
Abstract
The marbled crayfish (Procambarus virginalis) represents a very recently evolved parthenogenetic freshwater crayfish species that has invaded diverse habitats in Europe and in Madagascar. However, population genetic analyses have been hindered by the homogeneous genetic structure of the population and the lack of suitable tools for data analysis. We have used whole-genome sequencing to characterize reference specimens from various known wild populations. In parallel, we established a whole-genome sequencing data analysis pipeline for the population genetic analysis of nearly monoclonal genomes. Our results provide evidence for systematic genetic differences between geographically separated populations and illustrate the emerging differentiation of the marbled crayfish genome. We also used mark-recapture population size estimation in combination with genetic data to model the growth pattern of marbled crayfish populations. Our findings uncover evolutionary dynamics in the marbled crayfish genome over a very short evolutionary timescale and identify the rapid growth of marbled crayfish populations as an important factor for ecological monitoring.
Similar content being viewed by others

Introduction
Parthenogenetic reproduction, i.e., the development of embryos from unfertilized eggs, has been described in many groups of animals1. Arthropod examples include parthenogenetic lineages of silkworms (Bombyx mori), water fleas (Daphnia pulex), weevils (Otiorhynchus scaber), and stick insects (Timema spp.)2,3,4,5. Complete genome sequences from parthenogenetic animals have presented unique features, and it has been suggested that parthenogenetic genomes evolve toward effective haploidy, as predicted by the Meselson effect6,7. However, these processes require parthenogenesis over millions of years. Also, a comparative analysis of 24 parthenogenetic genomes revealed a substantial degree of diversity in their features, which reflects the considerable biological differences among the corresponding species8.
Marbled crayfish (Procambarus virginalis) are obligate parthenogenetic freshwater crayfish that were first described in the German aquarium trade9,10. The species is a recent descendant of the slough crayfish (Procambarus fallax), which is an abundant, sexually reproducing freshwater crayfish species in peninsular Florida and southern Georgia11. Marbled crayfish may have originated from P. fallax through an evolutionary very recent macromutation, which resulted in instant reproductive isolation12. All known animals can likely be traced back to an initial population that was founded in 199510. As such, the evolutionary age of the species appears to be ~25 years.
Parthenogenesis can cover a variety of reproductive mechanisms13. Automixis is characterized by the fusion of meiotic products, with the production of genetically distinct offspring due to genetic recombination. Apomixis, on the other hand, is characterized by a bypass of meiotic recombination, resulting in clonal progeny that represents an exact copy of the maternal genotype. Early reproductive studies, in combination with microsatellite analyses, suggested that marbled crayfish utilize apomictic parthenogenesis, which results in genetically uniform offspring14,15. The absence of meiosis during oocyte maturation was also concluded from histological studies of oocyte nuclei16. More recently, it has been described that the first meiotic division is defective in marbled crayfish, but the chromosomes recover and proceed through the second meiotic division17. While this finding is consistent with apomictic parthenogenesis, the absence of meiotic recombination events in marbled crayfish remains to be confirmed.
A de novo draft assembly of the marbled crayfish genome with a genome size of approximately 3.5 Gbp and >21,000 genes has been published recently18. It is characterized by a triploid AAB genotype, with a duplicated “A” haplotype resulting from autopolyploidization and a heterozygous “B’“ haplotype presumably resulting from fertilization18. Comparative whole-genome sequencing (WGS) of animals from various aquarium lineages and from wild populations suggested that the marbled crayfish meta-population represents a single, genetically homogeneous clone18. Genetic variants are generated by the ongoing accumulation of natural mutations18, but their numbers are greatly limited by the extremely young evolutionary age of the species.
Due to their overall robustness and aesthetic appeal, marbled crayfish have become popular and widely distributed aquarium pets19. In parallel, the animals have increasingly colonized wild habitats. It is generally assumed that these populations were founded by anthropogenic releases and thus represent offspring of a single animal. This is exemplified by the situation in Madagascar, where the animals were first detected around 200620,21, and have now populated an area of 100,000 km2 that encompasses many diverse habitats18,22. Isolated wild populations have also been described in Germany and in several other European countries23,24,25,26,27,28,29, but their genomic variation has not been analyzed yet.
