
Abstract
Super-resolution fluorescence microscopy plays a crucial role in our understanding of cell structure and function by reporting cellular ultrastructure with 20–30 nm resolution. However, this resolution is insufficient to image macro-molecular machinery at work. A path to improve resolution is to image under cryogenic conditions. This substantially increases the brightness of most fluorophores and preserves native ultrastructure much better than chemical fixation. Cryogenic conditions are, however, underutilised because of the lack of compatible high numerical aperture objectives. Here, using a low-cost super-hemispherical solid immersion lens (superSIL) and a basic set-up we achieve 12 nm resolution under cryogenic conditions, to our knowledge the best yet attained in cells using simple set-ups and/or commercial systems. By also allowing multicolour imaging, and by paving the way to total-internal-reflection fluorescence imaging of mammalian cells under cryogenic conditions, superSIL microscopy opens a straightforward route to achieve unmatched resolution on bacterial and mammalian cell samples.
Similar content being viewed by others


Introduction
To fully understand the cellular function, we must combine cellular ultrastructure information with knowledge of protein interaction networks at the molecular level. Super-resolution fluorescence microscopy has underpinned our understanding of interacting molecular networks in cells. Techniques such as structured illumination microscopy (SIM)1, stimulated emission depletion (STED) microscopy2, and single molecule localization microscopy (SMLM) (including stochastic optical reconstruction microscopy (STORM)3, photoactivated localization microscopy (PALM)4, and fluorescence photoactivated localization microscopy (FPALM)5) image cellular processes at resolutions in the 20–100 nm range. However, when the goal is to ascertain structure–function relationships, the challenge is to improve resolution by at least ~2-fold, making it comparable with the typical size of interacting macro-molecular units (~10 nm).
Theoretically, resolution can be improved substantially by exploiting SMLM techniques under cryogenic conditions. In SMLM, resolution depends on the precision with which individual molecules can be localized6,7. This depends on the number of photons emitted by the sample, which substantially increases under cryogenic conditions, the number of photons collected by the objective lens, and its numerical aperture (NA)6,7. NAs >1 and preferably >1.4 are required to achieve high resolution, but these need immersion fluids to couple the sample to the objective. Liquid media freeze at cryogenic temperatures, so dry objectives (NA ≤ 0.9) are mostly used in this case, with the disadvantage of lower resolution. If this could be overcome, the use of cryo-fixation could become routine in super-resolution microscopy, with the added benefit that rapid freezing is more effective than chemical fixation at preserving ultrastructure and minimizing artefacts, as demonstrated by electron microscopy (EM)8.
There are several examples in the literature of super-resolution under cryogenic conditions. The increased photon yield under cryogenic conditions allowed Kaufmann et al.9 to compensate for the low NA of the objective to achieve sub-diffraction limited resolution (~125 nm) on green fluorescence protein (GFP)-labelled samples. Liu et al.10 attained ~46 nm resolution using DRONPA, a protein 2.5 times brighter than GFP. Other set-ups relied on custom-built stages to incorporate cryofluids (immersion fluids that remain liquid at cryogenic temperatures); Nahmani et al.11 achieved ~35 nm, close to the resolution currently possible at room temperature in cell samples with simple and/or commercial set ups, by using a water-immersion objective and a methanol/propanol mixture as the immersion fluid (NA = ~1.2). A similar approach was taken by Faoro et al.12, but this set-up was not applied to super-resolution microscopy. Highly specialized cryo-STORM systems functioning at liquid helium temperature have yielded ~Angstrom localization precision in isolated molecules13,14. However, these complex, custom-built cryo-stages have not yet been employed in cell imaging.
We have solved this challenge using super-hemispherical solid immersion lenses (superSILs), truncated balls made of solid materials of high refractive index that fill the gap between the objective and the sample, eliminating the requirement for coupling fluids15,16,17. By using a superSIL to couple the sample to a dry objective, the effective NA of the latter is enhanced up to the value of the refractive index of the superSIL. We and others previously demonstrated the enhanced resolution of superSIL microscopy at room temperature18,19,20,21,22. The breakthrough here was the realization that the high NA delivered by superSILs is eminently compatible with cryogenic conditions and particularly suited to STORM techniques, for which, to the best of our knowledge, they have not been used before despite the critical dependence of STORM on NA.
We describe a STORM set-up that uses solid immersion technology and cryogenic conditions to achieve 12 nm resolution on bacterial cells. Importantly, this was achieved using a low-tech set-up that can be implemented by any laboratory. By achieving NAs >1.45, we also remove the barrier to total internal reflection fluorescence (TIRF) imaging of mammalian cells under cryogenic conditions. Because our set-up outperformed in some respects a much more expensive state-of-the-art STORM system at room temperature, we conclude that superSIL microscopy could become the method of choice for straightforward exploitation of nanoscale cell imaging in microscopy at any temperature.
Results
A cryo-compatible superSIL-based STORM microscope
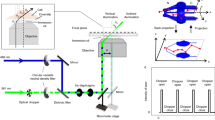
The key components are shown in Fig. 1. The optics employed to deliver and collect light consist of a 0.55 NA dry objective (Mitutoyo 100× Plan Apo SL Infinity Corrected) and a 1 mm diameter cubic zirconia superSIL (effective NA = 2.17). An achromatic doublet lens (200 mm focal length) was used as a tube lens before the EMCCD detector camera (Andor, iXon+ DU-897) (Supplementary Fig. 1). The superSIL was mounted into the central hole of a platinum disk using a thermally conductive cryo-adhesive (Loctite Stycast 2850 FT) (Supplementary Fig. 2). SuperSIL assemblies are robust and inexpensive (~US$20), and can be reused by cleaning the surfaces.

Schematic of superSIL microscope. a Side-view (top of the panel) and top-view (centre of the panel) of superSIL assembly. The side view shows the location of the sample plane (aplanatic surface of the superSIL). The photo of an assembly is at the bottom of the panel. b Schematic of key components in the microscope. An upright microscope configuration was employed. The blue shading illustrates the Köhler illumination laser beam and the green shading indicates the fluorescence emission. (For more info, see Supplementary Figs. 1–3)
The superSIL assembly is compatible with the standard EM grid-holder of an off-the-shelf liquid nitrogen (LN2)-cooled cryo-stage (Linkam, CMS-196). Samples were adhered to the flat surface of the superSILs and plunge-frozen together in liquid ethane (Supplementary Fig. 3). The convex surfaces of the superSILs faced upwards to the objective, and the flat surfaces faced downwards (Fig. 1b). SuperSIL assemblies thus play at cryogenic conditions the role played by liquid immersion media at room temperature. The assemblies were mounted on top of the brass bridge in the cryo-stage and translated in XY directions for fine positioning. The cryo-stage is self-contained with a built-in LN2 reservoir, and can be easily integrated into any conventional upright fluorescence microscope. Frozen samples were kept at 77 K by the LN2 vapour surrounding the brass bridge of the cryo-stage.
