
Abstract
Dromedaries have been essential for the prosperity of civilizations in arid environments and the dispersal of humans, goods and cultures along ancient, cross-continental trading routes. With increasing desertification their importance as livestock species is rising rapidly, but little is known about their genome-wide diversity and demographic history. As previous studies using few nuclear markers found weak phylogeographic structure, here we detected fine-scale population differentiation in dromedaries across Asia and Africa by adopting a genome-wide approach. Global patterns of effective migration rates revealed pathways of dispersal after domestication, following historic caravan routes like the Silk and Incense Roads. Our results show that a Pleistocene bottleneck and Medieval expansions during the rise of the Ottoman empire have shaped genome-wide diversity in modern dromedaries. By understanding subtle population structure we recognize the value of small, locally adapted populations and appeal for securing genomic diversity for a sustainable utilization of this key desert species.
Similar content being viewed by others

Introduction
As one of the most recently domesticated animals (~3000–4000 years before present; ybp), the dromedary (Camelus dromedarius) has a special position in human migration and trading1. Its physiological adaptations to harsh and dry environments allowed humans to traverse hostile lands such as deserts like no other livestock, including the horse2. Reversing a historian’s observation “the sea unites rather than divides”3 to the desert, dromedaries facilitated the expansion of civilizations1,4,5. Their superior and unique features predestined dromedaries for the use as military animals, and for the advance of international trading along ancient caravan routes, such as the Silk Road and Incense routes6,7. Interspecific hybridization between dromedaries and the closely related two-humped Bactrian camels (Camelus bactrianus) produced even more robust and enduring animals with an aptitude for the extreme climatic conditions of the Silk Road8,9. Nowadays, first generation hybrids and their backcrosses are valued in many countries for increased milk or wool production10, as well as in famous camel wrestling events11. These remarkable commercial networks in human history facilitated domestic animal exchange across large geographical distances and acted as gene-flow corridors, not only for camels but also for other livestock12. While camels were the chosen animals for transportation, horse movements along the complete network of the Silk Road mainly occurred in forms of tributes and gifts12,13.
The early progenitors (Protylopus) of camelids emerged in the North American savannah during the Eocence (~45 Mya). After their split into New (Lamini) and Old World (Camelini) camels around 15 Mya, the ancestors of the Old World camels crossed into Eurasia via the Bering Land Bridge (~6.6 Mya) and further diverged into one- and two-humped camels (reviewed in Burger et al.14). Early-domestic dromedaries (second and first millennium before Common Era; BCE) cohabited the coastal Southeast of the Arabian Peninsula for nearly one millennium together with wild specimens, which did not survive the beginning of the Common Era (CE)15,16,17,18,19. The early dispersal of modern dromedaries from the Arabian Peninsula to the Levant, North Africa, South Asia, and finally to Australia (introduced in the late 19th century)20 was followed by cross-continental back-and-forth movements along historic trading routes. This led to a blurring of genetic stocks21 culminating in a panmictic dromedary population at the mitochondrial DNA level1. Previous studies using a limited number of microsatellite markers and mtDNA detected only weak population structure in the global dromedary population1,20,22. As few genomic studies have been completed with the dromedary, this major ungulate species has been left out of the livestock genomic revolution. However, two draft reference genomes at the scaffold level have been released23,24, as well as a draft reference genome at the chromosome level25, which will facilitate further genomic investigations.
We wanted to understand how human-induced migration patterns and historic demographic changes might have influenced population structure in the global dromedary population. We sequenced 22,721 SNP markers to overcome the limitations of previous studies using not more than 20 microsatellites. With a global dataset including samples spread over three continents, we describe effective migration rates of modern dromedaries that follow their human-driven dispersal along ancient trading routes7. Understanding subtle population structure, which has been shaped by past and recent demographic events, will help in recognizing the value of small populations and securing genomic diversity for a sustainable utilization of this important livestock species in a globally changing world.
Results and discussion
We performed double-digest restriction site associated DNA (ddRAD) sequencing on 122 dromedary DNA samples from 18 countries (Supplementary Data 1) representative of the species distribution range. We included one Bactrian camel to test for potential interspecific hybridization, as this continues to be a widespread practice in Central Asia that might have started as early as pre-Roman times11. Higher numbers of reads mapping to the Bactrian camel were detected in three individuals from Iran and in six from Kazakhstan (see “Methods”), and we decided to remove these samples from downstream analysis due to potential introgression from Bactrian camel (Supplementary Data 2). After stringent filtering for genotype and individual missingness, minor allele frequency and relatedness, the final dataset consisted of 95 dromedaries and 22,721 SNPs present in at least 75% of the individuals.
