
Abstract
The helper CD4+ T cell-type 17 (Th17) cells and regulatory CD4+ T cells (Tregs) are balanced through numerous molecular regulators, particularly metabolic factors, and their alteration causes immune dysregulation. Herein, we report that peroxisome proliferator of activated receptor-alpha (Pparα), a lipid metabolism regulator, suppresses Th17 differentiation. We demonstrated that Pparα ablation improves Th17 and pro-Th17 factor HIF-1α by enhancing the expression and nuclear localization of NFκB-activator IκB kinase-alpha (IKKα). Unexpectedly, we found that IKKα directly interacts with RORγt and enhances the expression of Il17a gene. Meanwhile, IKKα also interacts with Foxp3, leading to the post-translational regulation of Foxp3 by elevating its proteasomal degradation, and influencing Th17 development. Pparα deficiency leads to enhanced Th17 development in vivo and is associated with enhanced pathology in a murine experimental autoimmune encephalomyelitis (EAE) model. Overall, our data indicate that Pparα may serve as a potential therapeutic target for autoimmune and inflammatory diseases.
Similar content being viewed by others


Introduction
Naïve CD4+ T cells develop into highly specialized T helper (Th) subsets when activated in the presence of lineage-associated cytokines1. Among these Th subsets, Th17 cells lineage is characterized by the expression of the transcription factor retinoic-acid related orphan receptor-γt (RORγt) and effector cytokines interleukin-17A (IL-17A) and IL-17F2. Meanwhile, Tregs are characterized by the expression of the transcription factor, forkhead-box P3 (Foxp3), and suppressive capacity of immune responses3. While performing the opposing functions, both Th17 and iTreg lineages share elements of their differentiation pathways, including the cytokine TGF-β and the initial expression of Foxp3 in the early stages of development4,5,6. Understanding the molecular mechanisms responsible for differentiating T cells towards one or the other fate is crucial as these mechanisms are highly associated with immune dysregulation and the onset of multiple diseases, including cancer and autoimmune diseases.
A growing body of work has revealed that, in addition to cytokines, metabolic factors also play an important part in determining the fate of naïve CD4+ T cells upon differentiation7,8. For instance, the ability of naïve T cells to uptake and utilize glucose is critical for the transition from naïve T cell precursors to effector T cells which are capable of rapid growth and production of effector molecules7,9. Similarly, molecular mechanism underlying the glucose uptake in T cells indicates that the expression level of Glut1, a plasma membrane glucose transporter in hematopoietic cells, is at lower levels in naïve T cells and is enhanced upon activation of T cells10,11. This increased expression of Glut1 and glucose uptake in activated T cells depends on activating the PI3K/Akt signaling pathway. Meanwhile, activation of PI3K–Akt pathway also triggers the mTOR, the mechanistic target of rapamycin, to facilitate the glycolytic metabolism for cell proliferation and growth7,12.
Similarly, HIF-1α, a transcription factor responding to hypoxia, regulates glycolysis to influence glucose uptake13,14. It has been investigated that mTOR regulates the HIF-1α to mediate the T cell metabolism. Likewise, the absence of HIF-1α in CD8 and Th17 cells mitigates the expression of metabolic genes, including Glut114,15,16,17. Chemical or genetic disruption of glucose metabolism and their associated pathways converts newly activated T cells from pro-inflammatory effector T cell fates (Th1, Th2, Th17) to an induced (i)Treg-fate18,19,20, which preferentially rely on mitochondrial respiration and lipid oxidation to meet their distinct metabolic needs18,20,21. Tregs have been identified to rely on mitochondrial lipid oxidation for their growth and expansion primarily due to the activation of AMP-activated kinase (AMPK), which is critical for metabolic processes and exerts opposing effects to that of mTOR20. Meanwhile, AMPK regulates T cell immunity by serving important roles in regulating fatty acid oxidation (FAO) and inhibiting de novo fatty acid (FA) production22. Peroxisome proliferator of activated receptor (Ppar) encompasses an important family of molecules that consists Pparα, Pparg, and Pparδ, and is known to play various roles in lipid metabolism, FAO, and immune regulation23. Previous studies found that PPARα agonist selectively suppresses the Th17 development during the progression of autoimmune diseases24,25. Similarly, Pparα agonist can ameliorate the severity of EAE in mice by suppressing Th1 and Th17 development26. Pparα was also recently found critical for a specialized subpopulation of adipose tissue-resident Tregs with a remarkable influence on metabolism27. Despite the apparent importance of Pparα across various immune cells, the role played by these enzymes in T cell differentiation is incompletely understood.
Among PPAR family, Pparα, a well-known fatty acid modulator, regulates the T cell immune responses, including the onset of T cell-mediated autoimmune diseases in a gender-specific manner24,28,29. Additionally, the role of Pparα has been suggested as an anti-inflammatory mediator in several immune cell types24,30,31,32,33. However, the precise contribution of Pparα in the differentiation of naïve CD4+ T cells to specific T effector lineage with pro- or anti-inflammatory potential remains unknown. Therefore, suspecting the rigorous role of Pparα in determining the effector T cell fate, we designed this study to investigate the mechanism underlying the PPARα-dependent differentiation of naïve CD4+ T cells to specific T effector cells. Briefly, we investigated that the upregulation of Pparα in activated CD4+ T cells inhibits the expression of IKKα. Meanwhile, pharmacological inhibition of Pparα leads to the IKKα-dependent degradation of Foxp3 and elevation of IL17a in an IKKα/RORγt-dependent manner.