SuperSILs can increase the resolution of STORM
The best theoretical localization precision value (σ), and therefore resolution, achievable in STORM imaging is effectively determined by
where \(S \approx \frac{{0.75\lambda }}{{\pi NA}}\) is the standard deviation of a Gaussian function approximating the point spread function (PSF) of the microscope, N is the number of photons, and λ the imaging wavelength6,7. We speculated that the intrinsic properties of superSIL optics, namely their light collection efficiency23,24 and high NA, together with the increased photon budget at cryogenic conditions10, could deliver unprecedented resolution without requiring custom-made cryo-stages or other cumbersome, highly-specialized systems.
The reason for the enhanced light collection efficiency of superSILs is the electromagnetic coupling to optically dense material via evanescent fields, described in detail by Yoshita et al.24. To illustrate this, we carried out simulations using the known fraction of light collected in a microscope from an isotropic light source25:
where NA is the numerical aperture of an objective lens, and n is the refractive index of immersion medium. Fundamentally, the collection efficiency is determined by α which is half the maximal light collection angle (Fig. 2a).

Comparison of collection efficiency in superSIL and standard objectives. a Models and simulations of collection efficiency in conventional fluorescence microscopy using dry objective lenses and superSIL microscopy. Schematics of ray propagation from a single fluorophore (blue dots) in conventional (top left) and superSIL microscopy (top right). The fluorescence emission distribution is depicted by the blue curves. Collection efficiency versus NAs from simulations is plotted in the graph at the bottom, in which the solid and dashed curves illustrate conventional and superSIL microscopy cases, respectively. b Representative images of 100 nm diameter fluorescent beads taken using the superSIL, 50 × 0.9 NA objective and 100 × 0.55 NA objective on the same microscope platform with identical filters, camera settings and laser illumination power. Scale bars: 2 µm. c The box chart showing the distribution of intensities from multiple beads fitted with Gaussian profiles, corrected for changes in laser power density arising from the different magnifications. The box extends from the lower to upper quartile values of the data, with a black line at the median. The median of intensity was 5573, 3162 and 862 in the case of superSIL, 0.9 NA and 0.55 NA objective, respectively. The whiskers extend from the box to show the range of data falling within the 1.5× inter-quartile range from the quartiles
In optical microscopes using dry objective lenses, collection efficiency increases rapidly with higher NAs (Fig. 2a, black solid curve), with a theoretical maximum value of 50% at α = 90°, i.e. 2π solid angle, in the case of an isotropic fluorescence dipole emitter (Fig. 2a, blue curves in the conventional fluorescence microscopy illustration). For example, the theoretical collection efficiency is 8% and 28% with NAs of 0.55 and 0.9, respectively. In practice, the maximum collection efficiency of dry objectives is limited to ~30% because severe aberrations arise from off-axis rays with larger NAs26. When coupled with a superSIL, a dry objective lens (i.e. NA ≥ 1/n, where n is the refractive index of the superSIL material) can collect light propagating with a maximum 90° polar angle23,24. This means that collection efficiency is effectively determined by the directional fluorescence emission pattern of a dipole emitter at a dielectric surface (Fig. 2a, blue curves in the superSIL microscopy illustration). A fluorophore in close proximity to a dielectric surface emits many more photons towards the dielectric medium due to stronger electromagnetic coupling to the medium with higher refractive index via an evanescent field27. Considering a fluorophore is in direct contact with the flat surface of a superSIL and there is no reflection loss at the surfaces, we simulated the collection efficiency versus effective NAs, determined by the refractive indices of the superSIL materials (Fig. 2a, black dashed curve). The theoretical collection efficiency in the superSIL microscope is 91%, which is 11.4-fold and 3.3-fold larger than those in the microscope using 0.55 NA and 0.9 NA dry objectives.
The theoretical predictions of collection efficiency from the three NAs were tested experimentally using the set up in Fig. 1 under otherwise identical conditions. The results show that the superSIL collected 6.5-fold and 1.8-fold more photons compared to 0.55 NA and 0.9 NA dry objectives (Fig. 2b, c). This is in agreement within errors with the predicted values (Fig. 2a). The discrepancy comes from three sources: (1) Reflection loss from superSIL surfaces; (2) The smaller portion of fluorescence emission directed into the superSIL as the samples, i.e. 100 nm beads, were effectively 50 nm away from the surface. (3) More fluorescence emission collected in the conventional microscope measurement because the coverslips introduced directional emission at the glass–air interface. Nevertheless, we validate in our set-up the previously predicted higher photon collection efficiency of superSIL. This means that, according to Eq. (1), the enhancement in photons collected by the 2.17 NA superSIL would increase the achievable resolution by ~\(\sqrt N = 2.5 - {\mathrm{fold}}\) and ~\(\sqrt N = 1.3 - {\mathrm{fold}}\) compared to objectives with NAs of 0.55 and 0.9, respectively.
STORM resolution is also inversely proportional to the NA (Eq. (1)). In the case of a superSIL-dry objective combination, the effective NA is: \(NA_{eff} = n^2NA\), subject to \(NA_{max} = n.\) The maximum \(NA_{eff}\) is equal to the refractive index of the superSIL employed (2.17 in this case). Given this, one would expect enhancements in resolution of 3.9-fold and 2.4-fold from the larger NA of the superSIL system compared to standard dry objectives with NAs = 0.55 and 0.9, respectively.
Combining the two sources of resolution enhancement, namely the larger NA and the increase in photon collection, given Eq. (1), one would expect the localization precision that can be obtained using the combination of superSIL and dry objective to be ~10-fold and ~3-fold better than that obtainable with objectives with NAs of 0.55 and 0.9, respectively.
SuperSIL images plunge-frozen bacteria at 12 nm resolution
The first step to characterize the performance of the superSIL microscope was measuring the size of the PSF in the focal plane of object space. We acquired 500 photoluminescence images of the sparse cubic zirconia defect spots present on the flat surface of the superSILs (Supplementary Fig. 4). These intrinsic defects are single point emitters28, located at the aplanatic plane of the superSIL, which is the position with the fewest optical aberrations18. The full width at half maximum (FWHM) of the PSF of these images therefore gives an estimation of the best achievable wide-field, diffraction-limited, lateral resolution29. Representative images and statistical results show a PSF size of 153 ± 14 nm (Fig. 3a, c), in agreement with that previously reported19. The ~25% discrepancy between experiment and theory (118 nm predicted using the Houston criterion30 given an effective NA of 2.17) arises from the inefficiency of collecting rays propagating at angles approaching 90° from the optical axis18.

