. If aligned by the C-termini, the N-termini of ScMet18 and MmMet18 are 44 angstroms apart. f ScMet18 and MmMet18 are aligned by their C-termini and shown in the CTC-DNA2 complex cryo-EM density5. Neither structure is a good fit, indicating that additional conformational rearrangement of Met18 must occur.
ScMet18 is similar by sequence (25% identity) and by structure to MmMet185 (Figs. 2e and S1). ScMet18 has a curved S-shaped structure comprised of 23 HEAT-repeat (HR) units; each HR contains two α-helices that are connected by loops (Figs. 2d and S4c). In total, there are 50 helices in ScMet18: four 310 helices and 46 α-helices in ScMet18. MmMet18 also has 23 HR units (Figs. 2e and S4c) and 46 α-helices. The S-shape solenoid of ScMet18 forms a left-handed superhelical structure with a length of 110-112 Å across (Fig. 2d). The curvature of ScMet18 differs from what has been observed previously with the MmMet18 protein in complex with DmCia2-DmCia1 (Fig. 2e and 2f); there is a 44 Å shift in the N-terminus of ScMet18 compared to MmMet18 (Fig. 2e).
ScMet18 tetrameric state is more apparent in grids prepared using the chameleon
To address the preferred orientation issues that we observed with the Vitrobot-prepared cryo-EM grids (Fig. S6a), we prepared grids of ScMet18 (SUMO-tag was cleaved) using a chameleon, a commercially available piezoelectric cryo-EM plunger that is based on the SpotitOn plunger developed by Carragher and co-workers18. We collected a dataset on a Talos Arctica (totaling 463 micrographs) and processed this dataset applying D3 and C1 symmetry to generate Maps 3 and 4, respectively. Map 3, a map showing the hexameric state of ScMet18, is at 8.46 Å resolution (Figs. S7–S8). This map compares well to Maps 1 and 2 showing 2-fold and 3-fold symmetry. The lower resolution can be attributed to the lower number of particles and higher pixel size used during data collection and processing. Despite the different plunging method used, preferred orientation was still an issue, albeit much less of an issue (Fig. S6b).
To our surprise, the data collected from the chameleon sample had tetramers present in addition to the hexamers observed in the Vitrobot dataset. The tetramer contains 2-fold symmetry and was refined to 12.77 Å (Figs. S7–9 and Table 2). Both the hexamer and the tetramer have similar overall dimensions and their chains have similar curvatures (Figs. 2a and S9), despite the fact that the tetramer contains two fewer chains. As a result, the tetramer is less tightly packed than the hexamer, especially around the more exposed N-termini (Fig. S9), since its protomers are shifted away from one another as compared to their relative orientation in the hexamer (Figs. 2a and S9).
With the discovery that ScMet18 can form a tetramer, we went back to re-analyze the Vitrobot-prepared samples, suspecting that tetrameric states were also present, but that there were not enough intact tetramers for robust class averages to be generated. Since we previously observed that chameleon-prepared samples suffer less from particle denaturation than those prepared on a Vitrobot19, we speculate that our ability to readily visualize the tetrameric state in the chameleon-prepared samples is due to less particle denaturation. The protomers of the tetrameric state appear to be less tightly packed (Fig. S9) than protomers of ScMet18 in the hexameric state, and thus the ScMet18 tetrameric state may be more prone to issues of particle denaturation. Short-lived and/or thermodynamically unstable states can provide valuable insight into protein assembly/disassembly processes but are hard to capture experimentally. The chameleon may prove beneficial in allowing for the visualization of such structural snapshots.
Pulldown assays suggest that conserved residues at the N-terminus of ScMet18 are involved in client protein binding
Previous CTC structure determinations and biochemical studies indicated that the conserved N-termini of MmMet18 and HsMet18 (HEAT-repeat (HR)2,3,4,5,6,7,8; Interpro domain IPR029240, Fig. S4c) are associated with client protein binding, and the final HR of Met18 is associated with Cia2 binding5. However, co-immunoprecipitation studies using truncated Met18 variants and various clients have generated conflicting information about which HR is involved in binding various clients14,20. Furthermore, Met18 also contains a second conserved domain comprising HR14-22 (MMS19, C-terminal domain; IPR024687, Fig. S4c). Currently, the function of this region of Met18 is not known; however, most of the interchain hydrogen bonding interactions in the ScMet18 hexamer involve these conserved regions, whereas the poorly conserved regions, HR1 and HR9-13, contribute little to oligomer formation (Fig. S4c). These data support the proposal that these conserved regions of Met18 are most important for their ability to mediate protein-protein interactions between the CTC and clients.