Results
Genetic ablation of Pparα triggers Th17 development
In line with the potential role of PPARα in T cell fate determination, we first investigated the expression of Pparα in different subsets of CD4+ Th cells. Our results indicated that the expression of Pparα was up-regulated in different CD4+ Th cells upon αCD3/αCD28-dependent activation of naïve CD4+ T cells (Fig. 1a). To further validate the effect of Pparα on the population of immune cell subsets, we collected lymphocytes from the spleen and thymus, and evaluated the population of different immune cells. It was determined that deficiency of Pparα didn’t change the population of CD11b+ Gr1+ myeloid-derived suppressor cells, CD11b+ CD11c+ dendritic cells, CD19+ B cells, CD8+ T cells and NK1.1+ natural killer cells (Supplementary Fig. 1a, b). We then investigated the effects of Pparα deficiency on naïve T cell proliferation and acquisition of effector cytokine expression. Naïve CD4+ T cells from WT and Pparα KO mice cultured under non-polarizing (Th0) conditions showed comparable proliferation by CFSE dilution (Supplementary Fig. 1c). Interferon-γ (IFN-γ) expression by these WT and Pparα KO cells was also comparable, while IL-2 production did appear enhanced in the absence of Pparα (Supplementary Fig. 1c). To further assess the effects of Pparα on Th differentiation, we activated naïve CD4+ T cells from WT and Pparα KO mice under different Th subset-generating conditions in vitro. Pparα KO CD4+ T cells displayed increased IL-17A expression under the Th17 polarizing condition than WT CD4+ T cells. In contrast, the development of Th1 and Th2 subsets remains intact in Pparα KO mice (Fig. 1b). Enhanced Th17 development in CD4+ T cells isolated from Pparα KO was associated with increased transcripts levels of Il17a and Il17f (Fig. 1d). These data suggest that Pparα plays a specific role in dampening Th17 differentiation.

a Differential transcript levels of PPAR subtypes in different Th subsets. Ppara (left) Pparg (middle) Pparδ (right) expression after 24 h of skewing of naïve CD4 + T cells (CD4+ CD25- CD62Lhi) under Th0, Th1, Th2, Th17 and Treg polarizing conditions. b Representative flow-plots of IFN-γ, IL-13, IL-17A, and Foxp3 expression in naïve CD4+ T cells isolated from wild-type (WT) and Ppara knock-out (KO) mice polarized for 72 h. Naïve CD4+ T cells from each group were cultured under different polarizing conditions as described in the method section. c Representative flow plots of IL-17A and Foxp3 of CD4+ T cells after 72 h of Th17 polarization under varying concentrations of gemfibrozil (top) and fenofibrate (bottom). d Il17a (left) and Il17f (right) transcript levels after 72 h of Th17 polarization of WT and Pparα KO CD4+ T cells. e CD4+ T cells from WT and Pparα KO mice were cultured under Th17 polarizing conditions for 24 h along with varying concentrations of gemfibrozil (left) and fenofibrate (right), and Il17a transcript levels were assessed by qRT-PCR. Results are shown as the means ± SEM representative of five independent experiments. Unpaired student t-test were used for statistical analysis representing *P < 0.05, **P < 0.01, and ***P < 0.001.
Pharmacological activation of Pparα reduces the Th17 development
The activation of Pparα requires the binding of ligands to modulate gene transcription34. Known natural ligands for Pparα include Leukotriene B (LTB) and unsaturated fatty acids35. Synthetic ligands, such as fenofibrate and gemfibrozil, often used as anti-diabetic drugs to modulate triglyceride levels in diabetic patients, can activate Pparα36. Evidence indicated that fenofibrate inhibited cellular proliferation; however, gemfibrozil, which had similar effects on IL-17 up-regulation, did not affect the proliferation. To further strengthen the effect of Pparα on Th17 cell development, we investigated whether synthetic Pparα ligands can modulate the Th17 development. Naïve CD4+ T cells from WT mice were activated under Th17 polarizing conditions in the presence of gemfibrozil and fenofibrate. After 72 h, the flow cytometric data indicated that the level of Il17a expression decreased in a dose-dependent manner (Fig. 1c). However, both gemfibrozil and fenofibrate didn’t affect the expression of Foxp3 (Fig. 1c). Furthermore, Il17a transcript levels in developing Th17 cells from WT mice were decreased by varying concentrations of gemfibrozil and fenofibrate (Fig. 1e), demonstrating transcriptional regulation of Il17a by Pparα. As expected, Pparα KO Th17 cells expressed higher levels of Il17a compared to WT Th17 cells, and the higher level of Il17a expression was maintained under various doses of gemfibrozil and fenofibrate. This data suggests that pharmacological targeting of Paprα through gemfibrozil and fenofibrate inhibits the development of Th17 cells.
Loss of Pparα assists the formation of IKKα/RORγt and IKKα/Foxp3 complexes to modulate the expression of Il17a
To investigate the molecular basis of Pparα-dependent suppression of Th17 development, we analyzed known regulators of Th17 development. It is known that phosphorylation of signal transducer and activator of transcription (STAT3), downstream of IL-6 signaling, is critical for Th17 development15,37. Pparα KO CD4+ T cells showed comparable STAT3 phosphorylation and Il17a levels compared to WT CD4 + T cells (Supplementary Fig. 2a, b). This lack of effect on STAT3 activation contrasts with the results of Pparα KO38. Expression of RORγt (gene name: Rorc) and RORα (gene name: Rora), master regulators of the Th17 lineage2,39, was also assessed in CD4+ T cells from WT and Pparα KO mice. Interestingly, despite the elevated IL-17 production seen in KO-derived T cells, we observed a slight increase in the expression of RORγt, while the expression of RORc was not affected in the CD4+ cells isolated from the Pparα KO mice (Fig. 2a). Also, in line with our previous findings15, hypoxia-inducible factor-1 (HIF-1α, gene name: Hif1α) expression was higher in the enhanced Th17 population lacking Ppara (Fig. 2a). Since HIF-1 levels can be stabilized, even under normoxic conditions, by TCR signaling-dependent transcriptional activation and NFKB activity, it is possible that the enhanced Hif1α level we found could be the result of enhanced NFKB signaling in the absence of PPARα, and that the enhanced IL-17 could be due to Hif1α elevation. Supporting this, we found an elevated NFKB activity in the absence of Pparα, which did not result from elevated Hif1α levels. We found increased expression of Chuk, conserved helix-loop-helix ubiquitous kinase, which is also known as an inhibitor of NFKB kinase subunit alpha (IKKα), after the loss of Pparα (Fig. 2a, b).