The clonality of marbled crayfish genomes presents unique challenges and requirements for the analysis of genomic variation. Marbled crayfish populations are groups of individuals that coexist spatially and temporally, but appear to miss classical attributes of panmictic populations, like random mating of individuals and active recombination. Hence, the population genetic structure of clonal marbled crayfish cannot be characterized by allele frequency dynamics, as the underlying processes are typical for sexually reproducing populations30,31. Our study addresses these issues and provides information about the emerging genetic differentiation within the marbled crayfish population.
Results
Survey of wild marbled crayfish populations in Europe
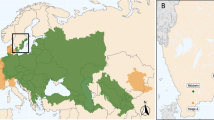
For a systematic genetic comparison of marbled crayfish populations in Europe, we surveyed known populations. This revealed that some reports had described transient appearances of single animals or groups of animals (probably from recent anthropogenic releases), while others described wild populations that have now stably persisted over several years. We restricted our study to stable wild populations and confirmed the taxonomic identity of the animals by genetic authentication of multiple specimens from 15 populations and from nine different European countries (Supplementary Table S1). Our results thus provide a systematic survey of stable wild marbled crayfish populations in Europe (Fig. 1).

The map was generated with QGIS 3.2.0, locations of specific populations are indicated by red dots (see Supplementary Table S1 for details).
WGS of wild marbled crayfish populations
While marbled crayfish can be considered as a single genetic clone, they also show a limited number of genetic polymorphisms in their nuclear genomes that result from the accumulation of genetic mutations within lineages over time18. Population-specific patterns of polymorphisms allow the analysis of the genetic relationships between populations. We, therefore, obtained WGS data for ten animals, representing ten populations in eight countries (Supplementary Table S2), as well as for four additional animals from a single wild population. The average mapping coverage was >30x for most samples (Supplementary Table S2), thus permitting detailed genetic analyses.
Comparative genome analysis
Comparative analysis of marbled crayfish genomes was based on the identification and pairwise comparison of single-nucleotide variant (SNV) patterns (see “Methods” for details), using the genome sequence of a specimen from the oldest known aquarium lineage of marbled crayfish (founded in 1995) as a reference. The numbers of SNVs ranged from 5722 to 6007. This is an order of magnitude below the frequency of SNVs in clonal daphniids32 and likely reflects the young evolutionary age of marbled crayfish. A corresponding distance tree showed high genetic similarity among the wild European populations of marbled crayfish and their more distant relationship to the ancestral aquarium lineage (Fig. 2A). Parallel analysis of mitochondrial genomes showed identical sequences for all marbled crayfish specimens and a clear separation from the reference mitochondrial genome of the sexually reproducing mother species, P. fallax. (Supplementary Fig. S1). These findings confirm the monoclonality of marbled crayfish from established wild populations across Europe and also suggest emerging genetic differentiation within the meta-population.

A Distance tree. Scale bar: 500 counts, representing 500 SNV from the set of common SNVs for all samples. The oldest known and likely foundational aquarium lineage of marbled crayfish is included as a reference (red star). B Bar plots illustrating the number of unique and shared SNVs. 6–9 and 2–5 indicate the number of SNVs that are shared by 6 to 9 and 2–5 animals, respectively. C Genome-wide analysis of variant allele frequency (VAF). Lines represent the average VAF per window of 10,000 heterozygous positions for each of the ten reference specimens. Variant allele frequencies are highly similar for all specimens and do not provide any evidence for loss of heterozygosity. The red line indicates the reference genome.
Further analysis showed that the proportion of unique, partially shared, and fixed polymorphisms were similar for all representative individuals (Fig. 2B). In addition, the number of unique SNVs was consistently low (Fig. 2B), suggesting minimal numbers of specific mutations for each population. Finally, we also addressed the possibility of low levels of recombination in marbled crayfish. Genome-wide analysis of variant allele frequency (VAF) failed to reveal any evidence for loss of heterozygosity (Fig. 2C). These results are consistent with the notion that marbled crayfish reproduce by apomictic parthenogenesis14,15 and suggest that genetic variation in marbled crayfish is driven by natural mutation.
Incipient genetic separation of marbled crayfish populations
To characterize the genomic variation of a single population, we analyzed WGS datasets from five animals from the type locality (Lake Reilingen, Germany). After processing the dataset through the previously established pipeline (Supplementary Table S2), a set of 3635 shared SNVs was identified in the five genomes, which showed a distinct overlap with the set of 2607 shared SNVs in the nine genomes from other European populations (Fig. 3A). Principal component analysis (PCA) on the complete set of SNVs from these specimens showed that the five samples from Lake Reilingen were clearly separated from the other samples (Fig. 3B), which is consistent with the close genetic relationship of animals from specific populations.