Moderate genome-wide diversity and low population structure
With 22,721 SNPs, we estimated expected (HE = 0.27 ± 0.17; mean ± SD) and observed (HO = 0.25 ± 0.17) heterozygosities in the global dromedary population (npop = 17; nind = 95). Separating the samples according to their continental origins, both Asian (nind = 49, HE = 0.27 ± 0.17/HO = 0.25 ± 0.17) and African dromedaries (nind = 46, HE = 0.26 ± 0.17/HO = 0.25 ± 0.18) showed similar genomic diversity. The mean HE (t = −2.2641, df = 45,398, P = 0.02) and inbreeding coefficients (t = −2.5159, df = 43,024, P = 0.01) were higher in Asian than African dromedaries, but mean HO (t = −1.2791, df = 45,385, P = 0.2) was not different between continents, according to Welch′s t test. Complete diversity and inbreeding values are given in Supplementary Table 1. In comparison with other domestic species, i.e., sheep (HE = 0.22–0.32)26 or cattle (HE = 0.24–0.30)27, we consider the genome-wide diversity in dromedaries as moderate at the best. Several bottlenecks during the last glacial period (see demographic analysis below, and Fitak et al.24) and during domestication left modern dromedaries with a minimum of only six maternal lineages1 and limited genome-wide diversity. This will have implications on future intensification of breeding and genomic selection in dromedaries from regions with increasing desertification.
In general, the genome-wide differentiation within the global dromedary population was very low. Analysis of Molecular Variance (AMOVA) revealed that most of the variation, ~94.3%, is explained within individuals (Supplementary Table 2). Allelic richness (AR) was similar between countries (AR = 0.25–0.27) with exception of Kenya which was lower (AR = 0.21). The pairwise fixation index between African and Asian individuals was very low (FST = 0.006; P < 0.001), and indices between dromedaries from different countries (if significant at all) were lowest in geographically close populations (e.g., Libya/Algeria: FST = 0.0002) and increased with geographic distance (Pakistan/Tunisia: FST = 0.0328) (Supplementary Table 3).
We screened for loci deviating from neutraliy using BayeScan 2.128 and identified sixteen FST outliers to be putatively under selection (false discovery rate (FDR) <0.05) between African and Asian dromedaries (Supplementary Fig. 1). We found it reasonable to investigate the biological functions of those genes harboring the SNPs as they might relevant for the adaptation of dromedaries to their respective environments. We found SNPs in two genes, CALN1 and TREM1, which are responsible for calcium ion binding and amplifying inflammatory responses triggered by bacterial and fungal infections, respectively (scaffold:SNP-location; JWIN01030783.1:128274 and JWIN01033764.1:729703). In addition, we examined (potentially linked) regions 200 kbp upstream and downstream of the FST-outlier loci and detected fifty-three genes related to a number of biological functions (Supplementary Data 3). Interestingly, around one fifth of the detected genes had functions related to the immune system hinting to an adaptive process in response to different pathogens in the respective environments. Other protein coding gene functions were related to pathways such as circadian rhythm, (ga)lactose, metabolism, reproductive or various cellular and developmental processes. A full list of genes is presented in Supplementary Data 3. Signatures of selection related to photoperiod, metabolism, immunity and growth have also been observed in chicken29, sheep30, and cattle (TGFB331).
To understand the low genome-wide differentiation in dromedaries across their global range, we investigated population structure and admixture between populations. We projected the genetic variation of each dromedary on the first three axes inferred from a principal component analysis (PCA) and incorporated continental information (Africa/Asia) for each sample (Fig. 1). Principal component 1 (PC1) clearly separated African from Asian dromedaries, while PC2 and PC3 split Kenyan individuals from the rest of Africa and identified a single population from Saudi Arabia grouping closer to African than Asian individuals, although showing some cross-continental admixture (Fig. 1 and Supplementary Fig. 2). This separated population belongs to a specific breed, Hadhana, and is one of the twelve recognized dromedary ecotypes in Saudi Arabia, limited to mountain regions in the South of the Arabian Peninsula, Al-Baha32. In this case, the geographic accessibility might have an important role in the observed genetic distinctiveness. A possible explanation for the close relationship of Hadhana and African dromedaries might be the historic sea route from Jiddah in Saudi Arabia to Aydhab and Port Sudan. On the western coast of the Red Sea existed a trading route connecting the Horn of Africa to Petra and Damascus via Port Sudan, Aydhab and Myos Hormos, near today’s Kosseir (Fig. 2)6,7. In general, the Asian dromedary population showed higher genetic variability, although the genetic variation explained by the three first axes was rather low with only 5.3% (Fig. 1). While this could be a sign for ancestral variation (the Arabian Peninsula was a center of domestication1, we cannot discard the hyphothesis of post-domestication movements of camels or multiple origins of the founder populations as this would have left similar signals in the genomes.

Principal component (PC) analysis based on 22,721 SNPs to represent populations pre-classified according to phylogenetic clustering (Supplementary Fig. 2), Africa (AF), Asia (AS), Hadhana (HD), and Kenya (KE). Elipses refer to the distribution of individuals within groups. The first three PCs explain 5.3% of the total variation and are shown in a PC1–PC2 and b PC1–PC3.

