
Abstract
The branchpoint (BP) motif is an essential intronic element for spliceosomal pre-mRNA splicing. In mammals, its sequence composition, distance to the downstream exon, and number of BPs per 3´ splice site are highly variable, unlike the GT/AG dinucleotides at the intron ends. These variations appear to provide evolutionary advantages for fostering alternative splicing, satisfying more diverse cellular contexts, and promoting resilience to genetic changes, thus contributing to an extra layer of complexity for gene regulation. Importantly, variants in the BP motif itself or in genes encoding BP-interacting factors cause human genetic diseases or cancers, highlighting the critical function of BP motif and the need to precisely identify functional BPs for faithful interpretation of their roles in splicing. In this perspective, we will succinctly summarize the major findings related to BP motif variations, discuss the relevant issues/challenges, and provide our insights.
Similar content being viewed by others
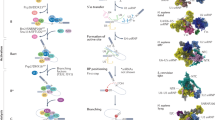


Introduction
Of the essential intronic sequences for splicing, the branchpoint (BP) motif perhaps has the most variations in terms of sequence composition, distance to the downstream exon, and the number of BPs per 3′ splice site (Fig. 1). Failure of proper BP recognition due to genetic variants of the motif itself or the trans-acting factors that recognize it causes human genetic diseases or cancer1,2,3. However, accurately predicting, validating, and interpreting the functional BP has been complicated by the variations of the BP motifs.

The BP location (up to 400nt) from the intron end and number of BPs (three BPs are shown as an example with details for the BP2 chosen for lariat formation here) per 3′ splice site are depicted. The SF3B1 (horseshoe) is shown in the pre-A complex (oval) before clamping on the U2-BP motif helix during PRP5-facilitated BP proofreading. Green boxes: exons.
In spliceosomal RNA splicing, the BP is mostly an adenosine1, which is located largely within 10–60nt from the downstream exon4,5, with the peak position at about 25nt in humans4,5,6. Initially, it is bound by the branchpoint binding protein (BBP, also known as splicing factor 1, SF1), after the U1 snRNP binding the 5′ splice site and the U2AF35/65 (U2AF1/U2AF2) heterodimer to the 3′ AG and polypyrimidine tract7. Then, facilitated by the ATP-dependent DEAD box helicase UAP56/DDX39B8, BBP/SF1 is displaced by SF3B1 from the U2 snRNP9, recruited via their interactions with the U2AF65/U2AF28,9,10. As this shift occurs, the U2 snRNA forms base pairs with the branchpoint motif, which is further stabilized as SF3B1 wraps around the BP region. The process is also coordinately regulated by the BP upstream anchoring site11, cis-acting splicing enhancers and silencers and trans-acting factors like serine/arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNP)12,13. These stepwise processes collectively control the 3′ splice site recognition and regulate alternative pre-mRNA splicing.
The BP motif plays an essential role by attacking the first nucleotide of the intron (the 5′ guanine) to form a 2′, 5′-phosphodiester bond, resulting in a lariat intermediate5,14,15. For this to occur, the BP adenosine needs to be bulged from the helix formed by the BP motif and U2 snRNA. Yeast Hsh155 (SF3B1 in humans) interacts and eventually clamps the helix through its HEAT domain, converting the pre-A to the A complex during spliceosome assembly16,17. This transesterification reaction is conserved from yeast to humans but the BP motif sequence, location, and particularly the number of BPs per 3′ splice site may vary among different introns and species.
Here we will briefly summarize the variations of the BP motifs in biology and diseases, related issues in BP identification, and provide our views to address the issues in future BP studies.
BP motif variations, advantages, diseases, and challenges
BP motif variations
The variations of the BP motifs appear to increase on an evolutional scale. For instance, in fungi and protists, variation of the BP motif is relatively less than that in metazoans particularly humans18, as revealed by massive lariat RNA sequencing (RNA-Seq), iCLIP-Seq (individual-nucleotide resolution UV crosslinking and immunoprecipitation - RNA sequencing), and/or consensus motif prediction1,4,5,14,18,19,20. Unicellular yeasts have a consensus motif TACTAAC (underlined: BP adenosine), multicellular fungi Ascomycetes CT(G/A)AC, and humans YTNAY (Y: pyrimidine C or T; N: any nucleotide)1,18,21, with increasing sequence degeneration, although TACTAAC remains the preferred motif in humans22. In mammalian cells, the BP adenosine can be replaced by cytidine or guanosine in some introns1,19,23.