A Venn diagram demonstrating the level of overlap of shared SNVs between the Lake Reilingen and other European populations. From the complete set of 7858 polymorphic sites, 3635 were shared among the Lake Reilingen specimens, and 2607 were shared among the other European specimens. B Principal component analysis (PCA) of multiple specimens from Lake Reilingen (red dots) and other European populations (blue dots).
In subsequent steps, we integrated the European WGS datasets with previously published WGS datasets from Madagascar18 and a newly generated dataset from Madagascar (Supplementary Table S2). After data processing and variant calling for the common dataset, we defined a set of 7537 SNVs from the European (N = 10) and Malagasy (N = 5) samples (Fig. 4A). These analyses indicated a lower number of shared polymorphisms for the European population (Fig. 4A), which is likely influenced by the higher number of European samples in the analysis. Furthermore, the considerable number of shared polymorphisms between European and Malagasy animals (Fig. 4A) likely reflects the distance of the wild populations from the ancestral aquarium lineage, which provided the reference genome sequence18. Furthermore, PCA showed clear segregation into two clusters according to the geographic region of origin (Fig. 4B). The visualization of SNV patterns also suggested the presence of defined clusters with population-specific variation patterns for the European and Malagasy populations (Fig. 4C), consistent with the existence of population-specific genetic signatures. Together, these findings illustrate the emerging differentiation of the marbled crayfish genome in geographically separated populations.

A Venn diagram demonstrating the level of overlap of shared single-nucleotide variants (SNVs) between the European and Malagasy populations. B Principal component analysis (PCA) of representative specimens from ten European and five Malagasy populations. C Heatmap of SNV patterns, based on the 8912 common polymorphic sites. The color represents the average number of SNVs per site, as indicated. The clustering reflects the specific geographical SNV patterns.
Finally, we also annotated the complete set of SNVs (N = 16,564) that was identified in our analyses. The results showed that the majority (74%) were located in intergenic regions and only 4% were located in coding regions (Supplementary Table S3). Moreover, the analysis of base changes showed a higher abundance of G to A and C to T transitions (Supplementary Table S4). Similar patterns have also been observed in other parthenogenetic arthropods33,34.
Size estimate and growth simulation of a marbled crayfish population
It is known that marbled crayfish populations can grow rapidly18, but population size estimates are currently not available. We, therefore, sought to apply our genetic analysis towards a better understanding of marbled crayfish population growth. Bathymetric mapping of Lake Reilingen (Fig. 5A) was used for planning a mark-recapture survey35 and revealed a maximum depth of 17 meters (Fig. 5B). In total, 18 traps were deployed (Fig. 5C), which were baited and checked daily for ten consecutive days. During this time period, a total of 394 animals was collected (Supplementary Table S5). 96% of these animals exceeded the trapping size cutoff of >6 cm (Supplementary Fig. S2), which was determined by the mesh width of the traps. After data processing (Supplementary Fig. S3), Schnabel’s formula was applied, resulting in a population size estimate of 22,962 (SE = 3613) sexually mature animals >6 cm. Further sampling and data analysis (see “Methods” for details) resulted in a total population size estimate of 192,000 (SE = 67,000) animals. The high error of the latter estimate is largely due to the difficulty to accurately determine the number of small animals in the lake. Nevertheless, the population size and corresponding population density is consistent with data reported for the closely related freshwater crayfish species P. fallax in Florida36.

A Picture of Lake Reilingen, a small (9 ha) lake in Germany with an established marbled crayfish population. Picture provided by Sina Tönges. B Bathymetric map of Lake Reilingen, generated with Deeper Smart Sonar software. Water depths are indicated by color, North is indicated by the arrowhead. C Positions of the 18 traps used for the population size estimate. Scale bar: 100 m. Map data: Google, Maxar Technologies. D Marbled crayfish population growth kinetics simulations. The horizontal dashed line represents the genetic variability of 1e+09 in the current Lake Reilingen population. Colored lines show the predicted genetic variability for exponential, logistic, and Allee growth models (see “Methods” for details). Dashed colored lines indicate error margins (SE).