SuperSIL resolution characterization and imaging of E. coli cells under cryogenic conditions. Representative images of sub-diffraction limited objects from a the superSIL microscope and b the specialist system. Scale bar: 0.5 µm. c Box charts of FWHM of the PSFs for the superSIL and specialist systems showing the 25th and 75th percentiles of the data points. The thick line with a square at the center shows the mean value; the thin line in the box shows 50th percentile. The mean of FWHM was 153 ± 14 and 328 ± 86 nm in the case of superSIL and 0.9 NA objective, respectively. The whiskers show the standard deviation. d ATP-binding cassette (ABC) transporter protein PH1735 fused with EGFP in E. coli cells imaged in wide-field superSIL microscopy and e superSIL STORM. f Localization precision histogram from the image in e. g The enlarged image of the region of the cell indicated by the red dashed border box in e. h The enlarged image of the small region in the cell indicated by the orange dashed border box in g. i Line profile of the cross-section of two PH1735 protein clusters indicated by the cyan dashed lines in h. j Line profile of the cross-section of two PH1735 protein clusters closer to each other indicated by the red dashed line in h. k Line profiles of the cross-section of two adjacent single molecules (magenta and green) indicated by the magenta and green dashed lines in h. l FRC curve revealing the 12 nm resolution in the region shown in h. Scale bars: 1 μm (d, e), 100 nm (g) and 20 nm (h)
For comparison, we evaluated the PSF of a standard off-the-shelf cryo-compatible system, namely an FEI CorrSight fluorescence microscope with a standard dry objective (ZEISS EC Plan-Neofluar 40×/0.9 Pol M27), similar to that used by Kaufmann et al.9. A representative image and the statistical analysis of the PSF measurement using sub-diffraction limited 100 nm fluorescent beads (ThermoFisher, TetraSpeck) are shown in Fig. 3b, c. These results report a mean PSF size of 328 ± 86 nm, consistent with the theoretical value of 290 nm at the wavelength of 515 nm, according to Houston criterion30.
To explore the resolution improvement that could be achieved by combining superSIL microscopy and STORM we compared its performance with that of the CorrSight. We imaged in LN2 vapour, plunge-frozen Escherichia coli cells expressing the ATP-binding cassette (ABC) transporter PH1735, a putative multidrug transporter31, fused to enhanced GFP (EGFP), which displayed robust single molecule blinking behaviour at cryogenic temperature (Supplementary Fig. 5A, 5B). Representative wide-field and STORM images from the superSIL set up are shown in Fig. 3d, e. Images from the off-the-shelf system are shown in Supplementary Fig. 5C.
To reach quasi-equilibrium between fluorescent on and off states of the EGFP molecules, we delivered the same 1.1 kW cm−2 laser power density at 488 nm wavelength to the sample plane of both the superSIL and the off-the-shelf systems. Interestingly, the substantially larger photon delivery and collection efficiency of the superSILs allowed us to use a laser power of 5.55 mW, whilst for specialist cryo-STORM imaging we required a laser power of 60 mW. Thus, cryo-superSIL not only unlocks the potential to perform nanoscale STORM under cryogenic conditions, but enables the use of low-power, low-cost lasers.
Relevant to comparing their performance in STORM imaging, the superSIL and FEI CorrSight systems used different emission filters. The superSIL set-up employed a 512/25 nm bandpass filter, which transmits 57% of the fluorescence from EGFP (Supplementary Fig. 6A). The off-the-shelf system employed a 496 nm long pass filter that transmits 87% of the fluorescence (Supplementary Fig. 6B). Also relevant, the superSIL and FEI CorrSight systems used different detectors, an EMCCD in the superSIL microscope and a scientific CMOS (sCMOS) camera (Hamamatsu, ORCA-Flash4.0 V2) in the FEI CorrSight. We calibrated the detector settings used to image E. coli and found the EMCCD/sCMOS photon detection efficiency ratio was 1.2:1 (Supplementary Fig. 6E), showing that the EMCCD detector is 20% more efficient at the settings employed.
The ratio of localization precision between the superSIL and the FEI CorrSight settings predicted by Eq. (1) is (NA\(\sqrt N\))/(NA′\(\sqrt {N\prime }\)). Substituting the 2.17 NA and 0.9 NA′ values of the two systems, the number of photons (N and N′) collected by the lenses, and considering the different filter transmissions and detector responses, the localization precision delivered by the superSIL set-up in the conditions described above should be ~2.8-fold better than that of the FEI CorrSight. The localization precision we obtained with the superSIL system is σ = 7.7 ± 3.2 nm (Fig. 3f) and that obtained with the CorrSight is σ′ = 35.7 ± 9.4 nm (Supplementary Fig. 5C, D). The latter value is consistent with that previously achieved in comparable conditions9. These results are consistent within errors with theory predictions.
We note that in the case described above, the localization precision enhancement delivered by superSIL entirely depends on its larger NA. This is because, instead of the long pass filter used in combination with the 0.9 NA of the FEI CorrSight, a 25 nm bandpass filter was required to minimize the chromatic aberration of the superSIL (choices of emission filters were discussed and demonstrated in earlier work18,19). This means that the increase in collection efficiency of the superSIL with respect to the 0.9 NA objective (1.6-fold) is cancelled out by reduced filter transmission (0.65-fold). Therefore, in one colour imaging, when long pass filters can be used, one would expect a ~2.4-fold improvement of resolution using superSILs.
To illustrate the resolution of the cryo-superSIL system, in Fig. 3g we show an expanded view of the area within the red box in Fig. 3e. The orange box in Fig. 3g was further expanded in Fig. 3h, showing single PH1735-EGFP molecules embedded in the membrane together with larger features. The profiles of fluorescence intensity across the areas marked in Fig. 3h by dashed lines reveal two features separated by ~65 nm (Fig. 3i), two features separated by ~16 nm (Fig. 3j), and two pairs of molecules separated by ~12 and ~15 nm (Fig. 3k). This resolution is consistent with the observed localization precision of 7.7 nm. We also evaluated resolution using the Fourier Ring Correlation (FRC) method32. We used a fixed correlation threshold equal to 1/7 ≈ 0.143, the most appropriate for localization microscopy images, and identified a spatial frequency threshold valued at 0.084 nm−1 (Fig. 3l), revealing an FRC resolution of 12 nm. PH1735 is a homodimeric ABC transporter (thus, fused with two EGFP molecules), and these features probably belong to a full transporter. Taking into account the linker length between the PH1735 and EGFP, the measured separation of 12–15 nm probably represents a nucleotide-free inward-open transporter, which is consistent with distances measured on other ABC transporters such as McjD33. To our knowledge, the combination of the <8 nm localization precision and 12 nm resolution here demonstrated is the highest to date on intact cells. Our ability to resolve individual proteins in a dimer shows that the resolution of cryo-superSIL STORM is adequate to probe macro-molecular organization in cells.