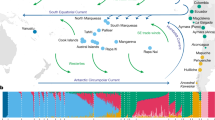
EEMS plot representing the posterior mean of effective migration rates (m) (on a log10 scale) across space. With this normalization, significantly higher than the overall average rates are represented in blue (“corridors”) and significantly lower than the overall average rate (“barriers”) are represented in brown. Zero corresponds to the overall mean migration rate. Samples are represented by diamonds and the size is proportional to the number of sampling. Approximate coordinates are used. Ethiopian Highlands and Arabian Desert are highlighted with white lines. Black lines represent historical network of caravan routes, i.e., Incense and Silk roads6,7 and main trans-Saharan gold trade networks36 (adapted from2).
We next inferred potential ancestry and admixture among Asian and African dromedaries using unsupervised genetic clustering in ADMIXTURE33 (Fig. 3). Based on the lowest cross-validation error, the best clustering solution was 1 (Supplementary Fig. 3), which suggests a panmictic dromedary population and reflects the low genetic differentiation of 0.6% among individuals from different continents. Increasing the numbers of potential ancestral populations (K) from two to seven confirmed the already observed differentiation between African and Asian dromedaries (K = 2), the clustering of the Saudi Arabian Hadhana breed with Africa (K = 4), the separation of Kenyan and Hadhana individuals, and the higher number of distinct clusters on the Asian continent (K = 7). We find a more homogenous gene pool in African animals with the exception of the East African group1 (Figs. 1, 2 and Supplementary Fig. 2), represented in our dataset by the two Kenyan dromedaries. This can be a consequence of a random founder effect followed by lack of gene flow due to geographical, physiological (e.g., Trypanosome infestation) and/or cultural barrier, i.e., dromedaries in East Africa were dominantly used for milk production rather than transport or riding1. There is a need to proceed with comprehensive analyses about the potential nature of natural and/or anthropogenic obstacles for gene flow between East African and other dromedaries.

Admixture analysis showing the proportion of potential ancestral populations (K = 2; 4; 7) for each individual (single bar). The geographical origins for each sample are represented below the bars in two-digit international country codes, where the middle line divides the first half (African countries) from the second half (Asian countries). Hadhana population is depict in cyan blue (K = 7).
Effective migration rates along ancient caravan routes
To formally test our qualitative observations of weak population structure among African and Asian dromedaries (Figs. 1, 3), we visualized the global spatial population structure using the Estimated Effective Migration Surfaces (EEMS) method34. Based on a stepping-stone model, EEMS detected a corridor of significantly higher effective migration rates than the overall mean along the Mediterranean coast, connecting the Northern parts of Africa and the Arabian Peninsula until the border of the Arabian Desert (Fig. 2). This pattern shows a continuous gene flow throughout the coastal dromedary populations, and a lower than average migration in the inland desert populations. A known trading route which fits this observed effective migration pattern bordered the Mediterranean coast connecting Northwestern Africa to the North of the Arabian Peninsula from where caravans traveled toward Southern Asia along the Silk Road (Fig. 2)1,7. The introduction of the dromedary into Northern Africa via the Sinai from Roman Egypt started in the early first millennium BCE and intensified in the Ptolemaic period6,15. From there, dromedaries migrated along the Mediterranean coast, as archeological evidence dates their presence in Northwest Africa to the fourth to seventh century CE (Late Antiquity/Early Middle Ages)1,6. Even earlier dispersal of taurine cattle along the Northern coast of Africa and through the Mediterranean sea to Europe was described during the Bronze age35.
It is clear that camels, unlike other domesticated species, were able to penetrate deep into the Saharan desert and to connect trans-Saharan cultures. West Sahara belonged to an Islamic trading network classified as one of the major gold suppliers in the ninth to tenth centuries CE36 (Fig. 2). Tadmekka, a territory located in the Southwestern Saharan desert and governed by the Tuareg, was operational by the eighth century CE and was one of the earliest towns established in the region where cross-Saharan camel caravans traded37. These trades prolonged at least until the fourteenth century when Timbuktu, which similar to Tadmekka hosted large groups of Islamic traders, engaged in coin-based exchange economies across the Sahara36.
While modern dromedaries along the western part of the Silk Road are still well connected today, the Iranian and Afghanistan deserts seem to present obstacles of effective migration. As EEMS assumes uniform migration rates the observed “barriers”, could however be assessed as areas of lower population density with fewer migrants exchanged per generation, producing an effective “barrier” to gene flow34. The three main parallel itineraries of the Incense Routes through the Arabian Desert connecting the South of the Arabian Peninsula with the Levant7 also showed lower than average migration rates (Fig. 2), which could be interpreted as lower population density or potential sampling gaps. These trading routes were essential during historical periods, not only for exchanging luxury products (e.g., incense or gems), but for trading everyday local products38.
The strongest barrier detected in our dataset concerned dromedaries from the Horn of Africa, which had the lowest genetic effective migration rates (Fig. 2). Geographical isolation due to the Ethiopian highlands, which might disrupt gene flow with northern populations, and the Golf of Aden would be the most likely explanation for the observed pattern. Genetic differentiation of livestock populations in East Africa has been described previously1,22,39.