Finally, we used our WGS datasets from Lake Reilingen to make inferences about the growth model of the population. In a first step, the SNV dataset of the four sequenced specimens from the population was used to estimate the total genetic variation occurring within the population (N = 192,000 animals). Subsequently, we applied three different growth models (logistic growth, exponential growth, and growth based on the Allee effect) to simulate the population growth, based on a range of physiological parameters for marbled crayfish reproduction (see “Methods” for details). We then calculated the probabilistic total genetic variability for the populations with different simulated growth models considering a well-established arthropod reference mutation rate37. Further analysis showed that the genetic variability with a simulated exponential growth intersects with the variability of the current Lake Reilingen population after 3.7 (±0.3) years (Fig. 5D). These findings suggest that marbled crayfish populations can have a very rapid initial growth phase until they reach saturation due to habitat resource limitations.
Discussion
The analysis of asexual genomes can inform on important evolutionary aspects of asexual reproduction8. Our study provides a genetic analysis of the marbled crayfish, an evolutionary very young species with a monoclonal population structure10,18. We confirm overall monoclonality and the absence of recombination in the marbled crayfish genome, consistent with the notion that marbled crayfish reproduce by apomictic parthenogenesis14,15. We also uncover the emergence of geographically stratified genotypes and provide inferences about the rapid growth of local colonies.
Clonal genomes present unique challenges for their analysis. We provide an approach for the analysis of clonal population patterns in the absence of allele frequency dynamics by using WGS for the detection of SNVs. In the context of the low genomic diversity within the populations, we applied PCA to detect the emerging divergence. The small number of population-specific mutations is consistent with the young age (<20 years) of the European and Malagasy populations, respectively20,21,23.
The lack of sexual reproduction and genetic recombination limits genetic variation and is often considered to constrain phenotypic diversity and environmental adaptation. However, the general-purpose genotype model suggests that asexuality can be evolutionarily successful through the selection of generalist lineages with large phenotypic plasticity that can occupy a variety of ecological niches38,39,40. Key examples are provided by bdelloid rotifers and darwinulid ostracods, which represent ancient generalist asexual organisms with wider geographical distribution and broader ecological tolerance than their sexual parent species. Clonal variation has been demonstrated in darwinulid ostracods and has been attributed to mutation accumulation over long evolutionary timeframes41,42. The analysis of marbled crayfish genomes will facilitate our understanding of the mechanisms of asexual genome diversification and adaptation. This could conceivably include the preservation of high heterozygosity levels, the selection of specific genetic variants, and changes in genome regulatory mechanisms43,44.
In conclusion, our study describes 15 stable populations of marbled crayfish from various European countries. It is very likely that a large number of additional populations has not been identified yet. This can be explained by the recent emergence of the animals, their nocturnal activity and the lack of awareness about their existence. eDNA-based methods45 might facilitate the detection of additional populations in the future and thus uncover the full extent of the marbled crayfish spread. Of note, our study also revealed a considerable number of animals in a relatively small habitat. The rapid growth of marbled crayfish populations likely represents a key factor in the emerging economic development of the animals in Madagascar22,46. Importantly, this rapid growth also suggests that marbled crayfish populations should be monitored to determine their impact on freshwater ecosystems.
Methods
DNA extraction and WGS
Animals were collected from various European wild populations (Supplementary Table S1) and from previously described populations in Madagascar18,22. Genomic DNA was isolated and purified from abdominal muscular tissue using a Tissue Ruptor (Qiagen), followed by proteinase K digestion and isopropanol precipitation. The quality of isolated genomic DNA was assessed via agarose gel electrophoresis and/or via TapeStation (Agilent). PCR genotyping was performed using the mitochondrial cytochrome B and the nuclear Dnmt1 markers18. Whole-genome sequencing was performed on an Illumina HiSeq platform (DKFZ Genomics and Proteomics Core Facility).
Read preprocessing
After quality control using FastQC, reads were trimmed with trimmomatic v.0.3247. Low-quality bases and adapters were removed with a leading and trailing Phred score <3 and reads were quality filtered by using a base sliding window of size 4 and an average quality per base below 20. Finally, trimmed reads shorter than 40 bases were removed.
Mapping and duplicate removal
Trimmed reads were aligned to the marbled crayfish genome (assembly version 0.4)18 using Bowtie 2 (version 2.2.6) with default parameters48. The resulting alignments were processed and filtered for duplicates using samtools (version 1.3)49. Also, alignments on short contigs (≤10 kb) were discarded to avoid loss of time in processing fragmented sequences. Sequencing statistics for each individual are summarized in Supplementary Table S2.