To test the importance of residues in the N-terminal (HR4-6) and C-terminal (HR22-23) regions of ScMet18 for facilitating protein-protein interactions, we made the following residue substitutions: R144A in HR4; K187E in HR5; F217A in HR6; I973A in HR22; and R1010E, R1013A, and R1020E in HR23 and used pulldown assays to investigate ScMet18’s ability to bind the CTC subunit ScCia2 and the client protein ScLeu1, previously shown to bind to ScMet18 in the presence of both ScCia1 and ScCia213 (Figs. S4c and 3). Since ScCia2 and ScMet18 form a stable binary complex13, we began these studies by monitoring the ability of a strep-tagged ScCia2 bait to pulldown ScMet18, which also has an N-terminal His-SUMO tag to aid with heterologous protein expression and solubility. The C-terminal ScMet18 variants R1010E, R1013A, and R1020E did not pulldown with ScCia2 to a substantial extent, whereas the variants R144A, K187E, F217A, and I973A were able to be pulled down with ScCia2 in amounts comparable to that of wild type ScMet18 (Fig. 3b, c). To evaluate the interaction between ScMet18 (SUMO-tagged) and the ScLeu1 client, the ScCia1 and ScCia2 proteins were included because the full CTC is required to pull down Leu1. Thus, only the ScMet18 variants competent to bind ScCia2 bait could be tested for their ability to pull down the CIA client ScLeu1. Each of the N-terminal variants, R144A, K187E, and F217E, were defective in ScLeu1 binding as compared to wild-type ScMet18 and a C-terminal variant, I973A (Fig. 3c). These results are consistent with the proposal5 that the N-terminus of Met18 is involved in client protein binding and that the C-terminus contacts Cia2.

a Structure of protomer colored by conservation (top) with ribbon drawing (bottom). At the N-terminus, the most conserved residues include R144 and G185-F187. At the C-terminus, the most conserved residues include L1005-R1020. Conservation is colored from most conserved (magenta) to least conserved (light blue) in the maps. Conservation correlates to the size of the letters in the sequence logo (i.e., largest letters are most conserved). Boxed letters with residue numbers indicate the residues that were mutated in this study. Sequence logo was made using WebLogo (https://weblogo.berkeley.edu/logo.cgi). b ScMet18-ScCia2 interaction analysis. The indicated ScMet18 variant was mixed with Strep-tagged ScCia2 bait (*) and chromatographed through Streptactin resin. The Streptactin column input (left panel) and elution (right panel) fractions were analyzed by SDS-PAGE. c ScLeu1 interaction with the CTC. Experiments were performed as described in b except ScLeu1 and ScCia1 were included in addition to the indicated ScMet18 variant and the ScCia2 bait. Unedited images of b, c are shown in Fig. S10.
Notably, residues that appeared to be involved in binding Cia2 (R1010, R1013, R1020) are buried in both the hexameric (Fig. 4a) and tetrameric states (Fig. 4b, c) of ScMet18, indicating that ScCia2 cannot bind to ScMet18 without disrupting these higher order oligomeric states. Similarly, the N-terminal residues whose substitution was shown above to impair association with client protein ScLeu1 (R144, K187, F217) are partially buried in the hexamer and in two of the chains of the tetramer (Fig. 4). In the other two chains of the tetramer, these N-terminal residues are mostly exposed (Fig. 4c). Thus, we propose that the higher order oligomeric states of ScMet18 must disassemble to allow for the formation of a high-affinity active CTC complex.

a Hexamerization of ScMet18 appears to limit access to conserved N-terminal residues R144, K187, and F217 and block access to conserved C-terminal residues K1008, K1009, R1010, R1013, K1014, and R1020. One protomer is shown in sandy brown in the map (transparent blue). b, c Tetramerization of ScMet18 also limits access to conserved N-terminal residues R144, K187, and F217 and blocks access to conserved C-terminal residues K1008, K1009, R1010, R1013, and K1014 in the sandy brown protomer, and blocks access to conserved C-terminus residues K1008, K1009, R1010, R1013, K1014, and R1020 in the hot pink protomer. Conserved N-terminal residues R144, K187, and F217 of the hot pink protomer are more accessible than they are in any of the hexamer protomers.