Late Pleistocene population decline and medieval expansions
To complete our understanding of the moderate genome-diversity observed in the global dromedaries, we investigated the demographic history and inferred effective population size (NE) over time with an Extended Bayesian Skyline approach (EBS)40 (Fig. 4). As genetic differentiation between Asian and African dromedary populations was low (0.6%) but highly significant (P < 0.001), we tested whether the dromedary populations from the two continents experienced a similar demographic history. First, we investigated the global population and second, African and Asian individuals separately. With the latter approach, we accounted for a potential confounding effect of population structure for the inference of NE40. Due to the observed substructure in Kenyan and Hadhana individuals, we excluded these two populations from the continental groups.

EBSP for all dromedaries, with three independent runs per continent (Africa1, Africa2, and Africa3; Asia1, Asia2, and Asia3) and one for the global population (All). Solid lines represent median scaled effective population size and dashed lines represent 95% Highest Posterior Density (HPD) intervals. In each EBSP, 50 random ddRAD loci with at least four but not more than six SNPs across at least 75% individuals were used, and all EBSP runs were calibrated with a RAD locus-specific clock rate to calibrate the time scale. The lowest median effective population size for the different independent runs occurred 58,926 years before present (ybp) for the global population, 37,713, 26,869, and 38,802 ybp for African dromedaries and 41,923, 46,984, and 39,999 ybp for Asian dromedaries, respectively.
Irrespective of the continental origin, all inferences showed similar patterns of an initial population expansion from one million ybp until ~700,000 ybp (Fig. 4). Our genome-wide population approach confirmed previous NE estimates based on a single dromedary whole genome sequence23 using pairwise sequentially Markovian coalescent41. This population expansion coincides with two remarkable periods: the middle Pleistocene transition (1.25–0.70 million ybp) characterized by climatic cycles, and the Galerian Mammal Age (1.2–0.60 million ybp), which influenced the distribution and evolution of biota and resulted in some species being adapted to arid, cold climates42,43. Moreover, this timeframe also overlaps with the maximal diversity of the family Camelidae (early Galerian), supporting the adaptation of the dromedary ancestor to environmental changes with an expansion of its population during the middle Pleistocene transition23,44. Population expansion was followed by a drastic decline in NE beginning 700,000 ybp until the dromedary population collapsed during the last glacial period (LGP; 100,000–20,000 ybp)45. This is a finding shared by previous Old World9 and New World camelid23 studies and those focusing on Late Quaternary Megafauna46.
Conversely, no bottleneck was picked up by any of the EBSPs during the time scale when dromedaries are predicted to have been first domesticated ~3000–4000 ybp15,16. Previous BSP analysis using mtDNA likewise did not show a population decline during the time of domestication1. It is possible that the detection of a bottleneck with the EBSP analysis related to domestication has been superimposed by the drastic decrease in NE ending ~30,000 ybp (Fig. 4). Similar demographic changes were observed in alpacas23, where three population bottlenecks were detected throughout the cold conditions of the LGM in South America, yet no bottleneck was visible during the domestication period.
After the Pleistocene bottleneck, the dromedary population slowly increased until reaching a stable NE around 300 ybp, with a higher NE present within Asia than in Africa. Demographic inferences based on mtDNA sequences described slightly earlier expansion of the maternal lineages around 600 ypb1 associated with the rise of the Ottoman empire and the conquest of Constantinople (1453 CE), followed by the extension to Southern Asia and the Red Sea coasts47.
Conclusions
Our study shows that assessing the evolutionary history of species using genome-wide approaches allows detailed inferences of population structure, migration, and potential signals of environmental adaptation. As the movements of dromedaries parallel those of humans, knowledge on dromedary spatial genetic signatures also sheds light into past human history. We detected genetic admixture across continental populations (Asia and Africa), which highlights the strong anthropogenic influence on these animals.
Human history is marked with the efforts for overcoming obstacles, be they of geographical (mountains, sea, and deserts) or cultural nature. Domestic animals, and in particular camels, have been linked to this process of human development and were essential for its success. By establishing trading routes and reusing them over millennia, corridors of gene flow were opened that shaped genetic diversity and structure not only in dromedaries, but also in Bactrian camels48, and in many other (livestock) species49,50. With a genome-wide dataset we detected patterns of high effective migration in the global dromedary population and revealed pathways of dispersal after domestication, mirroring ancient caravan routes (Fig. 3). While these served as corridors, deserts, and highlands (due to lower population density) represented possible barriers to gene flow. As we estimated an overall mean migration rate, the existing gene flow (below average) between populations along the caravan roads leading through desert regions might have been masked by the high connectivity (above average) of the coastal populations and/or lack of representative samples. Filling in population sampling gaps in future studies will provide a deeper understanding of the gene flow and genetic structure between populations. It will allow a powerful quantification of the magnitude of genetic isolation barriers that may persist.