The locations of BP by its distance from the downstream exon may vary as well. The location is relatively fixed in some yeasts or protists such as Y. lipolytica and B. microti strain RI (six nucleotides upstream, A-6) and E. invadens strain IP1 (eight nucleotides upstream, A-8)18,24. However, in other species, it is widely distributed within 10–100 nt upstream from the 3′ end of the intron25. The variations in humans can be even larger; for instance, 838 possible branchpoints were detected at up to 400nt away from their downstream exons25, with long U-rich polypyrimidine tracts and an AG-exclusion zone (AGEZ) ≥100 nt upstream from the 3′ AG. The distant BP and related 3′ splice site arrangement likely play a role in the mutually exclusive alternative splicing25,26,27.
The number of BPs per 3′ splice site may also exhibit variation. For example, the upstream BP of the alpha-tropomyosin exon 3 was mapped to A-175 by primer extension but its mutation abolished only 50% of exon 3 inclusion, suggesting that other BPs used for the same 3′ AG and downstream exon inclusion25. Indeed, A-182 together with A-175 mutation abolished all exon 3 inclusion25. In fact, multiple BPs that could result in the use of the same 3′ splice site seem to be more prevalent (>60%) for human introns as revealed by lariat RNA sequencing1,19.
Thus, the branchpoint motif has evolved from a relatively fixed sequence of TACTAAC in unicellular yeast to the divergent YTNAY in humans. The location of branchpoints varies from relatively fixed positions (A-6 and A-8) in some protists to a wide range of locations (A-10 to A-400) upstream of the intron end. Moreover, the number of BPs for a 3′ splice site has evolved from one in yeasts and protists to mostly more than one BP in humans.
Besides the above variations in the regular splicing that joins exons into mature RNA, a suboptimal BP consensus motif CTNA with more distant BPs (A-29) is found in the recursive splicing of zero-nucleotide exons in long introns, in contrast to the consensus CTAAT with A-14–-26 in shorter introns in the Drosophila genome28,29,30.
The advantages of BP motif variations
Despite the evolutionary changes of the BP motifs, the majority of the U2 snRNA genes in fungi contain a GTAG motif (GΨAG in the U2 snRNA, Ψ: pseudouridine) that is complementary to the CTAAC BP sequence18. Here the BP adenosine, sandwiched between its flanking U2-pairing nucleotides but itself without a complementary base in the U2 snRNA, protrudes from the helix, facilitated by the pseudouridine31. In humans, about 65% of the 172 human U2 snRNA sequences collected in the Rfam database (RF00004) also contain the GTAG motif within 50nt of their 5′ ends. Moreover, 75% of the entire 16,770 U2 snRNA sequences and 93% of the 208 representative ones in the database contain the GTAG motif in the potential BP-pairing region. Therefore, in humans and likely in many other species as well, a conserved GTAG motif but not a motif evolved to be perfectly complementary to the YTNAY is present in the majority of the U2 snRNA genes. This non-Watson-Crick-complementarity leads to wobble pairing between U2 and the BP motif, which may result in a less stable helix during the SF3B1-clamping and PRP5-facilitated proof-reading before the A complex formation, providing plasticity for the selection of alternative BPs and/or splice sites. Consistently, BP motif sequences non-complementary to the U2 GTAG motif are more associated with alternative splicing18,32. Moreover, different BP locations also regulate alternative splicing33. Similarly, in recursive splicing, which requires (skipped) cryptic exons following its 3′AG34,35, the suboptimal BP motif and more distant BP, though apparently needed for the zero-nucleotide exon splicing like the 5′ splice sites36, may also facilitate the cryptic exon skipping. Therefore, the BP motif sequence and location variations likely contribute to alternative or cryptic splicing, a feature that could also increase transcriptome and proteome diversity. Furthermore, having multi-BPs per 3′ splice site for different tissues or developmental stages may not only suit the more complex cellular context of BP recognition but also help avoid detrimental effects of BP sequence changes on splice site choice for more resilience to genetic variations in evolution as pointed out previously19.