The addition of ScCia2 disassembles the higher order oligomeric states of ScMet18
To investigate which, if any, CTC proteins facilitate disassembly of the ScMet18 tetramer or hexamer en route to an active CTC complex, we turned to mass photometry (MP). Although higher order oligomeric states of ScMet18 dominate our cryo-EM data, we find that when ScMet18 is in solution, it is present as a mixture of monomers, dimers, tetramers, and hexamers. This mixture exists regardless of whether or not the SUMO tag is present (Fig. 5a, b). However, when ScCia2 is titrated into the solution of untagged ScMet18, higher order forms of ScMet18 disappear (Fig. 5c). Interestingly, ScCia1 and ScLeu1 do not show as dramatic of an effect on the higher order states of ScMet18. Whereas the hexameric state of ScMet18 remains apparent, the tetrameric state of ScMet18 does not appear to be present (Fig. 5d, e). It is perhaps not surprising that ScCia1 has less of an impact on ScMet18’s oligomeric state than ScCia2, given that Cia1 does not bind directly to Met185,13. It is more surprising that ScLeu1 does not appear to substantially change the oligomeric state of ScMet18 given that our data indicate direct binding. That being said, the binding studies with ScLeu1 and ScMet18 were performed in the presence of both ScCia1 and ScCia2, and it has been shown that high-affinity complex formation between CTC proteins and a client protein requires Cia1, Cia2, and Met185,13. Collectively, these data indicate that ScCia2, which serves to disassemble the ScMet18 hexamer, plays a role in preparing Met18 for high-affinity client binding.

a Control: presence of SUMO-tag does not change the oligomeric distribution of ScMet18. Like untagged ScMet18 (see panel b), SUMO-tagged ScMet18 is a mixed of monomers, dimers, tetramers, and hexamers. The molecular weights (MWs) calculated by MP for SUMO-tagged ScMet18 are 164 kDa, 256 kDa, 549 kDa, and 796 kDa, which agree well with the predicted MWs of the monomer (131 kDa), dimer (262 kDa), tetramer (524 kDa), and hexamer (786 kDa) of the SUMO-tagged ScMet18. Dotted lines correspond to ScMet18 monomer, dimer, tetramer and hexamer (left to right). b Untagged ScMet18 is a mixture of monomers, dimers, tetramers, and hexamers. The MWs observed are 157 ± 3 kDa, 236 ± 4 kDa, 472 ± 17 kDa, and 719 ± 14 kDa for untagged ScMet18, which corresponds to the calculated MWs of untagged ScMet18 monomer (118 kDa), dimer (236 kDa), tetramer (472 kDa), and hexamer (708 kDa), respectively. Five replicates are shown with each replicate in a different color. c Untagged ScMet18 in the presence of ScCia2 shows a loss of higher order oligomeric species. Experimental MWs are 195.4 ± 6 kDa and 269.3 ± 20 kDa. Three replicates are shown. d Untagged ScMet18 in the presence of ScCia1 retains higher order oligomers compared to ScMet18 in presence of ScCia2 (panel c). Five replicates are shown. e Untagged ScMet18 in the presence of ScLeu1 retains higher order oligomers compared to ScMet18 with ScCia2 (panel c). Five replicates are shown.
Discussion
Metallocofactor delivery can be highly specialized, with one protein delivering one cofactor to one client protein, or delivery can be less specialized. CTC proteins standout in that they deliver Fe-S clusters to more than 30 clients1,2. CTC client proteins perform divergent functions including amino acid and nucleotide biosynthesis, ribosome biogenesis, and DNA replication and repair. These clients are structurally distinct, raising the questions: how do the same three CTC proteins, Met18, Cia1, and Cia2, recognize clients of different sizes and shapes while avoiding holo-clients and nonclients? Also, what happens to the CTC in between ‘jobs;’ are CTC proteins protected from degradation so that they are ready for the next client, and if so, how does the CTC reassemble in the presence of a new client? Here, we have used a variety of biophysical methods to probe these questions and better understand the properties of Met18 that support its function.
Our work, along with previous studies5, provides insight into the question of how the CTC recognizes and accommodates vastly different client proteins. Previous studies have indicated that each CTC subunit (Met18, Cia1, Cia2) contributes to client identification, leading to a model that two or more, low affinity, relatively promiscuous interactions are leveraged by the CTC to bind its clients with high affinity and specificity5,14,20,21,22. Indeed, Kassube and Thomä demonstrated that high-affinity client binding is cooperative and requires the full CTC complex5. We have found previously that the full CTC leads to higher affinity Leu1 binding13, and we show here that residues in HEAT-repeats 4-6 of Met18’s conserved N-terminal region are required for that Leu1 binding. The residues, R144, K187, and F217, are found in the center of a large patch of conserved residues. Interestingly, this conserved patch is adjacent to the putative client binding sites predicted by low-resolution cryo-EM data on client-CTC complexes5, further corroborating the role of this region of Met18 in client protein recognition. With respect to the binding of a wide variety of client proteins, our studies highlight Met18’s flexibility and show that Met18’s curvature adapts to its binding partners, supporting the proposal that Met18’s conformational flexibility is key for the recognition of clients of various sizes and shapes. This role is not a new one for a HEAT-repeat protein. Karyopherins, the HEAT-repeat proteins involved in transporting molecules between the nucleus and cytoplasm, rely on flexibility for the binding and release of cargo during nuclear transport11.