SNV calling and polymorphic site calculation
The Bayesian genetic variant detector Freebayes was used for variant calling with parameters set for triploidy, minimum mapping quality of 30, and minimum base quality of 20, to exclude low-quality reads and bases from the analysis. Filtering was based on a minimum read depth of 20, a maximum read depth of 200, and a minimum Phred-scaled quality score of 30 for each sample, using gatk-4.1.3.050 and SnpSift51. In addition, SNV association with genetic features was analyzed using SnpSift51. Filtering for indels was performed by custom filtering pipelines. The comparative analysis included all P. virginalis WGS datasets, and a WGS dataset from the oldest known aquarium lineage sample (Heidelberg) as a reference18. Only sites that were covered in the reference were included in the analysis. To streamline the analysis in the triploid background, heterozygous sites in the reference were eliminated, which focused the analysis of polymorphic sites on non-heterozygous loci. Polymorphic sites were identified by the presence of a single-nucleotide substitution relative to the reference sequence. Other types of substitutions such as insertions or deletions were also discarded from the analysis.
Tree construction
The re-coded matrix was used for the calculation of distances between pairs of individuals followed by neighbor-joining tree estimation with default parameters using the Ape package52. Finally, an unrooted phylogenetic tree was generated using the Phytools package53 and default parameters.
For the mitochondrial tree, WGS reads were trimmed and mapped onto the P. virginalis mitochondrial genome reference sequence (GenBank KT074364.1). After filtering for quality parameters and duplicate removal, the alignment files were used for the generation of consensus sequences of mitochondrial genomes. The genetic divergence was calculated by multiple sequence alignment in Clustal Omega with default parameters. The phylogenetic tree was constructed based on maximum likelihood estimation using PhyML54 with default parameters.
Loss of heterozygosity analysis
The initial SNV dataset was used for the selection of heterozygous positions in the reference genome sequence. Sites were filtered for a Phred-scaled quality score >30 and a sequencing coverage >5×. This resulted in 250,453 heterozygous positions for VAF analysis. VAF per site was calculated as the ratio of the alternative allele read count to the total number of reads.
Population structure analysis
After variant calling, each genotype of the analyzed sample was permutated with integers from 0 (0/0/0) to 3 (1/1/1) according to the alternative allele dosage. A PCA for each set of markers was run using the R package PCAMETHODS55. The heatmap was generated based on the permutated matrix using the Pheatmap R package with default clustering parameters and a row aggregation parameter of kmeans_k = 200.
Population size estimate
The population size estimate was performed at Lake Reilingen (49.296893N, 8.544591E) in July 2018. A bathymetric map of Lake Reilingen was established using a Smart Sonar PRO + (Deeper) equipped with a GIS system and sonar. A total of 18 traps (mesh width: 1 cm) were deployed at various water depths (Fig. 5C and Supplementary Table S5), with an average distance of 105 m between traps. Traps were baited with rotten chicken meat and fish food every morning before 10:00. Traps were checked every 24 h, and all newly caught animals were marked with nail polish (each trap with a unique color), and released back at the catch site. This protocol was followed for 10 consecutive days to reduce confounding effects arising through animal reproduction, death, migration, and moulting.
To estimate the population size, Schnabel’s method35 was used (Supplementary Fig. S3A). As the mark-recapture saturation was not reached after 10 days, we used R to run a non-linear least square (NLS) regression to predict the catch size plateau (Supplementary Fig. S3B). More specifically, we used a negative exponential equation with two parameters:
where c is replaced by (1/e) to run the model on R (function AR.2 from the R package DRC). Furthermore, animal density per square meter was calculated from three sampling points of the lake through hand-catching with small nets. The size distribution of the hand-catches was also used to extrapolate the total number of animals from the number of sexually mature animals in the traps. Finally, numbers were corrected for the bathymetric profile of the lake: for a depth ≤4 m, covering 47,500 m2 of the lake and ten traps, each trap was assumed to be the sampling point for 4750 m2. For a depth of 5–10 m, covering 16,000 m2 and four traps, each trap was assumed to be the sampling point for 4000 m2. For a depth greater than 10 m, covering 29,000 m2 and four traps, each trap was assumed to be the sampling point for 7250 m2.