a Naïve CD4+ T cells (CD4+ CD25- CD62Lhi) from WT and Pparα KO mice were cultured under Th17 polarizing condition for 72 h, and Rorc, Rora, Hif1a, and Chuk transcript levels were assessed by qRT-PCR. b Naïve CD4+ T cells were isolated from WT and Pparα KO mice and cultured under Th17 polarizing conditions for 72 h. IKKα expression in naïve CD4+ T cells (left) or Th17 cells (right) from WT and Pparα KO mice was assessed by western blot. c Naïve CD4+ T cells from WT and Ppara KO mice were polarized under Th17 condition for 72 h and immunofluorescence staining was performed as described in the method section, and confocal imaging was acquired. IKKα alone (left); nucleus (middle), and merged view (right). d Naïve CD4+ T cells from WT and Pparα KO mice were polarized under Th17 condition for 72 h, and chromatin immunoprecipitation (ChIP) assay was performed by utilizing isotype-match IgG (grey bars), anti-IKKα (red bars), anti-RORγt (blue bars), and anti-phospho-histone H3 (yellow bars) antibodies. qRT-PCR was performed utilizing primers flanking putative RORγt binding region or non-binding region as described in the method section. e 293 T cells were transfected with Myc-tagged RORγt expression plasmids (Myc-RORγt), HA-tagged IKKα expression plasmids (HA-IKKα), or both. Co-immunoprecipitation assay was performed by utilizing anti-Myc antibody (IP: Myc) on cell lysates, and the western-blot of either HA or Myc is shown (IB). The input amount is also shown in the bottom. Results are shown as the means ± SEM of five independent trials except for picture b, where results are shown as the means ± SEM of three independent trials. Unpaired student t-tests were used for statistical analysis representing *P < 0.05, and **P < 0.01.
Considering the role of IKKα in mediating the Il17a phenotype40, we determined the protein expression of IKKα in the naïve CD4+ T cells and Th17 cells. We found that Pparα-/- substantially increased the protein expression of IKKα in both naïve T and Th17 cells (Fig. 2b; Supplementary Fig. 5). Furthermore, given that IKKα can translocate into the nucleus41, we tested whether enhanced IKKα expression levels in Pparα KO Th17 cells correlate with its nuclear localization. Naïve CD4+ T cells were isolated from WT and Pparα KO mice and cultured under Th17 polarizing conditions for 72 h. Immunofluorescence assay indicates a strong perinuclear localization of IKKα in the WT Th17 cells (Fig. 2c, top). In contrast, IKKα expression was uniformly distributed throughout the center of the nucleus in Pparα KO Th17 cells (Fig. 2c, bottom), suggesting its physical proximity to active chromatins. Furthermore, the chromatin immunoprecipitation (ChIP) assay determined that RORγt binding to its well-known Il17a regulatory region was comparable in both WT and Pparα KO Th17 cells (Fig. 2d, blue bars). Still, IKKα binding to the same regulatory region was enhanced in Th17 cells from Pparα KO mice (Fig. 2d, red bars). Meanwhile, enhanced phosphorylation of histone H3 H3Ser10 was associated with increased IKKα binding in Th17 cells from Pparα KO compared to WT mice (Fig. 2d, yellow bar). To further strengthen the epigenetic modification of RORγt in Th17 cells from Pparα KO mice, we carried assay to determine H3K27ac enrichment at the RORγt. The results indicated that H3K27ac enrichment was significantly increased at RORγt binding region in Th17 cells from Pparα KO mice (Supplementary Fig. 3a). This supported our findings suggesting the enhanced nuclear localization of IKKα in Pparα-/-. During Il17a transcriptional regulation, RORγt can be associated with various transcriptional complexes, including Hif1α and RORα15,39. Given that Il17a doesn’t contain strong PPREs, we further analyzed whether IKKα can be associated with RORγt (Fig. 2e; Supplementary Fig. 5). Co-immunoprecipitation data indicated that there is a strong binding between IKKα and RORγt (Fig. 2e, lane 4; Supplementary Fig. 6). Overall, these data demonstrated that IKKα binds to the promotor region of RORγt to form a transcriptional complex that activates the transcription of IL-17.
Meanwhile, considering the role of IKKα in regulating Foxp3, we speculated the potential interaction of Foxp3 with IKKα. To validate this, we first used increasing concentration of IKKα plasmid in the presence of MG132 (protease inhibitor). The results indicated that an increase in the concentration of IKKα plasmid leads to a decrease in the expression of Foxp3 (Fig. 3a; Supplementary Fig. 7). Meanwhile, co-immunoprecipitation assay indicated the interaction between Foxp3 and IKKα (Fig. 3b; Supplementary Fig. 8). Furthermore, to identify the domain of Foxp3 interacting with IKKα, we generated several truncations containing the functional domains of the Foxp3 (Fig. 3c), and performed co-immunoprecipitation. The results indicated that FLAG-IKKα binds with the N1 and C1 region of HA-Foxp3 (Fig. 3d; Supplementary Fig. 9). To further strengthen the relationship between IKKα and Foxp3 in native conditions, we conducted endogenous Co-IP using the plasmids overexpressing IKKα and Foxp3. The data indicated that IKKα and Foxp3 also interacted endogenously (Supplementary Fig. 3b) Thus, we established that an increase in IKKα can lead to a decrease in Foxp3 expression through post-translational modification in term of proteasomal degradation of Foxp3 by phosphorylation.

a 293 T cells were transfected with Foxp3 expression plasmids together with increasing concentrations of IKKα overexpression plasmid in the presence of vehicle or MG132 (protease inhibitor). After 48 h of incubation, the cells were lysed and immunoblotted for Foxp3 (top) and β-actin (bottom). b 293 T cells were transfected with HA-tagged Foxp3 expression plasmids (HA-Foxp3), Flag-tagged IKKα expression plasmids (Flag-IKKα), or both. Co-immunoprecipitation assay was performed utilizing anti-Flag antibody (IP: Flag) or anti-HA antibody (IP: HA) on cell lysates and western-blot of either HA (Foxp3) or Flag (IKK) is shown (IB). The input amount is also shown in the bottom. c Full-length and truncated protein domains of Foxp3 are shown in schematic diagrams. N1, N2 and N3 represent Foxp3 constructs missing C-terminus domains, while C1, C2 and C3 represent Foxp3 constructs missing N-terminus domains. d Truncated Foxp3 constructs tagged with HA were transfected into 293 T cells together with Flag-IKKα and co-IP was performed with anti-HA-antibody (IP: HA). Subsequent western blot analysis of either HA (Foxp3) or Flag (IKKα) is shown. Results are shown as the means ± SEM of three independent trials. Unpaired student t-tests were used for statistical analysis representing *P < 0.05, and ***P < 0.001.