To objectively verify the resolution of the superSIL under cryogenic conditions, we imaged plunge-frozen ATTO 647N-based calibration samples of DNA origami nanorulers (GattaQuant)34, illuminated with 11.17 mW laser power (λ = 642 nm), resulting in a power density of 2.2 kW cm−2. Representative wide-field and STORM images are shown in Fig. 4a, b. Individual nanoruler structures can be observed in the cryo-superSIL STORM image, demonstrating a localization precision of 10.1 ± 3.8 nm (Fig. 4c). An example of the mark-to-mark distances from the nanorulers in the image of 23 nm magnitude is shown in Fig. 4d.

Resolution evaluation of cryo-superSIL STORM using DNA origami nanorulers. Images of ATTO 647N nanorulers from (a) wide-field cryo-superSIL fluorescence microscopy and b cryo-superSIL STORM (Scale bars: 200 nm). A nanoruler image is shown in the magnified inset within the white border box in b. Scale bar: 10 nm. c Localization precision histogram from the cryo-superSIL STORM image. d Line profile of the cross-section of a nanoruler (green dots), indicated by the green dashed line in the magnified inset in b, and its Gaussian fit (green line)
Two-colour cryo-superSIL STORM imaging of bacterial cells
Two-colour imaging is required to study inter-molecular interactions. To investigate the possibility of two-colour imaging, we imaged plunge-frozen E. coli cells expressing the antibacterial peptide ABC transporter McjD fused with EGFP33,35,36,37, in which cell membranes were stained with the red fluorescent probe DiSC3(5). The DiSC3(5) dye is cationic and labels putative inner membrane nanodomains, i.e. the fluid lipid network38, which consists of negatively charged phospholipids.
We used laser powers of 1.1 and 2.2 kW cm−2 in the 488 and 642 nm channels, respectively. Two-channel imaging was sequential, with the same camera setting (30 ms exposure time per frame, 10 MHz 14 bit EM amplifier readout rate, 5.2× preamp setting and 300 EM gain). Features detected in both channels in wide-field fluorescence images were used as markers to align two-colour super-resolution images during image post-processing. As under cryogenic conditions, 25 nm bandpass emission filters were used to minimize chromatic aberration (Supplementary Fig. 6A). We note, however, that, unlike in one colour imaging, where long pass filters are possible, in two colour imaging 25 nm bandpass filters are commonly used in conventional set-ups. Thus we would expect no significant losses in resolution from the requirement of 25 nm bandpass filters in superSIL set-ups, at least in the shorter wavelength channel. This is confirmed in our set-up which gave localization precisions of 10.3 ± 3.8 nm in the yellow channel and 10.7 ± 3.4 nm in the red channel (Fig. 5b). Images reveal the distribution of McjD proteins (yellow) and the organization of the fluid lipid network (red) (Fig. 5a), the latter displaying perceptible helical structures in agreement with previous findings39. A high degree of segregation is observed between the McjD and the lipid network.

SuperSIL multi-colour imaging under cryogenic conditions. a McjD-EGFP (yellow) and membrane-DiSC3(5) (red) in E. coli cells imaged in (top) wide-field cryo-superSIL microscopy and (bottom) cryo-superSIL STORM. The overlays of two-colour images are shown in the right-side column in each row. Scale bar: 1 µm. b The histograms of localization precision of (top) EGFP and (bottom) DiSC3(5)
In these proof-of-concept experiments, different colour images were collected sequentially with the same camera, as the longitudinal axial chromatic aberration required refocusing when changing imaging colours. We are currently implementing modifications to the microscope by splitting the emission path to acquire the two colour images simultaneously, and also deploying fiducial markers, which should achieve image correlation accuracies comparable to those previously shown40.
Cryo-superSIL imaging of mammalian cells
Super-resolution microscopy under cryogenic conditions has been incompatible with TIRF illumination, because in cryogenic conditions the available objectives have NA ≤ 1.2, and to create the evanescent fields required by TIRF, NAs > 1.4 are required41,42. This has precluded the application of super-resolution microscopy to the investigation of cryo-frozen mammalian cells.
Similarly to liquid immersion objectives, the depth of focus of the combination of the superSIL and its partner dry objective lens is inversely proportional to the square of the effective NA29. This results in a narrow depth of focus, and thus similar properties to TIRF. We previously demonstrated TIRF imaging using superSILs but only at room temperature19. Under cryogenic conditions we found that the resolution of the superSIL changes by the approximately constant value of ±15% throughout the depth range of 10–100 nm (Supplementary Fig. 7). From this we conclude that the resolution of the superSIL-dry lens combination is maintained up to separations of 100 nm from the flat surface of the lens, a depth is comparable to that achievable by TIRF. This paves the way to investigate crucial processes in mammalian cells that require TIRF imaging under cryogenic conditions.
To verify in cells that the predicted resolution is maintained in the basal periplasmic section adjacent to the lens surface, we imaged plunge-frozen Chinese hamster ovary (CHO) cells expressing the epidermal growth factor receptor (EGFR), a key molecule in cancer research43. EGFR was labelled with Alexa Fluor 488 (Thermo Fisher). To extract areas of highest resolution from the wide-field images, we used a ‘rolling ball’ background reduction algorithm during image post-processing44. As shown in Fig. 6a, the TIRF-like appearance of the image is apparent45. A small EGFR cluster in the cryo-superSIL image showed an apparent width of 230 nm (Fig. 6b, c). Two adjacent clusters 197 nm apart are also clearly distinguishable (Fig. 6d, e). These values are indistinguishable to those predicted at depths from 10 to 100 nm (Supplementary Fig. 7), showing that conventional TIRF would be eminently possible at cryogenic temperatures using superSIL optics.