The BP motif variants that cause human diseases
Despite the biological advantages, a disastrous consequence caused by some BP motif variants is human disease. For example, a BP motif variant in the MSH2 gene causes skipping of exon 16 and results in inherited cancer37, or in the KCNH2 gene causes partial retention of intron 9 and the Long QT syndrome1,38. Moreover, mutations in BP trans-acting factor genes such as SF3B1 or U2 snRNA3 drive cancer development2,3. Thus, BP motif variations and their recognition by trans-acting factors have a critical threshold for proper BP function; beyond which the lariat of critical introns/genes will not form for splicing to occur. However, only 40 pathogenic BP motif variants have been found to cause splicing defects and genetic diseases in the last decades1, which is surprisingly low given the exceedingly large number of BPs in the genome. Considering the causative effects of BP dysfunction in diseases, understanding the role of BP motif variants and elucidating their functional impact are essential in dissecting RNA splicing regulation in the physiological and pathophysiological settings. It will also be important for therapeutic applications with small molecules targeting the BP binding proteins such as SF3B1 by spliceostatin A and its analogs or with synthetic lethal genes39,40.
Challenges of prediction, validation, and interpretation of functional BP motifs
Prediction of functional BPs for splicing and experimental validation of BP motif variants involving in disease have been challenging despite the conclusion reported in the original studies. BP prediction software such as BPP, LaBranchoR, or Branchpointer has an AUC (area under the true-versus the false-positive curve) ranging from 0.591 to 0.82 as measured by an independent bioinformatics test41. Unexpectedly, when selected Branchpointer-predicted BP motifs were mutated in mini-gene splicing reporters, splicing was not substantially disrupted as assayed by RT-PCR; instead, novel and unpredicted BPs were used as detected by lariat sequencing42. A more recent software BPHunter has integrated experimentally identified and computationally predicted BP data, reporting a much higher success rate that approaches 100% and have identified novel BP motif variations1. However, the number of tested BPs in that report is small (n = 40 BPs) and its false-positive and false-negative rates are not measured.
The imperfect bioinformatics prediction and the lack of most BP information from the exome sequencing approach in detecting BP motif variations are thought to contribute to the surprisingly low rate of pathogenic BP motif variants reported so far1. Contributions by alternative BP motifs have not been systematically confirmed in experiments.
In addition to the prediction issues, the indirect BP detection method by mutation/splicing reporter assays of spliced mRNA could easily miss the functional BP at a multi-BP-containing splice site, where different BPs could be used for the same 3′ splice site19,27. Here the production of a spliced mRNA only indicates there is at least a functional BP present in the intron, but it does not equate finding the functional BP used for the splicing. Moreover, the mini-gene splicing reporters may not represent the endogenous gene’s BP usage due to the deviation from its native sequence and the chromatin contexts. Additionally, the problem of using different cell lines from the original reports that may have different cell-specific splicing regulators, further complicates the interpretation.
Moreover, the consensus motif of the cryptic (alternative) BP used in SF3B1 mutant cells is more like the consensus of the insensitive than the canonical BP33. The formers contain a higher frequency of purines (YTRAY) while the latter has more enriched C (YTCAY) at the BP-1 positions. How the cryptic BP motifs are chosen and why so many splice sites remain unaffected in the presence of a SF3B1 mutation in these cells remain elusive.
Furthermore, the molecular mechanisms of the effects of deep intron single nucleotide polymorphism (SNP) variants on human traits or diseases remain to be fully understood. Unraveling the impact of these genetic variants on splicing, especially the BP motifs, would enable us to identify the root causes of these diseases, ultimately facilitating the development of therapeutic interventions.