Dromedaries and Bactrian camels are the most important livestock species in desert areas; their impact on land and water resources for food production is less than that of any other livestock. With increasing desertification and global climate change their importance will grow even more. For this reason, it is essential to understand the demographic history that has shaped modern dromedary populations. The genome-wide diversity present today, which we have characterized in this study, is a result of genetic shuffling due to historic and recent movements along trading routes. This constant mixing might have led to a unique genetic makeup that could make camels more resilient to global environmental changes, and that needs to be preserved.
Methods
DNA samples
We selected a total of 122 dromedary samples from a pool of previously extracted DNA collected during projects supported from the Austrian Science Foundation, FWF P24607-B25 (PI: P.B.) and the European Research Council, EU ENPI CBC MED PROCAMED I.B.1.1/493 (PI: E.C.) under all legal requirements. The samples originated from 18 countries, which were representative of the species’ distributional range. In view of potential hybridization between one- and two-humped camels known to occur specifically in Central Asian regions, we included a Bactrian camel to control for introgression and as outgroup for phylogenetic analysis. Detailed information about all samples is provided in Supplementary Data 1.
Library preparation, sequencing, and initial data filtering
Library preparation and sequencing was performed at IGA Technology Services (Udine, Italy), to generate genome-wide data from ddRADseq. In silico analysis of the C. dromedarius genome assembly (NCBI accession: GCA_000803125.1)24 highlighted SphI-BstYI as the best combination of restriction enzymes able to produce DNA fragments between 400 and 530 bp. ddRAD barcoded libraries were pooled and sequenced on an Illumina HiSeq 2500 in high output mode using 125 bp paired-end reads. Initial raw data analysis as well as SNP calling was performed by IGA Technology Services in-house bioinformatics pipeline. Briefly, all reads were trimmed to 110 bp, and quality controlled reads were aligned to the North African dromedary genome assembly (NCBI accession: GCA_000803125.1) using BWA-MEM51. ddRAD reads were then processed with Stacks v.1.3552. Out of nearly half a million RAD loci, 88,836 SNPs passed the imposed filtering criteria. The pstack module was run with a minimum coverage of 3 reads to call a haplotype, while SNPs were called using a bounded model to highest = 0.5 and alpha = 0.05. Cstacks and sstacks modules were run with default parameters. Population module was run requiring a minimum genotyping of 75% of individuals to score loci, along with a calling likelihood filtering threshold of −25.
The retrieved raw SNP data were stringently filtered for missing values with PLINK 1.0753, first excluding individuals with more than 25% missing genotypes (--mind 0.25), next setting a threshold of 0.01 for minor allele frequency (--maf 0.01) and finally, removing SNPs with a missing genotype rate of more than 25% (--geno 0.25). Due to high individual missingness, 13 samples were removed from the dataset as well as 35,013 SNPs after filtering for minor allele frequency and missing genotype rate. To exclude any potentially related individuals, a symmetric identical by state matrix was created with PLINK with a cutoff value of 0.05; one sample from Jordan (JO434) and three from Nigeria (NG852, NG877 and NG887) were removed from further analyses (Supplementary Data 1).
Considering the practice of dromedary and Bactrian camel crossbreeding especially in Central Asian countries10, we screened for potential hybridization with Bactrian camel present in the dataset. Paired-end ddRAD reads from all 122 dromedaries were simultaneously mapped to either the dromedary (NCBI accession: GCA_000803125.2)25 or Bactrian camel (NCBI accession: GCF_000767855.1)23 genomes using BBSplit v. 38.79 (https://sourceforge.net/projects/bbmap/), with the following settings: minratio = 1.0, ambiguous = toss, ambiguous2 = toss. We preprocessed these two genome assemblies with dustmasker v. 1.054, and the percent Bactrian camel was estimated as the number of reads that unambiguously mapped to the Bactrian camel genome divided by the total number of unambiguously mapped reads to both dromedary and Bactrian camel multiplied by 100. We removed three individuals from Iran (IR715, IR717, and IR719) and six from Kazakhstan (KZ888, KZ889, KZ890, KZ891, KZ892, and KZ893) (Supplementary Data 2) that had “far-out” values, which are those greater than the third quartile plus the interquartile range multiplied by three55.
Genome-wide summary statistics and population structure
SNPs were tested for Hardy–Weinberg Equilibrium and linkage disequilibrium using VCFTOOLS v0.1.1556 and as no SNP exceeded a FDR of 0.05, all were retained. For subsequent file conversions, PGDSPIDER version 2.1.1.357 was used. The expected (HE) and observed (HO) heterozygosities, AR, and inbreeding coefficients (FIS) were calculated with the R package Hierfstat v0.04-2258,59. We have used a parametric Welch t test implemented in R v.3.5.1 using the t.test function to compare mean HE, HO, and FIS between African and Asian dromedaries. Pairwise FST and AMOVA59 were analyzed with the program Arlequin 3.5.2.260.