Loss of Pparα aggravates the EAE injury by altering the Th17/Treg balance
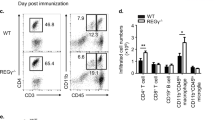
Finally, we utilized an EAE model and investigated whether Pparα deficiency can alter Th17 and Treg developmental balance in vivo. WT and Pparα KO mice were immunized with MOG peptide, and clinical paralysis was monitored and assessed for 22 days (Fig. 4a). Beginning from day 14, Pparα KO mice developed more severe paralysis, which was maintained rest of the experiment (Fig. 4a). On day 22, we isolated CD4+ T cells infiltrating the brain (CNS), and evaluated the expression of IL-17A, Foxp3 and IFN-γ expression by using flow cytometry (Fig. 4b). In association with more severe paralysis42, Pparα KO mice have significantly increased percentage of IL-17A+ IFN-γ+ double positive (DP) CD4+ T cells in the CNS compared to WT mice (Fig. 4c). At the same time, we observed a small increase in IL-17A single positive (SP) subset with a reduction in Foxp3 SP subset among CD4+ T cells in Pparα KO mice. The percentage of IL-17A+ Foxp3+ DP CD4+ T cells was significantly higher in Pparα KO mice (Fig. 4c), demonstrating enhanced IL-17A expression in the absence of Pparα43. In contrast, such difference was not seen in the Treg/Tconv ratio of CD4+ T cells from the spleen after stimulation (Supplementary Fig. 2c). To further analyze the expression of IL-17a, IKKα, and RORγt in the EAE-induced animal model, mononuclear cells were isolated from the brain of WT and Pparα KO mice on day 22 following induction of EAE, and qRT-PCR was performed. We found that the levels of IL-17A and IKKα were significantly upregulated. In contrast, the level of RORγt was not changed in Pparα KO mice as compared to WT mice (Fig. 4d). Meanwhile, the cytokine production of splenocytes isolated from WT and Pparα KO mice at day 22 after EAE induction was analyzed after stimulation with MOG35–55 peptide (MOGp35–55) or medium alone using the ELISA. The results indicated that the level of IL-17A was significantly increased in the splenocytes isolated from Pparα KO mice at 0, 2.5, and 10 μg/mL doses of MOGp35–55 compared to WT (Fig. 4e). However, the level of IL-10 was significantly decreased in cells isolated form Pparα KO mice at 0, 2.5 and 10 μg/mL doses of MOGp35–55 (Fig. 4e). Interestingly, we didn’t observe any change in the level of IFN-γ between Pparα KO and WT group (Fig. 4e). This was consistent with the finding that brain-resident MOG-specific T cells are associated with the development of EAE44.

a Clinical scores of WT and Pparα KO mice immunized with 100 ug MOG35-55 peptide (MOGp35–55) in complete Freund’s adjuvant. Clinical scores are shown as the means ± SEM of n = 6 mice per group. Unpaired student t-test were used for statistical analysis representing *P < 0.05. b Representative flow-plots of IFN-γ, IL-17A and Foxp3 expression in lymphocytes from the brain of WT and Pparα KO mice. c Foxp3+ IL17A+ (top) or IFN-γ+ IL-17A+ (bottom) double positive CD4+ T cells. d qRT-PCR analysis analyzed the expression of IL-17A, IKKα and RORγt from the mononuclear cells isolated from the brain of WT and Pparα KO mice on day 22 following the induction of EAE. e ELISA was performed to analyze the levels of IL-17A, IL-10 and IFN-γ in the splenocytes isolated from WT and Pparα KO mice at 22 days-induced EAE model after stimulation with MOGp35–55 or medium alone. In-vivo data are shown as the means ± SEM of n = 5 mice per group. For in-vitro experiments, results are shown as the means ± SEM of five independent trials. Unpaired student t-test were used for statistical analysis representing *P < 0.05, **P < 0.01, and ***P < 0.001.
Discussion
Many recent studies have recognized the role of nuclear receptors during Th17 and Treg development. Nuclear receptors, including PPARs, utilize unique modes of gene regulation called trans-repression and trans-activation45, which are different from other transcription factors. Rather than acting as a direct transcriptional activator or suppressor, PPARs regulate gene expression by recruiting epigenetic modifiers or interfering with other signaling pathways46. PPARα appears to follow this mechanism during Th17 development, recruiting co-repressors, silencing mediators for retinoid and thyroid receptors (SMRT), and nuclear receptor co-repressor (NcoR)25,47. Here, it has been illustrated that genetic ablation of Pparα can result in enhanced Th17 responses and exacerbated EAE. Meanwhile, we have found a modest increase in IL-17A expression in CD4+ T cells under Th17 polarizing conditions in Pparα KO mice.
Similar to Pparγ, other studies have found that Pparα can regulate Th17 responses gender-dependently29. Furthermore, gemfibrozil, a Pparα agonist, can enhance Th2 responses and thus ameliorates EAE symptoms48. Nevertheless, the mechanism of action of Pparα is not fully established, and to the best of our knowledge, our data present a novel molecular mechanism of Pparα regulating Th17. NF-κb signaling has been known to play roles in Th17 and Treg development49,50,51, yet its molecular mechanism has not been fully established. IKKα is a serine/threonine protein kinase member that mediates the classical and alternative NF-κb signaling pathway52. In the NF-κb dependent pathway, IKKα-mediated phosphorylation can signal proteasomal-mediated degradation of IκB or p100. Independent of the NF-κb pathway, IKKα also contains a nuclear localization signal sequence that allows its translocation and gene regulation by phosphorylation of histone H341,53. It has been known that Pparα can inhibit NF-κB-activation by suppressing IKKα activity54, and our finding provides mechanistic details of Pparα interaction with the NF-κB pathway. We found that Ppara KO naïve CD4+ T cells have elevated basal levels of IKKα expression compared to WT CD4+ T cells. Such difference was maintained after naïve T cells differentiated under Th17 polarizing conditions. Yet, it is not clear how Pparα inhibits IKKα expression levels in T cells, but a similar mechanism that regulates IκB expression may play a role55.