Lastly, RNA m6A modification at splice sites is emerging as a new layer of pre-mRNA splicing regulator. Splice site m6A methylation prevents binding of U2AF35/U2AF1 to inhibit RNA splicing43. Modification of U2 snRNA and U6 snRNA have also been shown to directly impact mRNA splicing44,45. SF3B1 protein expression can be regulated via ubiquitination, and is also phosphorylated by CDK11 during spliceosome transition from the B to Bact complex to control pre-mRNA splicing46,47. The BP upstream anchoring site and epigenetic factors among others also control or regulate splicing11,48. We can speculate that these modifications all could change BP selection to influence splicing, but little is known so far.
Perspectives about the challenges and issues
Based on the above BP motif variations and related issues, we suggest the following points to consider in studying functional BPs in splicing:
(1) Direct BP detection methods should be used after BP motif prediction (Fig. 2), such as lariat RT-PCR49, lariat sequencing5,21, primer extension15, or RNase protection assay50, instead of relying on detecting the spliced RNA alone. The direct detection eliminates false-negative results arising from alternative BP usage when the effects of BP motif variants are examined by spliced RNA only.

Each method is briefly described, together with a diagrammed lariat still attached to the downstream exon (green) before the 2nd transesterification. Oval: BP-interacting protein-RNA complex. Arrows: primers, dotted lines: sequence extension in reverse transcription (RT).
(2) Points to consider regarding discrepancies in detecting and validating BPs from transcriptome-wide lariat sequencing: (a) potential differences in multiple cellular contexts used; (b) indirect test of BP usage by using RT-PCR to detect spliced exons; (c) potential differences in BP usage in mini-genes versus endogenous genes; (d) hidden mutations affecting BP usage that may be acquired by the cancer cell during evolving genomic instability. These considerations are particularly important due to the temporal or spatial alternative BP usage in multi-BP introns19.
(3) The low rate of pathogenic BPs reported so far perhaps are also attributed by the alternative BP usage for the same exons, besides the prediction/detection issues. This is consistent with the observation that a smaller proportion of pathogenic but not other variants have been identified in the multi-BP than single-BP splice sites1. The candidate BPs can be experimentally tested by direct BP detection together with BP mutagenesis/splicing assays. It will be instrumental for identifying candidate BPs by clinical geneticists if an online genome-wide database of annotated alternative BPs of different tissues or developmental stages is available.
(4) The BP motif variations may also help explain how the cryptic BPs are chosen while many unaffected splice sites are also found in splicing factor mutants such as SF3B1 in cancer cells. Some mutated amino acid residues in SF3B1 add negative charges (e.g. the hotspot mutation K700E), causing conformational changes or affecting the interaction with SUGP151,52,53,54. This likely causes failure for the HEAT domain to clamp some BP motif-U2 snRNA helixes during PRP5-facilitated proofreading16. Particularly affected should be the U2 low-affinity BP motifs, e.g. the CTCAC that is more prevalent at the affected splice sites33. These sites are switched in mutants to the cryptic sites that are more enriched with the U2 higher-affinity CTGAC6,33. However, the latter high-affinity motifs are generally not used in the wild type; perhaps this was due to the overall weakened 3′ splice sites by a nearby splicing silencer (e.g. G-tracts)6, longer distance to the 3′ AG, and/or shorter/weaker Py, besides their deleterious effect of disrupting the mRNA sequences if used. Even more affected should be those 3′ splice sites with both U2 low-affinity motifs and single BPs or those with multi-BP motifs nearby but without the AGEZ25. Moreover, for those unaffected splice sites, they may have at least one strong U2-high-affinity BP motif, and those with multi-BPs, the mutant SF3B1 may have chosen alternative, U2 higher-affinity BPs without changing the spliced mRNA. In the latter case, an unaffected splice site in the mutant is not necessarily using the same BP as in the normal cell.
(5) One aspect of the effect of deep intron variants/SNPs on BP usage and cryptic splicing could be through increasing the BP motif base-pairing with the U2 snRNA or interaction with the BP-interacting proteins to enhance cryptic 3′ splice site usage. A consequence could be cryptic exon usage leading to frame shift and non-sense mediated mRNA decay37, particularly the cryptic exons of recursive splicing, which is also found in vertebrates including humans34,55.