Wide-field superSIL microscopy under cryogenic conditions. a CHO cells expressing EGFR labelled with Alexa Fluor 488. b A cluster of EGFR indicated by the red dashed border box in b. c The profile of the cross-section of the EGFR cluster (red dots) indicated by the red dashed line in b, and its Gaussian fit (red line). d Two adjacent EGFR clusters indicated by the magenta dashed border box in a. e The profile of the cross-section of the two adjacent EGFR clusters (magenta dots) indicated by the magenta dashed line in d, and their Gaussian fits (magenta line). Scale bars: 2 μm (a) and 0.2 μm (b, d)
It is worth noting that the depth of focus of a superSIL microscope depends on the refractive index of the superSIL material, allowing a degree of depth ‘tuning’. SuperSIL materials of lower refractive indexes (e.g. Quartz) could be used to increase the depth of focus approximately 2.2-fold (Supplementary Table 1). This is an important consideration to exploit superSIL imaging in correlative light and electron microscopy (CLEM), where cell lamella thickness is in the range of 50–300 nm46. A suitable technique for nanoscale resolution under cryogenic conditions, like cryo-superSIL STORM, can provide true complementarity between EM and fluorescence microscopy, crucial to realize the promise of CLEM in biology47.
Ambient superSIL STORM outperforms off-the-shelf systems
To compare with the resolution improvement at cryogenic conditions, we characterized superSIL microscope performance at room temperature. We first verified that the freezing procedure had not altered the spectral dispersion and thermal expansion properties of the CZ superSILs (Supplementary Fig. 8). As shown in Fig. 7a, the statistical results of PSF measurements from CZ defect spots confirmed that the resolution of the superSIL system at room temperature (153 ± 15 nm) is indistinguishable from that measured under cryogenic conditions (Fig. 3a, c).

SuperSIL resolution performance at room temperature. a Image of a point object (top). Scale bar: 0.5 µm. The box chart (bottom) of the full width half maximum (FWHM) measurements of images from point emitters. The boxes show the 25th and 75th percentiles of the data points. The thick line with a square at the center shows the mean value; the thin line in the box shows 50th percentile. The mean of FWHM was 153 ± 15 nm. The whiskers show the standard deviation. b Comparison of wide-field images of live McjD-EGFP in E. coli cells. The superSIL image (left) has ×471 magnification, while the specialist microscope (right) has ×100 magnification. For comparison the inset contains a ×4.71 scaled-up image of a cell. Scale bar: 5 µm, and 1 µm in the inset. c STORM images (top) and localization precision histograms (bottom) of live McjD-EGFP E. coli cells in superSIL STORM (left) and specialist STORM (right). The insets show wide-field fluorescence images of the cells. Scale bar: 1 µm. d Resolution evaluation of STORM imaging in the specialist microscope. Left: Schematic of DNA origami nanorulers labelled with ATTO 647N dye molecules. Right: image of a field of nanorulers obtained from wide-field (top left) and STORM (top right). Scale bar: 200 nm. The STORM image of a nanoruler is shown in the inset, indicated by the white-border box. Scale bar: 20 nm. Bottom left: Localization precision histogram from the STORM image of the nanorulers. Bottom right: Profile of the cross-section of the nanoruler (magenta dots) in the magnified inset, and its Gaussian fit (magenta line)
We compared the performance of the superSIL microscope with that of a state-of-the-art, off-the-shelf STORM microscope (ZEISS Elyra equipped with an Alpha Plan-Apochromat ×100/1.46 NA oil immersion DIC objective). To reach quasi-equilibrium between the fluorescent on and off states of the EGFP molecules, power densities of 0.349 and 1.1 kW cm−2 (λ = 488 nm) were delivered at the sample plane in the superSIL and off-the-shelf microscopes, respectively. The ~3-fold lower power density delivered to the sample in the superSIL microscope was required to avoid fast photobleaching. Both microscopes were equipped with the same EMCCD type (Andor, iXon+ DU-897). To compensate for lower power density and obtain similar image intensity as that of the off-the-shelf Elyra system, the linear EMCCD gain in the superSIL microscope was set 3-fold higher. In the superSIL set-up, we again used the 512/25 nm bandpass emission filter (57% transmission). In the Elyra system we used a 535/50 nm bandpass (64% transmission) (Supplementary Fig. 6A, C).
We imaged live E. coli cells expressing the antibacterial peptide ABC transporter McjD fused with EGFP33,35,36,37. Images of non-fluorescent E. coli cells expressing McjD transporter with no EGFP are shown in Supplementary Fig. 9. Typical wide-field images of EGFP-stained bacterial features from the superSIL and off-the-shelf microscopes are shown in Fig. 7b. Of note, the combination of a superSIL with a ×100 dry objective lens results in a magnification of ×471, nearly 5 times greater than a conventional ZEISS ×100 oil immersion objective.
Figure 7c shows representative STORM images and corresponding molecular localization precision histograms. The predicted localization improvement of the superSIL set-up with respect to the Elyra system predicted by (NA\(\sqrt N\))/(NA′\(\sqrt {N\prime }\)) under the above set-ups is 1.6. In agreement within errors with this prediction, the results reveal a ~2-fold enhancement in localization precision using the superSIL (15.4 ± 4.1 nm) compared with the off-the-shelf system (31.3 ± 9.3 nm). In this case, the higher 2.17 NA of the superSIL contributes 1.5-fold and the increase in photon collection 1.13-fold, with 0.06-fold being lost by the 25 nm bandwidth of the bandpass filter.
Comparison of localization precision obtained with the superSIL system at room temperature (15.4 ± 4.1 nm) (Fig. 7c) with that obtained under cryogenic conditions (7.7 ± 3.2 nm) (Fig. 3f) shows that the higher photon budget under cryogenic conditions improves resolution by ~2-fold, consistent with our results when comparing the two off-the-shelf systems we used as controls. Indeed, both off-the-shelf systems deliver similar localization precisions at room (31.3 ± 9.3 nm) and cryogenic (35.7 ± 9.4 nm) (Supplementary Fig. 5D) temperatures because of the lower NA of the dry objective required for the latter. Allowing for differences in filter throughput and detector efficiency, the localization precision delivered by the 1.46 NA oil immersion lens with a collection efficiency of 36% with respect to the 0.9 NA dry lens with collection efficiency of 28% should have been 2-fold better, according to (NA\(\sqrt N\))/(NA′\(\sqrt {N\prime }\)). Because we find approximately the same localization precision in both off-the-shelf systems, this shows that the enhanced photon budget afforded at cryogenic temperatures has compensated for the expected ~2-fold difference.