The hexameric and tetrameric forms of Met18 observed in our cryo-EM studies cannot be the forms of Met18 to which client proteins bind given that oligomerization partially buries R144, K187, and F217. Instead, we suggest that oligomerization could protect Met18 from degradation when the CTC is paused between clients and/or protect from interactions with nonclients or holo-clients. When a maturase has one job (deliver adenosylcobalamin to its client mutase)23, job completion and maturase degradation could go hand-in-hand, but for a maturase system with multiple clients, there is a logic to keeping the maturase available for a new client but not so available that nonclient or holo-protein interactions occur. Previous studies have demonstrated that degradation of the CTC subunits, particularly Met18 and Cia2, is regulated in part by CTC complex formation5,14,15. When the CTC is ‘working’, the close juxtaposition of Met18’s C-terminal ubiquitination sites and its Cia2 binding site is thought to prevent ubiquitination5. Our work provides a possible extension to this model; when the CTC is ‘paused’ between clients, the burial of Met18’s C-termini within a higher order oligomer should limit ubiquitination and allow Met18 to wait for the next client. The proposal that protein oligomerization/protein complex formation can provide protection from degradation is not new. For example, hexamerization of proinsulin shields it from protease digestion24. When released into the blood, the hexamer dissociates into active monomers that can be degraded24. Also, complex formation between WRB (tryptophan-rich basic protein) and CAML (calcium-modulating cyclophilin ligand), which together serve as an insertase for tail-anchored proteins, imparts greater stability25. When the WRB-CAML complex is unassembled, each subunit is differentially recognized and degraded25.
After decades with little to no structural data on CTC proteins, the glass ceiling has cracked. Structural snapshots (Fig. 6) are becoming available and key biochemical analyses are accompanying those snapshots. However, there is still much to do. For example, studies assessing the role of Met18 hexamerization/tetramerization (Fig. 6a) in protection of Met18 from ubiquitination and protein degradation will test hypotheses presented here. Although this work indicates that Cia2 shifts the Met18 oligomeric state equilibrium away from the higher order states toward ‘active’ states, molecular details for how Cia2 accomplishes this feat are lacking. Also, we have no structures of Met18 in a monomeric or dimeric state in the absence of Cia1 and Cia2, although we know these oligomeric states exist (Fig. 6b). Further, we do not know if client proteins bind to a monomeric or dimeric CTC (Fig. 6c). A crystal structure provides a view of a dimeric CTC (Fig. 6c), whereas EM studies indicate that a monomeric CTC may bind to client proteins (Fig. 6c)5, leaving the question of oligomeric state relevance open. Importantly, the CTC-primase structure shown in Fig. 6d cannot be the final state. As noted in reference5, the CTC-primase structure is not a competent state for Fe-S cluster transfer, given that the site to which the Fe-S cluster binds on the primase is 70-Å from the cysteine on Cia2 that is a proposed cluster ligand. It is likely that multiple states of a CTC-client complex exist: a state to which an Fe-S cluster is delivered, presumably by Nar1 (Fig. 1); a holoCTC-client protein state in which the cluster is bound to Cia2 or Cia2-Cia1; and another holoCTC-client protein state in which the cluster is bound to the client. Thus, although we now have a wealth of structural data compared to a few years ago, there are many more snapshots to be obtained in order for us to understand the fundamental, yet enigmatic, biological process of cytosolic iron-sulfur cluster assembly and delivery.

a–d The sandy brown Met18 protomer is shown in roughly the same orientation to facilitate comparison of the different observed structures. Other Met18 protomers are colored red-sox red, dodger blue, brown, purple, dark brown, and silver. Cia1 is magenta, Cia2 is green, and client proteins (primase and DNA2) are light pink.
Methods
ScMet18 mutagenesis and purification
The cloning of ScMet18 with an N-terminal His-tag followed by SUMO solubility tag (SUMOMet18) was created by amplifying the Met18 gene from genomic DNA using primers AV03 and AV0413. Met18 was inserted into a pRSF-Duet vector in between EcoRI and SalI sites via Gibson DNA assembly13. A SUMO coding sequence was inserted at the EcoRI using primers AV26 and AV2713. All mutations were introduced by Q5 mutagenesis kit (New England Biolabs) according to manufacturer’s instructions and confirmed by DNA sequencing (Genewiz).
