
Abstract
The internal organs embedded in the cavities are lined by an epithelial monolayer termed the mesothelium. The mesothelium is increasingly implicated in driving various internal organ pathologies, as many of the normal embryonic developmental pathways acting in mesothelial cells, such as those regulating epithelial-to-mesenchymal transition, also drive disease progression in adult life. Here, we summarize observations from different animal models and organ systems that collectively point toward a central role of epithelial-to-mesenchymal transition in driving tissue fibrosis, acute scarring, and cancer metastasis. Thus, drugs targeting pathways of mesothelium’s transition may have broad therapeutic benefits in patients suffering from these diseases.
Similar content being viewed by others

Introduction
Our internal organs and cavities are lined by a single continuous layer of epithelial cells known as the mesothelium that is derived from the embryonic mesoderm. It is the largest epithelial organ in the adult mammalian body1,2,3. The mesothelium covers several body cavities and their internal organs, including (but not limited to) the pericardium, encasing the heart, the pleural cavity, encasing the lungs, and the mesentery and peritoneum, encasing the various abdominal organs. The mesothelium also surrounds the internal reproductive organs in both males and females. The mesothelial membranes consist of a parietal (lining the body wall) and visceral (lining the internal organs) layer. The space in-between is filled with fluid, which acts to accommodate organ movement and decrease friction. Other well-known functions include protection against bacterial infections and their disseminations within the cavities, and to provide direct passage between the cavities and the internal organs4 (Box 1).
All vertebrate animals have a coelomic cavity that separates the outer and inner components of the body. It is formed as a result of a binary division of the lateral plate mesoderm. Through this division the coelom is lined by two different, but continuous tissue components5. During organ development, a cell population within these tissues acquires epithelial features, baso-apical polarization, and a basal lamina. It is at this point that we refer to these cells as the coelomic epithelium, the embryonic precursor of adult mesothelium. The coelomic epithelium significantly contributes to organ development, in particular by undergoing epithelial-to-mesenchymal transition (EMT). Through EMT, the coelomic epithelium contributes various cell types into the developing and growing organs, including fibroblastic cells and smooth muscle cells6. We and others have identified some of its distinguishing markers6, and used it to lineage trace the mesothelium’s embryonic and adult precursors. A subset of these cells are the likely self-renewing stem and progenitors of the underlying organ fibroblasts and smooth muscle6. In adult life, the same pathways that are involved in EMT during organ development and growth reappear during injury and organ disease, such as infarctions, ischaemia, fibrosis (developing within organs), adhesions (developing in-between organs, tethering them to one another), and cancer. We propose here that trunk organ injury and disease can be best understood in light of re-emergences of these cellular and molecular EMT programmes specific to mesothelial development. To illustrate this point we describe four organ systems: the heart, liver, gonadal system, and the lungs. We review the current literature on mesothelial cell involvement in normal development and growth of these organ systems, where a remarkable genetic overlap exists between different coelomic epithelial tissues. We then summarize the literature on injury and disease of these organ systems in adult life and show that these diseases recapitulate the normal embryonic expression of coelomic epithelial genes. We conclude with further examples of involvement of these genes in pathology of the abdominal wall (peritoneum) and in mesothelium-related cancers. As the pathologies across these organ systems are essentially characteristic of uncontrolled EMT progression, we deliberate on the impetus to exploit EMT for therapeutic gain and pose some pending questions and challenges relevant for the field.