The field of view in the superSIL images is relatively small, (maximum size of 17.4 × 17.4 μm2 in one frame), the result of high magnification and finite camera chip size. The usable field of view can be extended to 60 × 60 μm2 by scanning the sample in the lateral plane (Supplementary Fig. 10). Imaging becomes impractical beyond this area due to residual imaging aberrations17. Furthermore, it can be more difficult to handle samples, and more aberrations are present when imaging away from the surface.
Sample structure and labelling density can affect resolution48. To ascertain the ultimate localization precision obtainable at room temperature with the off-the-shelf Elyra STORM system, we imaged standard DNA origami calibration samples (GattaQuant), which carried ATTO 647N dye molecules ~20 times brighter than EGFP49. As shown in Fig. 7d, results revealed a localization precision of 17.6 ± 8.1 nm, in line with the manufacturer’s specifications. Interestingly, the best localization precision we could extract from the specialist system using ATTO 647N was no better than the 15.4 ± 4.1 nm returned by the superSIL system using the much dimmer EGFP fluorophore, so we conclude that the superSIL microscope outperforms the off-the-shelf STORM system. Moreover, the superSIL microscope costs ~20 times less than the specialist STORM system. A comparison of superSIL microscopy versus fluid immersion microscopy is summarized in Table 1.
Discussion
Our results show that by combining a superSIL and a low NA dry objective we have achieved our goal of improving 2-fold the resolution that can be achieved by STORM, using cell-friendly off-the-shelf equipment. Indeed, the localization precision and resolution attained are, to our knowledge, the best obtained to date on cell samples using a simple set-up. SuperSIL-based super-resolution is not limited to STORM, but can be combined with other established super-resolution imaging techniques such as SIM22 and STED20. Importantly, the superSIL’s high NA eliminates the barrier to combine cryo-imaging with TIRF, paving the way to the application of vitrification to super-resolution imaging in mammalian cells.
During characterization of the superSIL set-up, we found an unexpected bonus. The ultra-high NA of superSIL and its enhanced photon collection properties also deliver a better resolution at room temperature than the off-the-shelf STORM system. This means that, even if cryogenic conditions are unneccesary, e.g. when imaging live cells, a simple add-on based around the use of superSILs would enable any non-expert with a basic fluorescence microscope to achieve state-of-the-art super-resolution at low cost. In conditions where long pass filters can be used in conventional set-ups, the bulk of the increase in resolution that can be obtained from superSILs is due to the larger NA. This is because the increased photon collection of the superSIL will be cancelled out by the losses in the 25 nm bandpass filters required to minimize chromatic aberration. The latter is less likely to be an issue in multi-colour imaging, where bandpass filters are typically required in any case. We used superSILs in an upright microscope, but a simple modification of the superSIL assembly would easily enable the use of superSILs in inverted microscopes. The method therefore can overcome the costs and complexity inherent in specialist super-resolution set-ups by circumventing the use of expensive objectives, intricate multi-stage illumination paths, specialized sample stages, and high power lasers.
Given these advantages, we propose that low-cost superSIL technology has the potential to greatly extend the scope and the reach of super-resolution microscopy in cell biology. Furthermore, it has the potential to revolutionize cryo-CLEM, the use of which has been limited by the resolution mismatch between EM and super-resolution, largely restricting the optical microscopy element of the method to general identification of regions of interest. By delivering close to 10 nm resolution under cryogenic conditions, cryo-superSIL imaging is poised to bridge the resolution gap between fluorescence microscopy and EM, finally allowing sample registration at the nanoscale. Possible schemes for CLEM include imaging of cryogenically-sectioned samples by cryo-superSIL STORM and transmission EM, or the use of focus ion beam scanning EM to image and produce lamellae suitable for the cryo-superSIL microscope. Given the possibility of scaling down superSILs to very small and custom-variable sizes, it is possible to envisage these devices being incorporated into EM systems, allowing true correlative microscopy without the need to move the sample and relocate areas of interest.
In summary, the very high NA and efficient photon collection properties of superSILs deliver an unprecedented resolution to cell biology, simply and inexpensively. SuperSIL technology has the potential not only to transform super-resolution microscopy and CLEM, but also to increase the resolution attainable in any non-specialist laboratory at low-cost.
Methods
SuperSIL microscope
The light source was a laser beam combiner (Omicron, LightHUB), including 488 and 642 nm laser lines, and a 470 nm collimated LED (Thorlabs, M470L3-C5). For the single colour imaging of McjD-EGFP and PH1735-EGFP E. coli cells, the filter sets consisted of a 484 nm beam splitter (Semrock, FF484-FDi0) as dichroic filter, and a 512/25 nm bandpass filter (Semrock FF01-512/25-25) as emission filter. For the two-colour imaging of McjD-EGFP and membrane- DiSC3(5) E. coli cells, the filter sets consisted of quad-edge dichroic beam splitter (Semrock, Di01-R405/488/543/635-25×36) as dichroic filter and quad-band bandpass filter (Semrock, FF01-446/523/600/677-25) as emission filter, also used in the DNA origami nanoruler measurement.
SuperSIL assembly
The 1 mm diameter superSILs (Knight Optical Ltd., UK) were made of cubic zirconia (ZrO2), the cubic crystalline form of zirconium dioxide (ZrO2). The lenses’ refractive index is 2.17 and the Abbe number is 33.54 at the wavelength of 512 nm. This provides a high refractive index with medium dispersion suitable for superSIL microscopy. The assemblies were characterized by use of a coordinate measuring machine (OGP SmartScope ZIP 250 Coordinate Measuring Machine) to ensure the angle between the platinum disk and the superSIL’s flat surface is <1° (Supplementary Fig. 2B).
STORM data analysis
ZEISS ZEN software was used to process and render STORM images. In STORM image processing, various peak mask sizes were applied depending on pixel resolution and PSF size in each raw data set. The fit model was a two-dimensional Gaussian fit, and only single emitters from fluorophores were taken into account, whereas all multiple emitters were discarded. Following localization, displacements of molecules from drifts in the reconstructed images were corrected using feature detection and cross correlation. The counts from the raw images were first converted to the signal counts by deducting bias offset. Then the signal counts were converted to signal electrons by multiplying with the preamplifier gain. Finally, the signal electrons were converted to photon numbers by adding the detector electron-multiplying gain.