The plasmid for expression of wild-type SUMOScMet18 or the desired variant was transformed into Rosetta2(DE3), and protein expression was induced by autoinduction at room temperature. Autoinduction media (1 L) contained 6 g Na2HPO4, 3 g KH2PO4, 5 g NaCl, 10 mL 60% glycerol, 5 mL 10% glucose, 25 mL 8% lactose. For purification, cell paste was resuspended in Buffer A (50 mM Tris-HCl (pH 8), 100 mM NaCl, 5% glycerol and 5 mM β-mercaptoethanol (BME)) and lysed by sonication. Met18 was purified from the soluble extract using Ni-NTA resin, and it was exchanged into buffer B (50 mM Tris-HCl (pH 8), 100 mM NaCl, 1% glycerol, 1 mM BME) via gel filtration chromatography, and concentrated to 2.5 mg/mL (19 µM) for mass photometry studies13.
For removal of His-SUMO tag, His-tagged SUMO protease (2 mg or 0.074 µM) was added to SUMOScMet18 (25 mg or 0.191 µM) and incubated overnight at 4˚C. The mixture was passed over a Ni-NTA column and untagged ScMet18 was recovered from the flow through and concentrated. For cryo-EM analysis, samples were freshly prepared, avoiding freeze-thaw cycles, and diluted to the desired concentration in buffer C (50 mM HEPES, 100 mM NaCl, 1% glycerol, 1 mM BME).
Pulldown assays
For protein-protein interaction analysis, pulldown assays using double tagged ScCia2 (DTCia2, with N-terminal strep tag and C-terminal His tag), ScCia1 (HisCia1 with N-terminal His tag), SUMOScMet18 and ScLeu1 (HisLeu1 with N-terminal His tag) were performed as described12,13. Briefly, the strep-tagged ScCia2 bait (~2 µM) was mixed in equimolar amounts with the indicated prey proteins (SUMOMet18, HisCia1, and/or HisLeu1, as indicated) for 1 h at 4˚C in Buffer A. The mixture was batch absorbed to Strep-Tactin XT superflow resin (IBA), and then resin was collected, washed, and eluted according to manufacturer instructions. Fractions containing the highest concentration of protein were analyzed by SDS-PAGE in parallel with samples derived from Strep-Tactin input. In all cases, a negative control, in which the DTCia2 bait was omitted, and a positive control, utilizing wild-type ScMet18, were completed in parallel.
Mass photometry
Data were collected using AcquireMP (Refeyn Ltd) and analyzed using DiscoverMP (Refeyn Ltd). Each Mass Photometry (MP) measurement was performed for 60 sec. Samples were diluted in 50 mM Tris pH 8.0, 100 mM NaCl, 1% glycerol, and 5 mM BME to final concentrations of 0.050 µM SUMOScMet18. For SUMO-cleaved ScMet18, MP was conducted at 0.050 µM. Untagged ScMet18 in the following complexes: ScMet18-ScCia2, ScMet18-ScCia1, and ScMet18-ScLeu1 were all performed at 0.050 µM, as well. The molecular weights were obtained using contrast comparison with known molecular weights from mass standard calibrants measured the same day.
Vitrobot cryo-EM specimen preparation
The sample was prepared using a Quantifoil R 1.2/1.3 Au 300 mesh holey-carbon grid and plunged using a Thermo Fisher Scientific Vitrobot cryo-plunger. The grid was glow discharged at −15 mA for 1 min (PELCO easiGlow) before protein solution was applied. ScMet18 (untagged, His-SUMO tag removed by SUMO protease treatment) was diluted to a final concentration of 1.2 mg/mL (9 µM) in Buffer C (50 mM HEPES pH 7.5, 100 mM NaCl, 1% glycerol, and 1 mM BME) and 33 mM sodium acetate was added to the solution. The use of this buffer composition and this protein concentration was empirical; it allowed for good distribution of ScMet18 particles on grids, limiting the crowding of particles. ScMet18 (3 μL) was applied to the grid and was blotted for 6 sec before plunging into liquid ethane and transferring to storage buttons. The temperature and humidity were 10 °C and 95%, respectively.
Cryo-EM data collection for the 0°, 25°, and 40° tilted datasets
Data were collected at the University of Massachusetts Medical School at Worcester Cryo-EM Core Facility on a Thermo Fisher Titan Krios 300 kV electron microscope equipped with a Gatan GIF K2 camera at 130000x magnification across three collections. All three data collections were from the same protein preparation. Parameters for the 0° tilted dataset were as follows: 1.059 Å/pix (collected at super-resolution of 0.529 Å /pix), 45 frames, 1.102 electrons/ Å2 /frame dose, −0.7– −2.5 µm defocus range. This dataset contained 3968 frames. Parameters for the 25° and 40° tilted dataset were as follows: 1.059 Å /pix (collected at super-resolution of 0.529 Å /pix), 45 frames, 1.104 electrons/ Å2/frame dose, defocus range −0.8– −2.4 µm. The 25° tilted dataset contained 1783 frames, and the 40° tilted dataset contained 2280 frames (Table 1).