PPARs can directly bind to peroxisome proliferator response elements (PPREs) in proximal regulatory regions and regulate gene expression56. However, Il17a regulatory regions do not contain PPREs as previously reported25, leaving the mechanism of Pparα-dependent regulation of Th17 differentiation both unknown and potentially complex. Numerous studies have reported that IKKα-dependent phosphorylation of histone H3 is critical for IL-17A expression in CD4+ T cells40. We provide evidence that Pparα is an upstream regulator of IKKα expression and nuclear translocation during Th17 development. Furthermore, our data suggest that altered nuclear distribution of IKKα in Pparα KO Th17 cells in association with RORγt allows its transactivation of Il17a expression. Thus, it is plausible that Pparα may physically interact and sequester IKKα to the perinuclear region, inhibiting the interaction between IKKα and RORγt. Because RORγt is highly expressed in the Th17 subset, this proposed mechanism may also explain the specific effects of Pparα on Th17 differentiation.
The balance of Th17 and Treg is important for autoimmunity and immune tolerance15. However, the role of IKKα and Pparα in balancing Th17/Treg has not been understood. A previous finding indicates that IKKα deficiency in CD4 cells decreased their response to TCR stimulation and reduced the number of Tregs in peripheral lymph nodes and thymus57. Meanwhile, IKKα deficient Tregs showed failure in their ability to lower the expression of IFN-γ and elevation of IL-17 in naïve CD4 cells57,58. However, in our study, we observed an increased IKKα expression in the naïve CD4+ cells from Pparα KO mice cultured under Th17 polarization suggesting the role of IKKα in Th17 polarization. Meanwhile, we also found that forced expression of IKKα significantly lowers the expression of Foxp3, thus influencing the differentiation and function of Treg. Therefore, in accordance with previous findings40,57, our study urges that IKKα plays a critical role in the Th17 development and Th17/Treg balance by enhancing the transcription of IL-17 and proteasomal degradation of Foxp3. Interestingly, TGF-β regulates the differentiation of both Th17 and Tregs. In naïve T cells, TGF-β elevates the Tregs development, and inhibits Th17 development. However, TGF-β fails to induce Tregs under IL-6 conditions; instead, it promotes Th17 development by enhancing RORγt expression59. Thus, our study suggests that Pparα KO or inhibition of Foxp3 could enhance Th17 development. Meanwhile, further studies are required to investigate how inhibiting the expression of Pparα in immune cells can influence disease activity.
Besides the direct role of IKKα in the post-translational modification of Foxp3, as shown by proteasomal degradation of Foxp3 by IKKα in our study, IKKα also regulates the expression and function of Foxp3 in an NF-κB-dependent manner. However, in NF-κB-dependent regulation of Foxp3, c-Rel, a member of NF-κB, is particularly important for regulating the expression of Foxp3 at the transcriptional level60,61. Meanwhile, NF-κB and c-Rel also play an indispensable role in the development of Th17 cells50,62. However, we found that independent of NF-κB, IKKα can lead to the proteasomal degradation of Foxp3 and the transcription of IL-17. It was due to the nuclear translocation of IKKα in our Pparα KO mice. It has been previously observed that the nuclear translation of IKKα can also directly influence cell functions63. However, our study found that the absence of Pparα upregulates the IKKα, leading to post-translational changes in the Foxp3 through phosphorylation. Meanwhile, it needs further investigation to understand the cell-specific effect of Pparα in Treg and Th17 development.
Of note, we observed an increase in the Foxp3+ IL17+ and IL-17+ IFN-γ+ DP CD4 cells in the EAE model induced in Pparα KO mice. These DP T cells mediate the differentiation of Tregs and Th17 cells in the presence of cytokine milieu, i.e., IL-6 and TGF-β64,65. Increased DP cells act as a transient population and enhance the proinflammatory condition and disease severity during the pathogenesis of EAE in Pparα KO mice. A previous study also indicated that Foxp3 directly binds to the RORγt and regulates the activity and transcription of RORγt and IL-17 mRNA, respectively66. Conversely, an increase in double-positive cells further strengthens our hypothesis that IKKα, in the absence of Pparα, aggravates the progression of EAE by supporting the differentiation of Th17 through the proteasomal degradation of Foxp3.
Adoptive transfer of Tregs is a potent immunotherapy against various diseases67,68. It has been elucidated that the adoptive transfer of Tregs somehow prevents or decrease the severity of experimental autoimmune encephalomyelitis (EAE) and neurological symptoms69. Whereas the depletion of Tregs can aggravate the EAE70. However, we observed a little decrease in the population of Foxp3+ Tregs in Pparα KO mice induced with EAE. This was accompanied by the increase in Foxp3+ IL17+ and IL-17+ IFN-γ+ DP CD4 cells which alleviated the disease in KO mice.
Molecular regulators that regulate Th17 and Treg development have been identified71. TGF-β1 driven Foxp3 can inhibit RORγt activity in a dose-dependent manner and suppress Th17 development59,66, and its expression levels can affect Th17 development. HIF-1α can post-translationally modify Foxp3 expression during Th17 development while enhancing IL-17A expression15. Also, acetyl-CoA carboxylase 1 (ACC1) is required for fatty acid synthesis and optimal Th17 development72. IKKα’s interaction with RORγt presents another mechanism underlying the Th17/Treg developmental balance. Our results indicate that Pparα and IKKα are therapeutic targets for inhibiting the Th17 subset in major autoimmune diseases73. Overall, our study provides an interesting insights into Pparα and IKKα-dependent degradation of Foxp3 and transcriptional enhancement of Il17a (Supplementary Fig. 1d). It is suggested that during the activation of TCR signaling in naïve CD4+ T cell, there is an increase in the activity of Pparα. This activation of Pparα inhibits the expression and functions of IKKα. IKKα is involved in the proteasomal degradation of Foxp3 and the upregulation of Il17a by binding Foxp3 and RORγt, respectively. Thus, to the best of our knowledge, our results suggest a novel therapeutic approach to treating autoimmune and inflammatory diseases.