We applied BayeScan 2.128 to identify FST outlier loci putatively under of selection using default settings with a FDR61 of 0.05. To understand if SNPs putatively detected under selection were linked to significant biological pathways, we screened the respective RAD sequences for genes using the annotation of the new CamDro2 assembly25 and assessed their protein function with Genecards (http://www.genecards.org). To consider also regions in potential linkage disequilibrium, we included genes 200 kbp upstream and downstream of the SNPs under selection in the analysis.
Individual-based PCA was performed with Adegenet v2.1.1 using the s.class option to represent principal components of known groups. Furthermore we applied ADMIXTURE v1.333 to assess ancestry and possible structure among dromedary populations (i.e., countries of origin), using the lowest fivefold cross-validation error to choose the best number of clusters (K), from K = 1 to K = 10. To understand the phylogenetic relationship among individuals, we applied the Neighbor-Net method62 implemented in SplitsTree463, which is a neighbor-joining algorithm for constructing phylogenetic networks from a genetic (allele sharing) distance matrix created in PLINK.
Estimating effective migration rates
We used the software estimated effective migration surfaces (EEMS)34 to investigate effective migration patterns in the global dromedary population. Based on a stepping-stone model, and assuming that migration is symmetric, EEMS uses Markov Chain Monte Carlo to estimate the diversity and migration parameters and produces maps which represent the posterior mean of effective migration and effective diversity across space. We performed three distinct runs, each consisting of 10 million MCMC iterations, discarding the initial 5 million as burn-in and saving every 49,995 iterations for a grid with 500 demes. All runs reached convergence and results were similar across replicates. The habitat polygon was obtained using Google Maps API v3 Tool (http://www.birdtheme.org/useful/v3tool.html) and results were plotted using the R package rEEMSplots as suggested in Petkova et al.34.
Demographic analysis of the global dromedary population
We assessed the demographic history of the species employing a coalescent-based multi-locus analysis with variable loci using BEAST2 v. 2.5.164, setting the Coalescent EBS40 as a tree prior and following Huson and Bryant63. EBS analysis was conducted on the global population where we randomly selected 50 RAD loci containing at least four but not more than six SNPs across at least 75% of the individuals using a custom R script (https://github.com/jelber2/RAD-Scripts/blob/master/RAD_Haplotypes.R)65. We repeated the EBS analysis for African and Asian dromedaries separately, but due to slight population structuring, Kenya and Hadhana populations were excluded from this analysis. We ran EBS analyses three times per continent, with 50 random RAD loci used in each run. A RAD locus-specific clock rate (per generation) was estimated by calculating the average number of differences between dromedary and Bactrian camel sequences, dividing by the length of the RAD loci, taking the average among SNP classes (number of SNPs per RAD locus from 4 to 6), dividing by the split time between the Bactrian camel and dromedary of ~4.4 million ybp23, and using a dromedary generation time of 5 years1. Each EBS was run for 2,100,000,000 chains using the RAD locus-specific clock rate of 1.809442e-08 to calibrate the time scale.
Statistics and reproducibility
Sample size is outlined in Supplementary Data 1. We have used a parametric Welch t test implemented in R v.3.5.1 using the t.test function to compare mean HE, HO, and FIS, and data are expressed as mean and SD. Pairwise FST values were performed including a minimum of three individuals per countries and their significance levels are represented with “+”. AMOVA were analyzed with the program Arlequin. P values below 0.05 are considered as statistically significant for all statistical tests in this work. All analyses are reproducible with access to genetic data (see “Data availability”).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All sequence files (.cram) are deposited at the European Nuceotide Archive with the accession number PRJEB38954 (http://www.ebi.ac.uk/ena/data/view/PRJEB38954). In addition, SNP data (.map and .ped) can be downloaded from Dryad66 (https://doi.org/10.5061/dryad.kh189322q).
Code availability
Computer code and scripts for the various analyses are available at Dryad66 (https://doi.org/10.5061/dryad.kh189322q).
References
Almathen, F. et al. Ancient and modern DNA reveal dynamics of domestication and cross-continental dispersal of the dromedary. Proc. Natl Acad. Sci. USA 113, 6707–6712 (2016).
de Barros Damgaard, P. et al. The first horse herders and the impact of early Bronze Age steppe expansions into Asia. Science 360, 1–9 (2018).
Trevor-Roper, H. The Invention of Tradition: the Highland Tradition of Scotland (Cambridge University Press, Cambridge, 1983).
Ramadan, S. & Inoue-Murayama, M. Advances in camel genomics and their applications: a review. J. Anim. Genet. 45, 49–58 (2017).
Sala, R. The domestication of camel in the literary, archaeological and petroglyph records. J. Arid L. Stud. 26, 205–211 (2017).
Bulliet, R. W. The Camel and the Wheel (Columbia University Press, 1975).
Heiss, J. Caravans from South Arabia. in Camels in Asia and North Africa. Interdisciplinary Perspectives on their Significance in Past and Present (eds Knoll, E. M. & Burger, P.) 131–139 (Verlag der Österreichischen Akademie der Wissenschaften, 2012).