We evaluated the standard error (SE) by taking into account the uncertainties of the Schnabel estimate, of parameter a (from the negative exponential model above), of the surface covered by each trap (arbitrarily fixed SE = 10%), and the ratios of the considered populations relative to all measured populations (bootstrap, 10,000 replicates).
Population growth simulation
To simulate marbled crayfish population growth kinetics, the observed population estimates, a growth rate of 200 (±100) offspring per animal and per year, and a mutation rate of 3.6 ×10−9 per basepair and per year37 were used. Growth models included classical exponential growth, logistic growth, and growth based on a strong Allee effect. The classical exponential growth model describes a population size at any time point t by
and the population size
by initializing N(t0) = 1 for a clonal population with a growth rate of (exp(g) = 200 ± 100) per year. The logistic population growth model is described by
and the population size at time point ti+1- ti, where the maximum number of individuals was set to Nmax = 192,000 (SE = 34.98%) for the current population. The growth with strong Allee effect described by
where parameter A was chosen according to an established Allee threshold56 of −3.1.
For calculating the genetic variability of the Lake Reilingen population, the identified SNVs and the estimated mutation rate37 of the 3,511,656,756 bp marbled crayfish genome assembly18 were multiplied by population size estimates.
Statistics and reproducibility
This study is based on the analysis of WGS datasets of 19 P. virginalis specimens from 15 independent geographic locations. For one population (Lake Reilingen), five independent specimens were analyzed. Variant calling was performed on SNVs with a read coverage >20×. All statistical analyses and visualizations were performed using R (v3.5.0).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All sequencing data have been deposited as a NCBI BioProject (accession number PRJNA599283).
Code availability
Custom computer code is available on Github (https://github.com/OlenaMaiakovska/Population_Analysis_MC.git)57.
References
Suomalainen, E., Saura, A. & Lokki, J. Cytology and Evolution in Parthenogenesis (CRC Press, 1987).
Astaurov, B. L. Experimental alterations of the developmental cytogenetic mechanisms in mulberry silkworms: artificial parthenogenesis, polyploidy, gynogenesis, and androgenesis. Adv. Morphog. 6, 199–257 (1967).
Innes, D. J. & Hebert, P. D. N. The origin and genetic basis of obligate parthenogenesis in daphnia pulex. Evolution 42, 1024–1035 (1988).
Saura, A., Lokki, J. & Suomalainen, E. Origin of polyploidy in parthenogenetic weevils. J. Theor. Biol. 163, 449–456 (1993).
Schwander, T., Henry, L. & Crespi, B. J. Molecular evidence for ancient asexuality in timema stick insects. Curr. Biol. 21, 1129–1134 (2011).
Birky, C. W. Jr. Heterozygosity, heteromorphy, and phylogenetic trees in asexual eukaryotes. Genetics 144, 427–437 (1996).
Mark Welch, D. & Meselson, M. Evidence for the evolution of bdelloid rotifers without sexual reproduction or genetic exchange. Science 288, 1211–1215 (2000).
Jaron, K. S. et al. Genomic features of parthenogenetic animals. J. Hered. https://doi.org/10.1093/jhered/esaa031 (2020).
Scholtz, G. et al. Ecology: parthenogenesis in an outsider crayfish. Nature 421, 806 (2003).
Lyko, F. The marbled crayfish (Decapoda: Cambaridae) represents an independent new species. Zootaxa 4363, 544–552 (2017).
Martin, P., Dorn, N. J., Kawai, T., van der Heiden, C. & Scholtz, G. The enigmatic Marmorkrebs (marbled crayfish) is the parthenogenetic form of Procambarus fallax (Hagen, 1870). Contrib. Zool. 79, 107–118 (2010).
Vogt, G. et al. The marbled crayfish as a paradigm for saltational speciation by autopolyploidy and parthenogenesis in animals. Biol. Open 4, 1583–1594 (2015).
Schön, I., Martens, K. & van, Dijk P. Lost Sex. The Evolutionary Biology of Parthenogenesis (Springer, 2009).
Martin, P., Kohlmann, K. & Scholtz, G. The parthenogenetic Marmorkrebs (marbled crayfish) produces genetically uniform offspring. Naturwissenschaften 94, 843–846 (2007).
Vogt, G. et al. Production of different phenotypes from the same genotype in the same environment by developmental variation. J. Exp. Biol. 211, 510–523 (2008).