Frame alignment, defocus estimation, and micrograph assessment in SPHIRE of the 0°, 25°, and 40° tilted datasets
Each individual frame of dose-fractionated exposure was summed and aligned using MotionCor226. For the 0° tilted datasets, the defocus of the summed frames was estimated using CTER27. For the 25° and 40° tilted datasets, the defocus of the summed frames was estimated using GCTF28. Outputs from motion_cor2 and CTER were used to perform drift and CTF assessments within the SPHIRE software suite29. The final number of micrographs for the 0°, 25°, and 40° tilted datasets were 3612, 1646, and 2079, respectively, for a total of 7337 micrographs used for selection moving forward (Table 1).
Particle selection of the 0°, 25°, and 40° tilted datasets
For the 0-degree tilted dataset, ~1800 particles were picked manually from a subset of aligned movies. The neural-net automated particle picker Topaz30 was used to automatically pick particles from the entire dataset. Using a cutoff threshold of −2, the initial particle hits resulted in 172848 particles. For the 25-degree tilted dataset, ~1200 particles were picked manually from a subset of aligned movies. Topaz30 was used to automatically pick particles from the entire dataset. Using a cutoff threshold of −2, the initial particle hits resulted in 136153 particles. For the 40-degree tilted dataset, ~1600 particles were picked manually from a subset of aligned movies. Topaz30 was used to automatically pick particles from the entire dataset. Using a cutoff threshold of −3, the initial particle hits resulted in 114057 particles.
Frame alignments, map generations, and refinement in Relion 3.0 of the 0°, 25°, and 40° tilted datasets
Frame alignment using motion_cor2 was rerun on all three datasets within the Relion 3.0 software suite31. CTF estimation of the 0-degree tilt dataset was also rerun using CTFFIND432 on the output from Relion MotionCor2. A total of 418832 particles from Topaz were used to re-extract particles (box-size 330 pix) and perform initial reference-free 2D classification (mask diameter 200 Å for 25- and 40-degree tilt datasets, 190 Å for 0-degree tilted dataset). Reference-free 2D classification was performed separately for each dataset and 379779 particles were pooled to generate the ab initio reference-free 3D model applying no symmetry (C1) and D3 symmetry (Fig. S2). The D3 map was then subjected to 3D classification assigning two classes. Class 1 which had 171255 particles was then subjected to 3D-refinement with an initial low-pass filter of 50 Å. CTF refinement and Bayesian polishing were performed on this map, and another round of 3D-refinement and post processing was performed, resulting in a 3.33 Å resolution map (Map 1). The mask applied during post-processing was low-pass filtered by 15 Å with an initial binarization threshold of 0.006 extended by 5 pixels with a soft edge of 6 pixels.
The map was subjected to focused classification to resolve the 3-fold N-terminal density (Fig. S1). Focused masks were made in Chimera33 and Relion 3.031 and low-pass filtered by 15 Å with an initial binarization threshold of 0.004 extended by 5 pixels with a soft edge of 6 pixels. Using this mask, 3D classification was performed to classify the map into three classes. Class 3 of 44707 particles was subjected to reference-free masked 3D refinement and post-processing to obtain a final map of 3.57 Å resolution (Map 2). Maps 1 and 2 were combined using WARP Frankenmap17, and this combined map was used for model building and refinement.
Model building and refinement of the ScMet18 hexamer structure from the 0°, 25°, and 40° tilted datasets
Residues 138–1029 were manually built into the combined map using Coot34. The MmMet18 structure (PDB 6TC0) was docked into the map using Coot and used as a starting model for building residues 9-137. Multiple rounds of refinement were carried out in Phenix35 with model building in Coot34. The final ScMet18 hexamer model contains residues 9-225, 242–314, and 338–1029. Figures of the map and model were made using UCSF ChimeraX36. Coot, Phenix, and ChimeraX were licensed through the SBGrid Consortium operated out of Harvard Medical School37. Refinement and model statistics are summarized in Tables 1 and 2.
Chameleon cryo-EM specimen preparation and data collection
A specimen was prepared on a chameleon grid with 1.2 μm holes with 0.8 μm spacing. The grid was glow discharged at −12 mA for 210 s (PELCO easiGlow) before protein solution was applied. The concentration of untagged ScMet18 was at 2 μg/μL (15 µM) in 50 mM Tris pH 8.0, 100 mM NaCl, 1% glycerol, and 1 mM BME. After ScMet18 was applied to the grid, the grid self-wicked for 569 ms before plunging into liquid ethane and transferring to storage buttons. The temperature and humidity were 24 °C and 77%, respectively.