Wilson, R. T. The Camel (Longman, London, 1984).
Burger, P. A. The history of Old World camelids in the light of molecular genetics. Trop. Anim. Health Prod. 48, 905–913 (2016).
Faye, B. & Konuspayeva, G. The encounter between Bactrian and dromedary camels in Central Asia. in Camels in Asia and North Africa. Interdisciplinary Perspectives on their Significance in Past and Present (eds Knoll, E. M. & Burger, P.) 29–36 (Verlag der Österreichischen Akademie der Wissenschaften, 2012).
Çakırlar, C. & Berthon, R. Caravans, camel wrestling and cowrie shells: towards a social zooarchaeology of camel hybridization in Anatolia and adjacent regions. Anthropozoologica 49, 237–252 (2014).
Warmuth, V. M., Campana, M. G., Eriksson, A. & Bower, M. I. M. Ancient trade routes shaped the genetic structure of horses in eastern Eurasia. Mol. Ecol. 22, 5340–5351 (2013).
Liu, X. The Silk Road in World History (Oxford University Press, 2010).
Burger, P. A., Ciani, E. & Faye, B. Old World camels in a modern world—a balancing act between conservation and genetic improvement. Anim. Genet. 50, 598–612 (2019).
Uerpmann, H.-P. & Uerpmann, M. The appearance of the domestic camel in south-east Arabia. J. Oman Stud. 12, 235–260 (2002).
Uerpmann, H.-P. & Uerpmann, M. Archeozoology of camels in South-Eastern Arabia. in Camels in Asia and North Africa. Interdisciplinary Perspectives on their Significance in Past and Present (eds Knoll, E. M. & Burger, P.) 109–122 (Österreichischen Akademie der Wissenschaften, 2012).
Von den Driesch, A. & Obermaier, H. The hunt for wild dromedaries during the 3rd and 2nd millennia BC on the United Arab Emirates coast. Camel bone finds from the excavations at Al Sufouh 2 Dubai, UAE. in Documenta Archaeobiologiae Bd. 5—Skeletal series and their socio-economic context. (eds Grupe, G. & Peters, J.) 133–167 (Verlag Marie Leidorf GmbH, Rahden (Germany), 2007).
Grigson, C. Copper and Donkeys in the Early Iron Age of the Southern Levant: Timna Revisited. Levant 44, 82–100 (2012).
Grigson, C. The history of the camel bone dating project. Anthropozoologica 49, 225–235 (2014).
Spencer, P. B. S. & Woolnough, A. P. Assessment and genetic characterisation of Australian camels using microsatellite polymorphisms. Livest. Sci. 129, 241–245 (2010).
Jones, P. & Kenny, A. Australia’s Muslim Cameleers: Pioneers of the Inland, 1860s–1930s (Wakefield Press, 2010).
Mburu, D. N. et al. Genetic diversity and relationships of indigenous Kenyan camel (Camelus dromedarius) populations: implications for their classification. Anim. Genet. 34, 26–32 (2003).
Wu, H. et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 5, 1–9 (2014).
Fitak, R. R., Mohandesan, E., Corander, J. & Burger, P. A. The de novo genome assembly and annotation of a female domestic dromedary of North African origin. Mol. Ecol. Resour. 16, 314–324 (2016).
Elbers, J. P. et al. Improving Illumina assemblies with Hi-C and long reads: an example with the North African dromedary. Mol. Ecol. Resour. 19, 1015–1026 (2019).
Kijas, J. W. et al. A genome wide survey of SNP variation reveals the genetic structure of sheep breeds. PLoS ONE 4, 1–13 (2009).
Makina, S. O. et al. Genetic diversity and population structure among six cattle breeds in South Africa using a whole genome SNP panel. Front. Genet. 5, 1–7 (2014).
Foll, M. & Gaggiotti, O. A genome scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180, 977–993 (2008).
Rubin, C. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591 (2010).
Larrañaga, O. R. et al. Genomic selection signatures in sheep from the Western Pyrenees. Genet. Sel. Evol. 50, 9–21 (2018).
Gurgul, A. et al. Identification of genome-wide selection signatures in the Limousin beef cattle breed. J. Anim. Breed. Genet. 133, 264–276 (2016).
Faye, B., Abdallah, H. R., Almathen, F. S., Harzallah, B. D. & Al-Mutairi, S. E. Camel biodiversity. Camel phenotypes in the Kingdom of Saudi Arabia (FAO, Rome, 2011).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Petkova, D., Novembre, J. & Stephens, M. Visualizing spatial population structure with estimated effective migration surfaces. Nat. Genet. 48, 94–100 (2016).
Beja-Pereira, A. et al. The origin of European cattle: evidence from modern and ancient DNA. PNAS 103, 8113–8118 (2006).
Nixon, S., Rehren, T. & Guerra, M. F. New light on the early Islamic West African gold trade: coin moulds from Tadmekka, Mali. Antiquity 85, 1353–1368 (2011).