Sample preparation
Bacterial cell culture and staining: E. coli strain C43 expressing McjD fused with EGFP were prepared for imaging. Briefly, the overnight starter culture was diluted 1:100 in fresh LB media supplemented with 50 μg ml−1 kanamycin and grown at 37 °C until an optical density at 600 nm (O.D.600) of 0.6 was achieved. Then the expression of McjD was induced by adding IPTG (Isopropyl β-D-1-thiogalactopyranoside) to a final concentration of 1 mM at 25 °C overnight. The cells were spun down before freezing. The EGFP counts were measured using a Spectramax microplate reader (Molecular Devices) from 1 ml of culture re-suspended in 200 μl PBS (Thermofisher). For the analysis, the bacterial pellet was re-suspended in 2 ml of PBS to an optical density (O.D.600) of ~0.6 and kept on ice until plunge-freezing. For two-colour imaging, E. coli expressing McjD-EGFP were re-suspended in 250 μl PBS to O.D.600 ~ 0.6 and centrifuged at 10,000g for 5 min to wash out residues of culture medium. The pellet was then re-suspended in 25 μl of 100 nM DiSC3(5) in DPBS and incubated for 15 min on ice. 2.5 μl of sample were applied to the flat surface of each superSIL immediately prior to plunge-freezing.
Mammalian cell culture and staining: All reagents unless otherwise stated were from Thermo Scientific, UK. CHO cells expressing wtEGFR under an inducible Tet-ON promoter were grown in 5% CO2 in air at 37 °C in phenol-red free Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, 2 mM glutamine, 100 μg ml−1 hygromycin B and 100 μg ml−1 geneticin. All cells used were regularly tested for mycoplasma contamination. Cells were seeded at a density of 105 ml−1 on superSILS passivated with PEG-BSA nanogel as described previously50,51. Briefly, superSILs were etched with Piranha solution for 10 min, and thoroughly rinsed. Priming was performed for 5 min with Vectabond reagent (Vectorlabs) diluted 1:50 in acetone, followed by thorough rinsing. PEG-BSA nanogel was applied for 1 h at 37 °C, rinsed twice with PBS, capped with 20 mg ml−1 BSA in PBS for 1 h 37 °C, quenched with 1 M Tris pH 8.0 for 15 min, followed by three washes in PBS. Cells were cultured for 48 h, rinsed, subjected to nutrient starvation for 2 h at 37 °C to wash out EGFR ligands from the serum and then labelled with 5 nM EGF conjugated to Alexa Fluor 488 (Thermo Scientific) for 30 min at 4 °C. Clustering of EGFR was induced by a 5 min incubation at 37 °C prior to plunge-freezing.
DNA origami nanorulers: Samples were prepared according to manufacturer’s instructions. Briefly, superSILs were glow discharged for 120 s (negative) using a Quorum Q150T ES system, then washed 3 times with PBS, then immersed for 5 min in a solution of BSA-biotin 1 mg ml−1 in PBS and washed a further 3 times with PBS. Coating with neutravidin 1 mg ml−1 in PBS was also performed by immersion for 5 min, followed by three washes in PBS + 10 mM MgCl2 (immobilization buffer). 2.5 μl of DNA Origami diluted in immobilization buffer to a final concentration of 1:100 were applied to the flat surface of the superSILs, prior to blotting and plunge-freezing.
Plunge freezing: The superSILs were etched for 15 min with Piranha solution (3:1 concentrated sulphuric acid, 30% H2O2, both from Sigma-Aldrich) then rinsed with plenty of water and left to air dry for 1 h. SuperSILs were then glow-discharged for 120 s (negative) prior to sample loading and freezing using a Quorum Q150T ES system. Samples were frozen by plunge-freezing using FEI Vitrobot MKIV according to manufacturer’s instructions. The chamber was equilibrated to 4 °C, 95% relative humidity. Blotting was performed manually.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Code availability
ZEN software was used to process and render STORM images in this work. The software is commercially available from Carl Zeiss Ltd.
Data availability
All relevant data are available from the authors upon request.
References
Gustafsson, M. G. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 198, 82–87 (2000).
Hell, S. W. & Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782 (1994).
Rust, M. J., Bates, M. & Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793 (2006).
Betzig, E. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006).
Hess, S. T., Girirajan, T. P. & Mason, M. D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91, 4258–4272 (2006).
Thompson, R. E., Larson, D. R. & Webb, W. W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82, 2775–2783 (2002).
Shtengel, G. et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl Acad. Sci. USA 106, 3125–3130 (2009).
Kellenberger, E. et al. Artefacts and morphological changes during chemical fixation. J. Microsc. 168, 181–201 (1992).
Kaufmann, R. et al. Super-resolution microscopy using standard fluorescent proteins in intact cells under cryo-conditions. Nano Lett. 14, 4171–4175 (2014).
Liu, B. et al. Three-dimensional super-resolution protein localization correlated with vitrified cellular context. Sci. Rep. 5, 13017 (2015).
Nahmani, M., Lanahan, C., DeRosier, D. & Turrigiano, G. G. High-numerical-aperture cryogenic light microscopy for increased precision of superresolution reconstructions. Proc. Natl Acad. Sci. USA 114, 3832–3836 (2017).
Faoro, R. et al. Aberration-corrected cryoimmersion light microscopy. Proc. Natl Acad. Sci. USA 115, 1204–1209 (2018).
Weisenburger, S. et al. Cryogenic optical localization provides 3D protein structure data with Angstrom resolution. Nat. Methods 14, 141 (2017).
Furubayashi, T. et al. Three-dimensional localization of an individual fluorescent molecule with Angstrom precision. J. Am. Chem. Soc. 139, 8990–8994 (2017).
Terris, B., Mamin, H., Rugar, D., Studenmund, W. & Kino, G. Near-field optical data storage using a solid immersion lens. Appl. Phys. Lett. 65, 388–390 (1994).
Chen, R., Agarwal, K., Sheppard, C. J., Phang, J. C. & Chen, X. A complete and computationally efficient numerical model of aplanatic solid immersion lens scanning microscope. Opt. Express 21, 14316–14330 (2013).
Zhang, J., See, C. & Somekh, M. Imaging performance of widefield solid immersion lens microscopy. Appl. Opt. 46, 4202–4208 (2007).
Wang, L., Pitter, M. C. & Somekh, M. G. Wide-field high-resolution solid immersion fluorescence microscopy applying an aplanatic solid immersion lens. Appl. Opt. 49, 6160–6169 (2010).
Wang, L. et al. Highly confined surface imaging by solid immersion total internal reflection fluorescence microscopy. Opt. Express 20, 3311–3324 (2012).
Wildanger, D. et al. Solid immersion facilitates fluorescence microscopy with nanometer resolution and sub-Ångström emitter localization. Adv. Mater. 24, OP309-13 (2012).
Kim, W.-C. et al. Investigation on achieving super-resolution by solid immersion lens based STED microscopy. Opt. Express 25, 16629–16642 (2017).