(6) Beyond the above approaches to BP prediction/identification, deep learning methods such as SpliceAI and BigRNA56,57, combine more genome as well as transcriptome features including SNP and associated RNA-seq reads to predict variant-associated cryptic or alternative splicing events. These predicted events provide targets with likely splicing consequences for identifying pathogenic BP motif variants facilitated by other software such as BPHunter1.
There are still several key questions that remain to be explored. Are there any new trans-acting factors or RNA binding proteins (RBP) interacting with BPs yet to be defined? Are there novel disease-relevant BPs yet to be discovered? What will be an effective way to identify any of them? How are the alternative BPs chosen by trans-acting factors in cells? How to use specific BPs for therapy without off-target effects by small molecules? With the new developments of CRISPR-based gene editing/screening, now we should be able to test with relative ease the direct involvement of any BP in splicing by mutating the BP motif in the genome or to discover novel RBP factors for alternative/cryptic BP usage.
We hope the insights will help researchers to recognize the significance of accurately identifying and validating functional BPs for the proper interpretation of their roles in RNA splicing. Additionally, we wish to draw attention to the crucial link between BP motif variations and splicing factor mutants (such as SF3B1) in diseases. We believe that this aspect warrants careful consideration by researchers investigating disease pathogenesis associated with these splicing factors, as well as BP biology in general.
References
Zhang, P. et al. Genome-wide detection of human variants that disrupt intronic branchpoints. Proc. Natl Acad. Sci. USA 119, e2211194119 (2022).
Yin, S. et al. A murine model of chronic lymphocytic leukemia based on b cell-restricted expression of Sf3b1 mutation and Atm deletion. Cancer Cell 35, 283–296 e285 (2019).
Bousquets-Munoz, P. et al. PanCancer analysis of somatic mutations in repetitive regions reveals recurrent mutations in snRNA U2. Npj Genom. Med. 7, 19 (2022).
Taggart, A. J. et al. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 27, 639–649 (2017).
Mercer, T. R. et al. Genome-wide discovery of human splicing branchpoints. Genome Res. 25, 290–303 (2015).
Nguyen, H. & Xie, J. Widespread separation of the polypyrimidine tract from 3′ AG by G tracts in association with alternative exons in metazoa and plants. Front. Genet. 9, 741 (2018).
Wilkinson, M. E., Charenton, C. & Nagai, K. RNA splicing by the spliceosome. Annu. Rev. Biochem. 89, 359–388 (2020).
Fleckner, J., Zhang, M., Valcarcel, J. & Green, M. R. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 11, 1864–1872 (1997).
Gozani, O., Potashkin, J. & Reed, R. A potential role for U2AF-SAP 155 interactions in recruiting U2 snRNP to the branch site. Mol. Cell. Biol. 18, 4752–4760 (1998).
Berglund, J. A., Abovich, N. & Rosbash, M. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branchpoint region recognition. Genes Dev. 12, 858–867 (1998).
Gozani, O., Feld, R. & Reed, R. Evidence that sequence-independent binding of highly conserved U2 snRNP proteins upstream of the branch site is required for assembly of spliceosomal complex A. Gene Dev. 10, 233–243 (1996).
Sohail, M. & Xie, J. Diverse regulation of 3′ splice site usage. Cell. Mol. Life Sci. 72, 4771–4793 (2015).
Dvinge, H. Regulation of alternative mRNA splicing: old players and new perspectives. FEBS Lett. 592, 2987–3006 (2018).
Briese, M. et al. A systems view of spliceosomal assembly and branchpoints with iCLIP. Nat. Struct. Mol. Biol. 26, 930–940 (2019).
Ruskin, B., Krainer, A. R., Maniatis, T. & Green, M. R. Excision of an intact intron as a novel lariat structure during pre-messenger rna splicing invitro. Cell 38, 317–331 (1984).
Zhang, Z. et al. Structural insights into how Prp5 proofreads the pre-mRNA branch site. Nature 596, 296–300 (2021).