Nixon, S. Excavating Essouk-Tadmakka (Mali): new archaeological investigations of early Islamic trans-Saharan trade. Azania Archaeol. Res. Afr. 44, 217–255 (2009).
Gingrich, A. Honig und tribale Gesellschaft: Historischer Hintergrund, sozialer Gebrauch und traditionelle Erzeugung im südlichen Hijaz. in Tribale Gesellschaften der südwestlichen Regionen des Königreiches Saudi Arabien: sozialanthropologische Untersuchungen, Vol. 732 (eds Gingrich, A., Heiss, J. & Zötl, J.) 173–215 (Verlag der Österreichischen Akademie der Wissenschaften, 2006).
Gifford-gonzalez, D. & Hanotte, O. Domesticating animals in Africa: implications of genetic and archaeological findings. J. World Prehist. 24, 1–23 (2011).
Heled, J. & Drummond, A. J. Bayesian inference of population size history from multiple loci. BMC Evol. Biol. 8, 289 (2008).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Head, M. J., Pillans, B. & Farquhar, S. A. The Early—Middle Pleistocene Transition: characterization and proposed guide for the defining boundary. Episodes 31, 255–259 (2008).
Rook, L. & Martínez-Navarro, B. Villafranchian: the long story of a Plio-Pleistocene European large mammal biochronologic unit. Quat. Int. 219, 134–144 (2010).
Vislobokova, I. A. Main stages in evolution of Artiodactyla Communities from the Pliocene—Early Middle Pleistocene of Northern Eurasia: Part 2. Paleontol. J. 42, 414–424 (2008).
Yokoyama, Y., Lambeck, K., Deckker, P., De, Johnston, P. & Fifield, L. K. Timing of the last glacial maximum from observed sea-level minima. Nature 406, 713–716 (2000).
Stuart, A. J. Late quaternary megafaunal extinctions on the continents: a short review. Geol. J. 50, 338–363 (2015).
Kennedy, H. The Great Arab Conquests: How the Spread of Islam Changed the World We Live in (Da Capo Press, 2007).
Ming, L. et al. Whole-genome sequencing of 128 camels across Asia reveals origin and migration of domestic Bactrian camels. Commun. Biol. 3, 1–9 (2020).
Fages, A. et al. Tracking five millennia of horse management with extensive ancient genome time series. Cell 177, 1419–1435 (2019).
Edwards, C. J., Baird, J. F. & Machugh, D. E. Taurine and zebu admixture in Near Eastern cattle: a comparison of mitochondrial, autosomal and Y-chromosomal data. Anim. Genet. 38, 520–524 (2007).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Morgulis, A., Gertz, E. M., Schäffer, A. A. & Agarwala, R. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040 (2006).
Tukey, J. W. Exploratory Data Analysis, Vol. 2 (1977).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Lischer, H. E. L. & Excoffier, L. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012).
Goudet, J. HIERFSTAT,a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186 (2005).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application. Genet. Soc. Am. 131, 479–491 (1992).
Excoffier, L. & Lischer, H. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Bryant, D. & Moulton, V. Neighbor-Net, an agglomerative algorithm for the construction of phylogenetic networks. Mol. Biol. Evol. 21, 255–265 (2002).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Bouckaert, R. et al. BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15, e1006650 (2019).
Trucchi, E. et al. King penguin demography since the last glaciation inferred from genome-wide data. Proc. R. Soc. B Biol. Sci. 281, 1–8 (2014).
Lado, S. et al. Dryad repository for computer codes and scripts. Dryad https://doi.org/10.5061/dryad.kh189322q (2020).
Acknowledgements
We are very grateful to all veterinarian colleagues and camel owners for their agreements to submit sample aliquots for scientific purposes. We thank Steve Smith for lab support and Jukka Corander for general discussion. Samples collection was performed during the Austrian Science Fund (FWF) project number P24607-B25 (to P.B.) and the EU project EU ENPI CBC MED Project PROCAMED. B.1.1/493 (to. E.C.); ddRAD sequencing was performed in the frame of the EU project EU ENPI CBC MED Project PROCAMED. B.1.1/493 (to. E.C.); S.L. and J.P.E. acknowledge funding from the FWF project number P29623-B25 (to P.B.).
Author information
Authors and Affiliations
Contributions
S.L. performed analysis and wrote the first draft of the paper, J.P.E., A.D., D.S., E.T., E.C., and P.B. performed analyses, F.A., N.S., and M.H.B. contributed essential samples, E.C. and P.B. conceived and managed the project and wrote the paper. All authors provided valuable discussions, commented, and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing non-financial interests. Author D.S. is employed by the sequencing company IGA Technologies, Udine, Italy. All other authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lado, S., Elbers, J.P., Doskocil, A. et al. Genome-wide diversity and global migration patterns in dromedaries follow ancient caravan routes. Commun Biol 3, 387 (2020). https://doi.org/10.1038/s42003-020-1098-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-1098-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