Wang, L., Pitter, M. C. & Somekh, M. G. Wide-field high-resolution structured illumination solid immersion fluorescence microscopy. Opt. Lett. 36, 2794–2796 (2011).
Liu, Z. et al. High resolution, high collection efficiency in numerical aperture increasing lens microscopy of individual quantum dots. Appl. Phys. Lett. 87, 071905 (2005).
Yoshita, M., Koyama, K., Hayamizu, Y., Baba, M. & Akiyama, H. Improved high collection efficiency in fluorescence microscopy with a Weierstrass-sphere solid immersion lens. Jpn. J. Appl. Phys. 41, L858–L860 (2002).
Muller, M. Introduction to Confocal Fluorescence Microscopy, Vol. 69 (SPIE Press, Bellingham, WA, US, 2006).
Smith, W. J. Modern Optical Engineering: The Design Of Optical Systems, Vol. 4, 754 (McGraw-Hill Professional, New York, NY, US, 2007).
Hellen, E. H. & Axelrod, D. Fluorescence emission at dielectric and metal-film interfaces. J. Opt. Soc. Am. B 4, 337–350 (1987).
Rabouw, F. T. et al. Non-blinking single-photon emitters in silica. Sci. Rep. 6, 21187 (2016).
Born, M. & Wolf, E. Principles of Optics: Electromagnetic Theory of Propagation, Interference and Diffraction of Light 7th edition (Cambridge University Press, Cambridge, UK, 1999).
Den Dekker, A. & Van den Bos, A. Resolution: a survey. J. Opt. Soc. Am. A 14, 547–557 (1997).
Beis, K. Structural basis for the mechanism of ABC transporters. Biochem. Soc. Trans. 43, 889–893 (2015).
Nieuwenhuizen, R. P. J. et al. Measuring image resolution in optical nanoscopy. Nat. Methods 10, 557 (2013).
Bountra, K. et al. Structural basis for antibacterial peptide self-immunity by the bacterial ABC transporter McjD. EMBO J. 36, 3062–3079 (2017).
Rothemund, P. W. Folding DNA to create nanoscale shapes and patterns. Nature 440, 297 (2006).
Choudhury, H. G. et al. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc. Natl Acad. Sci. USA 111, 9145–9150 (2014).
Mehmood, S. et al. Structural and functional basis for lipid synergy on the activity of the antibacterial peptide ABC transporter McjD. J. Biol. Chem. 291, 21656–21668 (2016).
Romano, M. et al. Structural basis for natural product selection and export by bacterial ABC transporters. ACS Chem. Biol. 13, 1598–1609 (2018).
Strahl, H. & Errington, J. Bacterial membranes: structure, domains, and function. Annu. Rev. Microbiol. 71, 519–538 (2017).
Barák, I., Muchová, K., Wilkinson, A. J., O’toole, P. J. & Pavlendová, N. Lipid spirals in Bacillus subtilis and their role in cell division. Mol. Microbiol. 68, 1315–1327 (2008).
Bates, M., Huang, B., Dempsey, G. T. & Zhuang, X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science 317, 1749–1753 (2007).
Mattheyses, A. L., Simon, S. M. & Rappoport, J. Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 123, 3621–3628 (2010).
Axelrod, D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 89, 141–145 (1981).
Wee, P. & Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 9, 52 (2017).
Sternberg, S. R. Biomedical image processing. Computer 16, 22–34 (1983).
Mutch, L. J., Howden, J. D., Jenner, E. P. L., Poulter, N. S. & Rappoport, J. Z. Polarised clathrin-mediated endocytosis of EGFR during chemotactic invasion. Traffic 15, 648–664 (2014).
Lang, C., Hiscock, M., Dawson, M. & Hartfield, C. Local thickness and composition analysis of TEM lamellae in the FIB. Microelectron. Reliab. 54, 1790–1793 (2014).
De Boer, P., Hoogenboom, J. P. & Giepmans, B. N. Correlated light and electron microscopy: ultrastructure lights up! Nat. Methods 12, 503 (2015).
Fitzgerald, J. E., Lu, J. & Schnitzer, M. J. Estimation theoretic measure of resolution for stochastic localization microscopy. Phys. Rev. Lett. 109, 048102 (2012).
Dempsey, G. T., Vaughan, J. C., Chen, K. H., Bates, M. & Zhuang, X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat. Methods 8, 1027 (2011).
Tessler, L. A. et al. Nanogel surface coatings for improved single-molecule imaging substrates. J. R. Soc. Interface 8, 1400–1408 (2011).
Zanetti-Domingues, L. C., Martin-Fernandez, M. L., Needham, S. R., Rolfe, D. J. & Clarke, D. T. A systematic investigation of differential effects of cell culture substrates on the extent of artifacts in single-molecule tracking. PLoS ONE 7, e45655 (2012).
Acknowledgements
The authors thank John Collier for his continuous support and contributions to this project and Simon Nestead and David Drew for kindly providing them with the expression construct for PH1735. The authors also thank Daniel Axelrod at the University of Michigan for invaluable discussions and help. This work has been funded by Medical Research Council grants (MR/K015591/1) to M.L.M.-F. and (MR/N020103/1) to K.B.
Author information
Authors and Affiliations
Contributions
L.W. and M.L.M.-F. conceived and designed the research. B.B. and L.W. constructed the cryo-SIL microscope and implemented the measurements. S.A. and C.S. manufactured the superSIL assembly. L.C.Z.-D. and M.C.D. developed sample plunge freezing protocol. L.C.Z.-D., M.R., A.N.M. and S.R.N. prepared biological samples. K.B. supervised M.R. in the bacterial sample preparation. L.W., B.B., D.J.R., D.T.C. and M.L.M.-F. analyzed data. D.T.C., M.L.M.-F. and L.W. wrote the manuscript. All the authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Bateman, B., Zanetti-Domingues, L.C. et al. Solid immersion microscopy images cells under cryogenic conditions with 12 nm resolution. Commun Biol 2, 74 (2019). https://doi.org/10.1038/s42003-019-0317-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0317-6
This article is cited by
-
A systematic study on the use of multifunctional nanodiamonds for neuritogenesis and super-resolution imaging
Biomaterials Research (2023)
-
Cryo-electron tomography on focused ion beam lamellae transforms structural cell biology
Nature Methods (2023)
-
Contribution of advanced fluorescence nano microscopy towards revealing mitotic chromosome structure
Chromosome Research (2021)
-
Cryogenic superresolution correlative light and electron microscopy on the frontier of subcellular imaging
Biophysical Reviews (2021)
-
Cryo-nanoscale chromosome imaging—future prospects
Biophysical Reviews (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