Methods
Mouse strains
Ppara KO mice on C57/BL6 background were purchased from the Jackson Laboratory. Stat3 f/f; Cd4-cre mice were previously described. Ppara KO mice were crossed to Stat3 f/f; CD4-cre to generate DKO mice. 6–8 weeks-old male mice were used for experiments. All animal experiments complied with and were approved by the Johns Hopkins and SIAT Animal Care and Use Policy.
In vitro T-cell differentiation
Naïve CD4+ T cells (CD4+ CD25- CD62LHi) were sorted on a FACS Aria high speed sorter. The sorted cells were activated with plate-bound α-CD3 (10 ug/mL) and soluble α-CD28 (2 ug/mL) with the following polarizing conditions: Th1 (IL-12 (10 ng/mL), IL-4 (10 ug/mL)), Th2 (IL-4 (10 ng/mL), IFN-γ (10 ug/mL), IL-12 (10 ug/mL)), Th17 (IL-6 (10 ng/mL), TGF-β (1.25 ng/mL), IL-23 (10 ng/mL), IL-1β (10 ng/mL), IFN-γ (10ug/mL), IL-4 (10 ug/mL)), Treg (TGF-β (5 ng/mL), IL-2 (100 IU/mL)).
Flow cytometry
For extracellular staining, harvested cells were washed and incubated in PBS containing 1% FBS containing the below fluorochrome-conjugated antibodies in a U-bottom 96-well plate. For intracellular cytokine staining, harvested cells were re-challenged in PMA and Ionomycin in the presence of Golgi-Plug (BD Biosciences). After 5 hours of incubation, the cells were fixed/permeabilized (eBioscience) and incubated with antibodies. The following antibodies were used for the flow cytometric assay: IFN-γ PE, IFN-γ APC, IL-13 PE, IL-17 APC (BD Bioscience), IL-2 APC (BD Pharmingen), Foxp3 PE (eBioscience). The gating strategy used has been described in Supplementary Fig. 4. For cellular proliferation, Cell Trace CFSE cell proliferation kit (Invitrogen) was used per the manufacture’s manual.
Co-immunoprecipitation and Immunoblotting
293 T cells were transfected with the plasmids overexpressing IKKα, RORγt, Foxp3, and various fragments of Foxp3 using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA). Plasmids were co-transfected or transfected individually depending on the requirement for endogenous or exogenous Co-IP experiment. After 48 h of transfection, total protein was extracted using the RIPA lysis buffer. Proteins mixtures were then incubated with following antibodies: anti-HA (Sigma-Aldrich, 1:2000), anti-Flag (Sigma-Aldrich, 1:5000), or anti-Myc (Sigma-Aldrich, 1:2000) to bind with the protein complexes. Later, Dynabeads® Protein G (Invitrogen, Cat.No.100.04D) were utilized to pulldown the protein complexes. Finally, the samples were centrifuged to remove the supernatant, washed with IP buffer three times, eluted in SDS-loading buffer, and heated at 100 °C for 8 min.
Samples were then subjected to western blotting for separation using 10% SDS-PAGE. After the electrophoresis, proteins were transferred to a PVDF membrane which was ultimately blocked with 5% skim milk for 2 h. After blocking, membrane was incubated with respective primary antibodies for overnight. Next day, membrane was washed and incubated with HRP-conjugated secondary antibodies. After washing, membrane was exposed using ECL system. Protein sizes in blot images were calculated by resolving the pre-stained potein marker using electrophoresis. ImageJ was used to calculate the intensity of protein bands.
Small molecule inhibitors
Fenofibrate and Gemfibrozil were purchased from Sigma-Aldrich and dissolved in DMSO. Dilutions were made in DMSO before being added to cultures to make indicated final concentrations in culture.
EAE induction
Age- and sex-matched mice were immunized with 100 ug MOG35-55 peptide (MOGp35-55) in complete Freund’s adjuvant (Sigma-Aldrich) in the rear flank s.c. The mice were further challenged with 250 ng of pertussis toxin (List Biological) on day 0 and day 2. Paralysis in mice were monitored and scored as previously described criteria.
Quantitative real-time PCR
RNA was extracted using Trizol (Invitrogen) followed by cDNA synthesis reaction using SuperScript III (Invitrogen) in a 20 ul reaction/well. The same amount of RNA was used in each cDNA synthesis reaction measured by NanoDrop Spectrophotometer (ThermoScientific). The same volume of cDNA per sample was prepared for qRT-PCR analysis using SYBR Green (Pierce) and the indicated primers to assess transcript levels of each gene. The primer sequences were included in the Supplementary Table 1.
ELISA
The concentration of IL-17, IL-10, and IFN-γ from the splenocytes isolated from WT and Pparα KO mice at 22 days after EAE induction and stimulated with MOGp35–55 or medium alone. Samples were detected for respective cytokines using the ELISA kit. Values were detected using a micro-plate reader at OD450.
CFSE dilution assay
CD4+ T cells were isolated, as mentioned earlier. Cells were labeled with 1 mM CFSE (Molecular Probes) in PBS and incubated at 37 °C for 3 min. Ultimately cells were washed and seeded in a 96-well plate, which was previously coated with 10 mg/mL anti- IgG Fc-specific Ab along with 1 mg/mL anti-CD3 and 0.25 mg/mL anti-CD28 at 4 ̊C overnight. Cells were then harvested and analyzed for IL-2 and IFN-γ using flow cytometry.
Chromatin immunoprecipitation assay
Relative histone modification levels in Th17 cells from WT and Paprα KO mice were detected using in-vitro ChIP assay. Cells were cross-linked using disuccinimidyl glutarate (DSG) for 30 min followed by 1% methanol-free ultrapure formaldehyde for 10 min at RT. Glycine was then added for 5 min at 125 mM final concentration. After fixation, chromatin samples were sonicated with a Diagenode Bioraptor to generate small fragments of 200–1000 bp, with a peak of intensity of about 500 bp. The supernatant after centrifugation was diluted by ChIP dilution buffer and precleared with protein A–agarose beads, which was then immunoprecipitated. Chromatin was immunoprecipitated with antibodies against IgG, anti-IKKα, anti-RORγt, anti-p-H3K27ac, and anti-p-H3Ser10. The immunoprecipitated DNA was isolated by Universal DNA purification kit (TIANGEN, DP214-03). Purified DNA was analyzed by qRT-PCR with specific primers.