Data were collected at the MIT.nano Cryo-EM Facility on a Talos Arctica 200 kV electron microscope equipped with a Falcon3 camera at 92000x magnification. Parameters for the dataset were as follows: 1.5998 Å/pix, 14 frames, 3.804 electrons/ Å2 /frame dose, −1.3 - −3.4 µm defocus range. This dataset contained 483 frames (Table 1).
Chameleon cryo-EM data processing
Data were processed in cryoSPARC v3.2.038. Frame alignment was performed using Patch motion correction. CTF estimation was estimated using the CTFFIND4 wrapper32. Roughly 1000 particles were manually picked using the Manual Picker and these particles were subjected to 2D classification to obtain template for automatic picking using the Template Picker. The Template Picker picked 460492 number of particles, and these particles inspected and a final stack of 139569 number of particles were used for 2D classification. 87417 particles were used as inputs for 3D ab initio modeling into 3 models. Models 1 (30104 particles) and 2 (36582 particles) were subjected to one round of non-uniform Refinement to obtain final maps 4 and 3, respectively. Map 3 is at a final resolution of 8.46 Å, and map 4 is at a final resolution of 12.77 Å (Figs. S7 and S8).
Model building and refinement of the ScMet18 tetramer structure
Chains A, C, D, and F (each containing residues 9-225, 242-314, and 338-1029) from the 3.3 Å resolution structure of the ScMet18 hexamer were docked into the 12.8 Å resolution map (map 4) using Phenix Dock In Map35. One round of rigid-body real space refinement was carried out in Phenix35. Figures of the map and model were made using UCSF ChimeraX36. Coot, Phenix, and ChimeraX were licensed through the SBGrid Consortium operated out of Harvard Medical School37. Refinement and model statistics are summarized in Tables 1 and 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM density maps of the S. cerevisiae Met18 hexamer from Vitrobot sample preparation method, Map 1, Map 2, and the composite map, have been deposited to the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-42512, and EMD-42513, and EMDB-42510, respectively. The S. cerevisiae Met18 hexamer Map 3 from the chameleon sample preparation method has been deposited to the EMDB under accession number EMD-42514. The S. cerevisiae Met18 tetramer Map 4 from the chameleon sample preparation has been deposited to the EMDB under accession number EMD-42511. The S. cerevisiae hexamer and tetramer cryo-EM structures have been deposited to the Protein Data Bank (PDB) under accession numbers 8USP and 8USQ, respectively. Unedited gels from the CTC pulldown assays are found in Supplementary Fig. 10. All relevant data are available from the authors upon request.
References
Wachnowsky, C., Fidai, I. & Cowan, J. A. Iron-sulfur cluster biosynthesis and trafficking - impact on human disease conditions. Metallomics 10, 9–29 (2018).
Ciofi-Baffoni, S., Nasta, V. & Banci, L. Protein networks in the maturation of human iron-sulfur proteins. Metallomics 10, 49–72 (2018).
Lill, R. et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell Biol. 94, 280–291 (2015).
Stehling, O. et al. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 337, 195–199 (2012).
Kassube, S. A. & Thoma, N. H. Structural insights into Fe-S protein biogenesis by the CIA targeting complex. Nat. Struct. Mol. Biol. 27, 735–742 (2020).
Gari, K. et al. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 337, 243-245 (2012).
Balk, J., Aguilar Netz, D. J., Tepper, K., Pierik, A. J. & Lill, R. The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Mol. Cell Biol. 25, 10833–10841 (2005).
Weerapana, E. et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795 (2010).
Masselot, M. & de Robichon-Szulmajster, H. Methionine biosynthesis in Saccharomyces cerevisiae. Mol. Gen. Genet. 139, 121–132 (1975).
Prakash, L. & Prakash, S. Three additional genes involved in pyrimidine dimer removal in Saccharomyces cerevisiae: RAD7, RAD14 and MMS19. Mol. Gen. Genet. 176, 351–359 (1979).
Yoshimura, S. H. & Hirano, T. HEAT repeats - versatile arrays of amphiphilic helices working in crowded environments? J. Cell Sci. 129, 3963–3970 (2016).
Vo, A. T. et al. Defining the domains of Cia2 required for its essential function in vivo and in vitro. Metallomics 9, 1645–1654 (2017).
Vo, A. et al. Identifying the protein interactions of the cytosolic iron-sulfur cluster targeting complex essential for its assembly and recognition of apo-targets. Biochemistry 57, 2349–2358 (2018).
Odermatt, D. C. & Gari, K. The CIA targeting complex is highly regulated and provides two distinct binding sites for client iron-sulfur proteins. Cell Rep. 18, 1434–1443 (2017).
Weon, J. L., Yang, S. W. & Potts, P. R. Cytosolic iron-sulfur assembly is evolutionarily tuned by a cancer-amplified ubiquitin ligase. Mol. Cell 69, 113–125 (2018).