Kennedy, S. D., Bauer, W. J., Wang, W. & Kielkopf, C. L. Dynamic stacking of an expected branch point adenosine in duplexes containing pseudouridine-modified or unmodified U2 snRNA sites. Biochem. Biophys. Res. Commun. 511, 416–421 (2019).
Nguyen, H., Das, U. & Xie, J. Y. Genome-wide evolution of wobble base-pairing nucleotides of branchpoint motifs with increasing organismal complexity. RNA Biol. 17, 311–324 (2020).
Pineda, J. M. B. & Bradley, R. K. Most human introns are recognized via multiple and tissue-specific branchpoints. Gene Dev. 32, 577–591 (2018).
Damianov, A et al. The apoptotic splicing regulators RBM5 and RBM10 are subunits of the U2 snRNP engaged with intron branch sites on chromatin. bioRxiv https://doi.org/10.1101/2023.09.21.558883 (2023).
Qin, D., Huang, L., Wlodaver, A., Andrade, J. & Staley, J. P. Sequencing of lariat termini in S. cerevisiae reveals 5′ splice sites, branch points, and novel splicing events. RNA 22, 237–253 (2016).
Zhuang, Y. A., Goldstein, A. M. & Weiner, A. M. UACUAAC is the preferred branch site for mammalian mRNA splicing. Proc. Natl Acad. Sci. USA 86, 2752–2756 (1989).
Saini, H., Bicknell, A. A., Eddy, S. R. & Moore, M. J. Free circular introns with an unusual branchpoint in neuronal projections. Elife 8, e47809 (2019).
Irimia, M. & Roy, S. W. Evolutionary convergence on highly-conserved 3′ intron structures in intron-poor eukaryotes and insights into the ancestral eukaryotic genome. PLoS Genet. 4, e1000148 (2008).
Gooding, C. et al. A class of human exons with predicted distant branch points revealed by analysis of AG dinucleotide exclusion zones. Genome Biol. 7, R1 (2006).
Goux-Pelletan, M. et al. In vitro splicing of mutually exclusive exons from the chicken beta-tropomyosin gene: role of the branch point location and very long pyrimidine stretch. EMBO J. 9, 241–249 (1990).
Smith, C. W. J. & Nadalginard, B. Mutually exclusive splicing of alpha-tropomyosin exons enforced by an unusual lariat branch point location - implications for constitutive splicing. Cell 56, 749–758 (1989).
Duff, M. O. et al. Genome-wide identification of zero nucleotide recursive splicing in Drosophila. Nature 521, 376–379 (2015).
Burnette, J. M., Miyamoto-Sato, E., Schaub, M. A., Conklin, J. & Lopez, A. J. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics 170, 661–674 (2005).
Hatton, A. R., Subramaniam, V. & Lopez, A. J. Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon-exon junctions. Mol. Cell 2, 787–796 (1998).
Newby, M. I. & Greenbaum, N. L. Sculpting of the spliceosomal branch site recognition motif by a conserved pseudouridine. Nat. Struct. Biol. 9, 958–965 (2002).
Corvelo, A., Hallegger, M., Smith, C. W. & Eyras, E. Genome-wide association between branch point properties and alternative splicing. PLoS Comput. Biol. 6, e1001016 (2010).
Alsafadi, S. et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 7, 10615 (2016).
Sibley, C. R. et al. Recursive splicing in long vertebrate genes. Nature 521, 371–375 (2015).
Joseph, B., Kondo, S. & Lai, E. C. Short cryptic exons mediate recursive splicing in Drosophila. Nat. Struct. Mol. Biol. 25, 365–371 (2018).
Joseph, B., Scala, C., Kondo, S. & Lai, E. C. Molecular and genetic dissection of recursive splicing. Life Sci. Alliance 5, e202101063 (2022).
Casadei, S. et al. Characterization of splice-altering mutations in inherited predisposition to cancer. Proc. Natl Acad. Sci. USA 116, 26798–26807 (2019).
Crotti, L. et al. A KCNH2 branch point mutation causing aberrant splicing contributes to an explanation of genotype-negative long QT syndrome. Heart Rhythm 6, 212–218 (2009).