Vogt, G., Tolley, L. & Scholtz, G. Life stages and reproductive components of the Marmorkrebs (marbled crayfish), the first parthenogenetic decapod crustacean. J. Morphol. 261, 286–311 (2004).
Kato, M., Hiruta, C. & Tochinai, S. The behavior of chromosomes during parthenogenetic oogenesis in Marmorkrebs Procambarus fallax f. virginalis. Zool. Sci. 33, 426–430 (2016).
Gutekunst, J. et al. Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nat. Ecol. Evol. 2, 567–573 (2018).
Chucholl, C. Marbled crayfish gaining ground in Europe: the role of the pet trade as invasion pathway. in Freshwater Crayfish: Global Overview (eds Kawai, T. et al.) 83–114 (CRC Press, 2015).
Jones, J. P. G. et al. The perfect invader: a parthenogenic crayfish poses a new threat to Madagascar’s freshwater biodiversity. Biol. Invasions 11, 1475–1482 (2009).
Kawai, T. et al. Parthenogenetic alien crayfish (Decapoda: Cambaridae) spreading in Madagascar. J. Crust. Biol. 29, 562–567 (2009).
Andriantsoa, R. et al. Ecological plasticity and commercial impact of invasive marbled crayfish populations in Madagascar. BMC Ecol. 19, 8 (2019).
Chucholl, C. & Pfeiffer, M. First evidence for an established Marmorkrebs (Decapoda, Astacida, Cambaridae) population in Southwestern Germany, in syntopic occurrence with Orconectes limosus (Rafinesque, 1817). Aquat. Invasions 5, 405–412 (2010).
Lipták, B. et al. Expansion of the marbled crayfish in Slovakia: beginning of an invasion in the Danube catchment? J. Limnol. 75, 305–312 (2016).
Novitsky, R. A. & Son, M. O. The first records of Marmorkrebs [Procambarus fallax (Hagen, 1870) f. virginalis] (Crustacea, Decapoda, Cambaridae) in Ukraine. Ecol. Montenegrina 5, 44–46 (2016).
Patoka, J. et al. Predictions of marbled crayfish establishment in conurbations fulfilled: evidences from the Czech Republic. Biologia 71, 1380–1385 (2016).
Pârvulescu, L. et al. First established population of marbled crayfish Procambarus fallax (Hagen, 1870) f. virginalis (Decapoda, Cambaridae) in Romania. Bioinvasions Rec. 6, 357–362 (2017).
Deidun, A. et al. Invasion by non-indigenous freshwater decapods of Malta and Sicily, central Mediterranean Sea. J. Crust. Biol. 38, 748–753 (2018).
Ercoli, F., Kaldre, K., Paaver, T. & Gross, R. First record of an established marbled crayfish Procambarus virginalis (Lyko, 2017) population in Estonia. Bioinvasions Rec. 8, 675–683 (2019).
Charlesworth, B. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 10, 195–205 (2009).
Ellegren, H. & Galtier, N. Determinants of genetic diversity. Nat. Rev. Genet. 17, 422–433 (2016).
Munoz, J., Chaturvedi, A., De Meester, L. & Weider, L. J. Characterization of genome-wide SNPs for the water flea Daphnia pulicaria generated by genotyping-by-sequencing (GBS). Sci. Rep. 6, 28569 (2016).
Flynn, J. M., Chain, F. J., Schoen, D. J. & Cristescu, M. E. Spontaneous mutation accumulation in Daphnia pulex in selection-free vs. competitive environments. Mol. Biol. Evol. 34, 160–173 (2017).
Fazalova, V. & Nevado, B. Low spontaneous mutation rate and pleistocene radiation of pea aphids. Mol. Biol. Evol. 37, 2045–2051 (2020).
Krebs, C. J. Estimating abundance in animal and plant populations. in Ecological Methodology https://www.zoology.ubc.ca/~krebs/downloads/krebs_chapter_02_2020.pdf (2014).
van der Heiden, C. A. & Dorn, N. J. Benefits of adjacent habitat patches to the distribution of a crayfish population in a hydro-dynamic wetland landscape. Aquat. Ecol. 51, 219–233 (2017).
Liu, H. et al. Direct determination of the mutation rate in the bumblebee reveals evidence for weak recombination-associated mutation and an approximate rate constancy in insects. Mol. Biol. Evol. 34, 119–130 (2017).