Statistics and reproducibility
In-vivo data are shown as the means ± SEM of n = 6 mice per group. For in-vitro experiments, results are shown as the means ± SEM of three or more independent trials as represented in figure legends. The raw data related to this study has been presented in Supplementary Data 1. Data were analyzed and compared through the GraphPad Prism 8.0 by utilizing an unpaired Student’s t-test, and for the significance, p-value of <0.05 was acquired.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Supplementary Figs. 5–9 are the source uncropped Western blot images with size marker shown in this manuscript. The Supplementary Data 1 contain all the statistical source data for the graphs presented in the manuscript. All other data is available from the corresponding author upon reasonable request.
References
Zhu, J., Yamane, H. & Paul, W. E. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28, 445–489 (2010).
Ivanov, I. I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper. cells Cell 126, 1121–1133 (2006).
Lu, L., Barbi, J. & Pan, F. The regulation of immune tolerance by FOXP3. Nat. Rev. Immunol. 17, 703–717 (2017).
Dumitriu, I. E., Dunbar, D. R., Howie, S. E., Sethi, T. & Gregory, C. D. Human dendritic cells produce TGF-beta 1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. (Baltim., Md. 1950) 182, 2795–2807 (2009).
O’Garra, A., Stockinger, B. & Veldhoen, M. Differentiation of human T(H)-17 cells does require TGF-beta! Nat. Immunol. 9, 588–590 (2008).
Zheng, S. G. Regulatory T cells vs Th17: differentiation of Th17 versus Treg, are the mutually exclusive? Am. J. Clin. Exp. Immunol. 2, 94–106 (2013).
Shyer, J. A., Flavell, R. A. & Bailis, W. Metabolic signaling in T cells. Cell Res. 30, 649–659 (2020).
Buck, M. D., O’Sullivan, D. & Pearce, E. L. T cell metabolism drives immunity. J. Exp. Med. 212, 1345–1360 (2015).
Menk, A. V. et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 22, 1509–1521 (2018).
Chakrabarti, R., Jung, C. Y., Lee, T. P., Liu, H. & Mookerjee, B. K. Changes in glucose transport and transporter isoforms during the activation of human peripheral blood lymphocytes by phytohemagglutinin. J. Immunol. (Baltim., Md.: 1950) 152, 2660–2668 (1994).
Frauwirth, K. A. & Thompson, C. B. Regulation of T Lymphocyte metabolism. J. Immunol. 172, 4661–4665 (2004).
Wieman, H. L., Wofford, J. A. & Rathmell, J. C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol. Biol. Cell 18, 1437–1446 (2007).
Kim, J. W., Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006).
Finlay, D. K. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8 + T cells. J. Exp. Med. 209, 2441–2453 (2012).
Dang, E. V. et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784 (2011).
Shi, L. Z. et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Wohlfert, E. A., Nichols, F. C., Nevius, E. & Clark, R. B. Peroxisome proliferator-activated receptor gamma (PPARgamma) and immunoregulation: enhancement of regulatory T cells through PPARgamma-dependent and -independent mechanisms. J. Immunol. (Baltim., Md. 1950) 178, 4129–4135 (2007).
Atif, M. et al. Metabolic optimisation of regulatory T cells in transplantation. Front. Immunol. 11, 2005 (2020).
Michalek, R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4 + T cell subsets. J. Immunol. (Baltim., Md. 1950) 186, 3299–3303 (2011).
Deberardinis, R. J., Lum, J. J. & Thompson, C. B. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J. Biol. Chem. 281, 37372–37380 (2006).
Lee, J. et al. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. (Baltim., Md. 1950) 192, 3190–3199 (2014).
Choi, J. M. & Bothwell, A. L. The nuclear receptor PPARs as important regulators of T-cell functions and autoimmune diseases. Molecules cells 33, 217–222 (2012).
Dunn, S. E. et al. Peroxisome proliferator-activated receptor (PPAR)alpha expression in T cells mediates gender differences in development of T cell-mediated autoimmunity. J. Exp. Med. 204, 321–330 (2007).
Klotz, L. et al. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J. Exp. Med. 206, 2079–2089 (2009).
Kanakasabai, S. et al. Peroxisome proliferator-activated receptor delta agonists inhibit T helper type 1 (Th1) and Th17 responses in experimental allergic encephalomyelitis. Immunology 130, 572–588 (2010).
Cipolletta, D. et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553 (2012).
Daynes, R. A. & Jones, D. C. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2, 748–759 (2002).
Zhang, M. A. et al. Peroxisome proliferator-activated receptor (PPAR)α and -γ regulate IFNγ and IL-17A production by human T cells in a sex-specific way. Proc. Natl Acad. Sci. USA. 109, 9505–9510 (2012).
Jones, D. C., Ding, X. & Daynes, R. A. Nuclear receptor peroxisome proliferator-activated receptor alpha (PPARalpha) is expressed in resting murine lymphocytes. The PPARalpha in T and B lymphocytes is both transactivation and transrepression competent. J. Biol. Chem. 277, 6838–6845 (2002).
Kliewer, S. A. et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl Acad. Sci. USA 91, 7355–7359 (1994).
Lee, J. W. et al. Fenofibrate represses interleukin-17 and interferon-gamma expression and improves colitis in interleukin-10-deficient mice. Gastroenterology 133, 108–123 (2007).
Zhang, Y. et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 32, 377–391.e379 (2017).
Gebel, T., Arand, M. & Oesch, F. Induction of the peroxisome proliferator activated receptor by fenofibrate in rat liver. FEBS Lett. 309, 37–40 (1992).
Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications-a review. Nutr. J. 13, 17 (2014).
Tenenbaum, A. & Fisman, E. Z. Fibrates are an essential part of modern anti-dyslipidemic arsenal: spotlight on atherogenic dyslipidemia and residual risk reduction. Cardiovasc. Diabetol. 11, 125 (2012).
Harris, T. J. et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. (Baltim., Md.: 1950) 179, 4313–4317 (2007).