Maione, V., Cantini, F., Severi, M. & Banci, L. Investigating the role of the human CIA2A-CIAO1 complex in the maturation of aconitase. Biochim. Biophys. Acta Gen. Subj. 1862, 1980–1987 (2018).
Farnung, L., Ochmann, M., Engeholm, M. & Cramer, P. Structural basis of nucleosome transcription mediated by Chd1 and FACT. Nat. Struct. Mol. Biol. 28, 382–387 (2021).
Wei, H. et al. Spotiton and chameleon vitrification robots. Microsc. Microanal. 25, 1010–1011 (2019).
Levitz, T. S., Brignole, E. J., Fong, I., Darrow, M. C. & Drennan, C. L. Effects of chameleon dispense-to-plunge speed on particle concentration, complex formation, and final resolution: a case study using the Neisseria gonorrhoeae ribonucleotide reductase inactive complex. J. Struct. Biol. 214, 107825 (2022).
van Wietmarschen, N., Moradian, A., Morin, G. B., Lansdorp, P. M. & Uringa, E. J. The mammalian proteins MMS19, MIP18, and ANT2 are involved in cytoplasmic iron-sulfur cluster protein assembly. J. Biol. Chem. 287, 43351–43358 (2012).
Stehling, O. et al. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 18, 187–198 (2013).
Marquez, M. D. et al. Cytosolic iron-sulfur protein assembly system identifies clients by a C-terminal tripeptide. Proc. Natl Acad. Sci. USA 120, e2311057120 (2023).
Vaccaro, F. A. & Drennan, C. L. The role of nucleoside triphosphate hydrolase metallochaperones in making metalloenzymes. Metallomics 14, mfac030 (2022).
Goodsell, D. S. & Olsen, A. J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 29, 105–153 (2000).
Inglis, A. J., Page, K. R., Guna, A. & Voorhees, R. M. Differential modes of orphan subunit recognition for the WRB/CAML complex. Cell Rep. 30, 3691–3698 (2020).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Penczek, P. A. et al. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy 140, 9–19 (2014).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Moriya, T. et al. High-resolution single particle analysis from electron cryo-microscopy images using SPHIRE. J. Vis. Exp. 123, 55448 (2017).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. elife 7, e42166 (2018).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Goddard, T. D. et al. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Morin, A. et al. Collaboration gets the most out of software. elife 2, e01456 (2013).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Acknowledgements
We would like to thank Dr. Gyunghoon “Kenny” Kang for data processing advice and Dr. Patricia Feliciano for advice on model building. We would also like to thank Gerardo Perez Goncalvez, and Naike Ye, for help with mass photometry experiments and data processing, and providing SUMO protease for experiments, respectively. This work was supported by NIH grant GM126982 to C.L.D., NIH grant GM121673 to D.L.P.; NSF graduate research fellowships DGE-1247312 to M.D.M and DGE-1745302 to S.V.; a Howard Hughes Medical Institute Gilliam Fellowship to S.V.; and NIH T32 Training Grant 5T32GM007287 (S.V.) awarded to the MIT Biology Department. C.L.D is a Howard Hughes Medical Institute Investigator and Senior Fellow for the Canadian Institute for Advanced Research Bio-Inspired Solar Energy program. Sample optimization was carried out at the Brandeis University Cryo-EM facility. Cryo-EM data were collected on a Titan Krios at the UMass Medical School Worcester Cryo-EM Facility with the help of Dr. Kangkang Song and Dr. Chen Xu. Specimens prepared on the chameleon were imaged at the Automated Cryogenic Electron Microscopy Facility in MIT.nano on a Talos Arctica microscope with the help of Christopher Borsa, which was a gift from the Arnold and Mabel Beckman Foundation. The chameleon was purchased by C.L.D. using funds from the HHMI Transformative Technology 2019 Award.
Author information
Authors and Affiliations
Contributions
M.D.M., A.V. and S.V. purified ScMet18 for the cryo-EM experiments. S.V. carried out all the cryo-EM structural work, with the exception of the initial cryo-EM grid preparations, which were performed by EJB. M.D.M., S.K. and C.P. purified proteins for the pulldown assay, and performed pulldown assays. M.D.M. and S.V. conducted mass photometry experiments; S.V. analyzed the data. Structural studies were carried out under the supervision of CLD and biochemical analyses under the supervision of DLP. S.V. and C.L.D. wrote the manuscript with input from MDM and DLP and the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Ingrid Span and Tobias Goris.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vasquez, S., Marquez, M.D., Brignole, E.J. et al. Structural and biochemical investigations of a HEAT-repeat protein involved in the cytosolic iron-sulfur cluster assembly pathway. Commun Biol 6, 1276 (2023). https://doi.org/10.1038/s42003-023-05579-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05579-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