Stanley, R. F. & Abdel-Wahab, O. Dysregulation and therapeutic targeting of RNA splicing in cancer. Nat. Cancer 3, 536–546 (2022).
North, K. et al. Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat. Biotechnol. 40, 1103–1113 (2022).
Leman, R. et al. Assessment of branch point prediction tools to predict physiological branch points and their alteration by variants. BMC Genomics 21, 86 (2020).
Canson, D. M. et al. The splicing effect of variants at branchpoint elements in cancer genes. Genet. Med. 24, 398–409 (2022).
Mendel, M. et al. Splice site m(6)A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell 184, 3125–3142.e3125 (2021).
Ishigami, Y., Ohira, T., Isokawa, Y., Suzuki, Y. & Suzuki, T. A single m(6)A modification in U6 snRNA diversifies exon sequence at the 5′ splice site. Nat. Commun. 12, 3244 (2021).
Goh, Y. T., Koh, C. W. Q., Sim, D. Y., Roca, X. & Goh, W. S. S. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 48, 9250–9261 (2020).
Hluchy, M. et al. CDK11 regulates pre-mRNA splicing by phosphorylation of SF3B1. Nature 609, 829–834 (2022).
Han, C. J. et al. SF3B1 homeostasis is critical for survival and therapeutic response in T cell leukemia. Sci. Adv. 8, eabj8357 (2022).
Zhang, J., Zhang, Y. Z., Jiang, J. & Duan, C. G. The crosstalk between epigenetic mechanisms and alternative RNA processing regulation. Front. Genet. 11, 998 (2020).
Suzuki, H. et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 34, e63 (2006).
Vogel, J., Hess, W. R. & Borner, T. Precise branch point mapping and quantification of splicing intermediates. Nucleic Acids Res. 25, 2030–2031 (1997).
Zhang, J. et al. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol. Cell 76, 82–95 (2019).
Canbezdi, C. et al. Functional and conformational impact of cancer-associated SF3B1 mutations depends on the position and the charge of amino acid substitution. Comput. Struct. Biotechnol. J. 19, 1361–1370 (2021).
Maji, D., Grossfield, A. & Kielkopf, C. L. Structures of SF3b1 reveal a dynamic Achilles heel of spliceosome assembly: Implications for cancer-associated abnormalities and drug discovery. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 194440 (2019).
Cretu, C. et al. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol. Cell 64, 307–319 (2016).
Hoppe, E. R., Udy, D. B. & Bradley, R. K. Recursive splicing discovery using lariats in total RNA sequencing. Life Sci. Alliance 6, e202201889 (2023).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e524 (2019).
Celaj, A. et al. An RNA foundation model enables discovery of disease mechanisms and candidate therapeutics. bioRxiv https://doi.org/10.1101/2023.09.20.558508 (2023).
Acknowledgements
We thank Yanzhong Yang of City of Hope and Andrey Damianov of UCLA for critical comments and suggestions. J.X. would like to thank the discovery grant support by the NSERC RGPIN-2022-05023 and the visiting professorship fund by Morgan Chu Endowment to City of Hope. L.W. acknowledges support from NCI (R01CA240910, R01CA282450).
Author information
Authors and Affiliations
Contributions
JX, LW, and RJL discussed, analyzed the literature, and wrote the manuscript together.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests. Jiuyong Xie is a Morgan Chu Visiting Professor at the Center for RNA Biology & Therapeutics, Beckman Research Institute at the City of Hope National Medical Center.
Peer review
Peer review information
Communications Biology thanks Juan Valcárcel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Manuel Breuer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Morgan Chu Visiting Professor at the Irell and Manella Graduate School and the Center for RNA Biology & Therapeutics, Beckman Research Institute, City of Hope National Medical Center, Duarte, CA 91010, USA
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, J., Wang, L. & Lin, RJ. Variations of intronic branchpoint motif: identification and functional implications in splicing and disease. Commun Biol 6, 1142 (2023). https://doi.org/10.1038/s42003-023-05513-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05513-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