Vandel, A. La parthénogenèse géographique. Contribution à l’étude biologique et cytologique de la parthénogenèse naturelle. Bull. Biol. Fr. Belg. 62, 164–281 (1928).
Baker, H. G. Characteristics and modes of origin of weeds. in The Genetics of Colonising Species (eds Baker, H. G. & Stebbins, G. L.) 147–172 (Academic Press, 1965).
Tilquin, A. & Kokko, H. What does the geography of parthenogenesis teach us about sex? Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 20150538 (2016).
Van Doninck, K., Schon, I., De Bruyn, L. & Martens, K. A general purpose genotype in an ancient asexual. Oecologia 132, 205–212 (2002).
Van Doninck, K., Schon, I., Martens, K. & Backeljau, T. Clonal diversity in the ancient asexual ostracod Darwinula stevensoni assessed by RAPD-PCR. Heredity 93, 154–160 (2004).
Gatzmann, F. et al. The methylome of the marbled crayfish links gene body methylation to stable expression of poorly accessible genes. Epigenetics Chromatin 11, 57 (2018).
Carneiro, V. C. & Lyko, F. Rapid epigenetic adaptation in animals and its role in invasiveness. Integr. Comp. Biol. 60, 267–274 (2020).
Mauvisseau, Q., Tönges, S., Andriantsoa, R., Lyko, F. & Sweet, M. Early detection of an emerging invasive species: eDNA monitoring of a parthenogenetic crayfish in freshwater systems. Manag. Biol. Invasions 10, 461–472 (2019).
Andriantsoa, R. et al. Perceived socio-economic impacts of the marbled crayfish invasion in Madagascar. PLoS ONE 15, e0231773 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Paradis, E., Claude, J. & Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
Revell, L. J. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223 (2012).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Stacklies, W., Redestig, H., Scholz, M., Walther, D. & Selbig, J. pcaMethods–a bioconductor package providing PCA methods for incomplete data. Bioinformatics 23, 1164–1167 (2007).
Johnson, K. E. et al. Cancer cell population growth kinetics at low densities deviate from the exponential growth model and suggest an Allee effect. PLoS Biol. 17, e3000399 (2019).
Maiakovska, O. & Legrand, C. OlenaMaiakovska/Population_Analysis_MC. Zenodo, https://doi.org/10.5281/zenodo.4110932 (2020).
Acknowledgements
We thank Esther Sciberras, Jeffrey Sciberras, Justin Formosa, and Daniela Latzer for sample collection. We also thank many colleagues and the Angelsportverein 1960 Reilingen e.V. for their support of the mark-recapture analysis at Lake Reilingen. F.E. acknowledges funding from the Estonian Ministry of Education and Research (institutional research funding project IUT 21-2 to Tiina Nõges) and from the Estonian Research Council (Mobilitas Pluss research project MOBJD29). This work was supported by funding from the Czech Science Foundation (project no. 19-04431S to A.K.).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
O.M. performed the data analysis and contributed to the paper writing; R.A. and S.T. designed and carried out the mark-recapture analysis; C.L. designed the population growth model; J.G. designed the SNV analysis; K.H. carried out genotyping experiments; L.P., R.N., A.W., A.S., A.D., F.E., and A.K. provided samples; F.L. conceived the study, and wrote the paper with contributions from other co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Maiakovska, O., Andriantsoa, R., Tönges, S. et al. Genome analysis of the monoclonal marbled crayfish reveals genetic separation over a short evolutionary timescale. Commun Biol 4, 74 (2021). https://doi.org/10.1038/s42003-020-01588-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-020-01588-8
This article is cited by
-
Development and application of haploid embryonic stem cells
Stem Cell Research & Therapy (2024)
-
Considerations for protein and amino acids in standardized reference diet for parthenogenetic marbled crayfish Procambarus virginalis model organism
Scientific Reports (2024)
-
Considerations for fatty acids in standardized reference diet for parthenogenetic marbled crayfish Procambarus virginalis model organism
Scientific Reports (2024)
-
Ecological importance of crayfish claws in consumption of mobile benthic prey
Aquatic Sciences (2024)
-
Studying phenotypic variation and DNA methylation across development, ecology and evolution in the clonal marbled crayfish: a paradigm for investigating epigenotype-phenotype relationships in macro-invertebrates
The Science of Nature (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