Wang, L. H. et al. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity 20, 205–218 (2004).
Yang, X. O. et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39 (2008).
Li, L. et al. Transcriptional regulation of the Th17 immune response by IKK(alpha). J. Exp. Med. 208, 787–796 (2011).
Birbach, A. et al. Signaling molecules of the NF-kappa B pathway shuttle constitutively between cytoplasm and nucleus. J. Biol. Chem. 277, 10842–10851 (2002).
Korn, T. et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 13, 423–431 (2007).
Yang, X. O. et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29, 44–56 (2008).
Sonobe, Y. et al. Chronological changes of CD4(+) and CD8(+) T cell subsets in the experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Tohoku J. Exp. Med. 213, 329–339 (2007).
Ricote, M. & Glass, C. K. PPARs and molecular mechanisms of transrepression. Biochimica et. biophysica acta 1771, 926–935 (2007).
Rosenfeld, M. G. & Glass, C. K. Coregulator codes of transcriptional regulation by nuclear receptors. J. Biol. Chem. 276, 36865–36868 (2001).
Klotz, L. & Knolle, P. Nuclear receptors: TH17 cell control from within. FEBS Lett. 585, 3764–3769 (2011).
Gocke, A. R. et al. Transcriptional modulation of the immune response by peroxisome proliferator-activated receptor-{alpha} agonists in autoimmune disease. J. Immunol. (Baltim., Md. 1950) 182, 4479–4487 (2009).
Chang, J. H. et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat. Immunol. 13, 481–490 (2012).
Chen, G. et al. The NF-κB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J. Immunol. (Baltim., Md. 1950) 187, 4483–4491 (2011).
Isomura, I. et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 206, 3001–3014 (2009).
Chen, Z. J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 7, 758–765 (2005).
Yamamoto, Y., Verma, U. N., Prajapati, S., Kwak, Y. T. & Gaynor, R. B. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423, 655–659 (2003).
Okayasu, T., Tomizawa, A., Suzuki, K., Manaka, K. & Hattori, Y. PPARalpha activators upregulate eNOS activity and inhibit cytokine-induced NF-kappaB activation through AMP-activated protein kinase activation. Life Sci. 82, 884–891 (2008).
Yamaguchi, T., Kimura, J., Miki, Y. & Yoshida, K. The deubiquitinating enzyme USP11 controls an IkappaB kinase alpha (IKKalpha)-p53 signaling pathway in response to tumor necrosis factor alpha (TNFalpha). J. Biol. Chem. 282, 33943–33948 (2007).
Kassam, A., Winrow, C. J., Fernandez-Rachubinski, F., Capone, J. P. & Rachubinski, R. A. The peroxisome proliferator response element of the gene encoding the peroxisomal beta-oxidation enzyme enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase is a target for constitutive androstane receptor beta/9-cis-retinoic acid receptor-mediated transactivation. J. Biol. Chem. 275, 4345–4350 (2000).
Chen, X. et al. IKKα is required for the homeostasis of regulatory T cells and for the expansion of both regulatory and effector CD4 T cells. FASEB J. Off. Publ. Federation Am. Societies Exp. Biol. 29, 443–454 (2015).
Chen, X. et al. TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J. Immunol. (Baltim., Md. 1950) 190, 1076–1084 (2013).
Zhou, L. et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240 (2008).
Joshi, N. S. et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295 (2007).
Ruan, Q. et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 31, 932–940 (2009).
Ruan, Q. et al. The Th17 immune response is controlled by the Rel-RORγ-RORγ T transcriptional axis. J. Exp. Med. 208, 2321–2333 (2011).
Margalef, P. et al. A truncated form of IKKα is responsible for specific nuclear IKK activity in colorectal cancer. Cell Rep. 2, 840–854 (2012).
Du, R., Zhao, H., Yan, F. & Li, H. IL-17+Foxp3+ T cells: an intermediate differentiation stage between Th17 cells and regulatory T cells. J. Leukoc. Biol. 96, 39–48 (2014).
Wei, G. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4 + T cells. Immunity 30, 155–167 (2009).
Ichiyama, K. et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J. Biol. Chem. 283, 17003–17008 (2008).
Duffy, S. S., Keating, B. A. & Moalem-Taylor, G. Adoptive Transfer of Regulatory T Cells as a Promising Immunotherapy for the Treatment of Multiple Sclerosis. Front. Neurosci. 13, 1107 (2019).
Mohammadi, S. et al. Adoptive transfer of Tregs: A novel strategy for cell-based immunotherapy in spontaneous abortion: Lessons from experimental models. Int. Immunopharmacol. 90, 107195 (2021).
Beyersdorf, N. et al. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J. Exp. Med. 202, 445–455 (2005).
McGeachy, M. J., Stephens, L. A. & Anderton, S. M. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. (Baltim., Md. 1950) 175, 3025–3032 (2005).
Hatton, R. D. & Weaver, C. T. Duality in the Th17-Treg developmental decision. F1000 Biol. Rep. 1, 5 (2009).
Berod, L. et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333 (2014).
Noack, M. & Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 13, 668–677 (2014).
Acknowledgements
This work was supported by the National Key R&D Program of China (2021YFC2400500), the National Natural Science Foundation of China (Grant 32170925), Shenzhen Science and Technology Program (KQTD20210811090115019), the Shenzhen Science and Technology Program (JCYJ2022081800807016), the startup fund of SIAT and CAS, the Natural Science Foundation of Chongqing Grant CSTB2022NSCQ-MSX1069, the Entrepreneurship and Innovation Support Program of Chongqing for overseas Scholars Grant CX2022118. The authors declare no competing financial interests.
Author information
Authors and Affiliations
Contributions
P.W. and F.P. designed the study. P.W., W.K., J.F., and Z.C. conducted the experiments. P.W. and F. P. analyzed the data and drafted the manuscript. F.P. critically revised the manuscript and supervised the study. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Guangyong Peng and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editor: Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, P., Kou, W., Fu, J. et al. Pparα knockout in mice increases the Th17 development by facilitating the IKKα/RORγt and IKKα/Foxp3 complexes. Commun Biol 6, 721 (2023). https://doi.org/10.1038/s42003-023-05104-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05104-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