Mesothelial cell EMT involvement in healthy organ development and growth
Heart development
The coelomic epithelium that covers the developing heart is derived from a group of progenitor cells near the venous pole of the heart known as the proepicardial organ, which in turn comes from a thick mass of cranial mesenchyme called the septum transversum. The precursors of the coelomic epithelium migrate to form a layer of tissue between the pericardium and the heart, termed epicardium. A subpopulation of these cells proliferates and undergoes EMT that allows them to invade the developing heart and colonize its internal spaces as subendocardial fibroblasts, and by encircling the developing coronary blood vessels, where they develop into smooth muscle cells7,8 (Fig. 1). Indeed, several independent lineage-tracing experiments using epicardial-specific Cre-LoxP transgenes, e.g. mesothelin (Msln), T-box transcription factor 18 (Tbx18), and Wilms’ tumour 1 (Wt1), have demonstrated that epicardial cells can differentiate into coronary smooth muscle cells, and fibroblasts (including pericytes)6,9,10,11,12,13. The importance of the epicardium in heart development has been further demonstrated with epicardial knockout models. From these studies, a number of genes have been identified that highlight epicardial EMT as a crucial factor in heart morphogenesis. TBX18 over-expression promotes EMT in cultured epicardial cells by inducing the expression of Slug14. The role of the transcription factor WT1 in epicardial development has been described in more detail. WT1 binds directly to the Snail and E-cadherin promoter sites, thereby either promoting or repressing transcription respectively12. Mice with a Wt1 deletion in the epicardium develop peripheral oedema, pericardial haemorrhage, and heart wall thinning12,15 resulting from impaired EMT16. In line with this, epicardial Wt1 knockouts have reduced epicardial expression of the key EMT activators Snail and Slug, and show reduced expression of the mesenchymal marker Vimentin12,16. This is paralleled by increased expression of the epithelial markers E-cadherin and cytokeratin17. WT1 is an upstream regulator of the canonical WNT signalling pathway that leads to activation of β-catenin. Epicardial-specific β-catenin-null mice have disturbed epicardial EMT, blunted invasion of the heart muscle, failed expansion of the sub-epicardial space, and impaired differentiation of epicardial cells into coronary smooth muscle16,18. EMT also fails in cardiac fibroblasts (that remain in the epicardium) in the absence of the β-catenin-binding partner TCF2119,20. Another WNT protein, WNT5A has also been implicated as an epicardial factor that promotes compact heart muscle growth21. Both Wnt5a and Wt1 mouse knockout hearts have thin compact heart muscle16. Another epicardial gene whose expression is dependent on WT1 and that is important for heart development is retinaldehyde dehydrogenase 2 (Raldh2), encoding the enzyme that controls retinoic acid availability. Wt1 knockouts show reduced RALDH2 expression, and like Wt1 knockout embryos, Raldh2−/− embryos, treated with exogenous retinoic acid to bypass early embryonic lethality22, exhibit myocardial hypoplasia and coronary vessel abnormalities. Retinoic acid supplementation also works to restore embryonic EMT and viability of Wt1 knockout hearts, suggesting a functional link between WT1 and RALDH2 (refs. 16,23). Like WT1, retinoic acid also regulates differentiation of epicardial cells24, thus the contribution of RALDH2 deficiency to the WT1 knockout phenotype is likely multifactorial. Differentiation of epicardial cells is also regulated by retinoid X receptor alpha (RXRα), as epicardial-selective knockouts have impaired EMT that disrupts coronary arteriogenesis and obviates myocardial compaction25. In this regard, other genes act in close cooperation with epicardial EMT. Gata4 (refs. 26,27) and its co-factor Friend of GATA 2 (Fog2)28,29 deficiency results in embryonic death in mice due to vascular defects and myocardial thinning26,30,31. Finally, epicardial Notch signalling is also directly involved in heart development. Mouse mutants defective in the Notch effector RBPjκ show premature EMT in the form of ectopic patches of alpha-smooth-muscle-actin (α-SMA) at the venous pole of the heart, but show no alterations in expression of WT1, TBX18, GATA4, and RALDH2 (ref. 32). This may indicate an EMT programme independent from WT1. Wt1-specific Notch1 knockouts display severe defects due to defective EMT, leading to embryonic lethality in some cases. Like Wt1 mutants, these mice develop a severe reduction and disorganization of the coronary vascular plexus, and dramatic reduction in the number of smooth muscle cells in the compact myocardium32. Thus, different Notch effectors may regulate different aspects of EMT, depending on the stage of development. Collectively, epicardial EMT is controlled by a plethora of factors, where WT1 appears to be the master regulator that sits atop of a hierarchy of gene loci required for EMT and proper heart formation.

Mesothelial involvement in embryonic development. Coelomic epithelial cells provide the bulk of fibroblastic and smooth muscle lineages of the body and critically support EMT during embryonic development of different organs. A common set of genes, in particular WT1, TBX18, MSLN, Notch1, GATA4, and their downstream effectors underlie these EMT programmes to regulate mesenchymal differentiation. Four examples are given: (1) heart, (2) liver, (3) gonads, and (4) the lungs, showing how the coelomic epithelium expands and ingresses to form tissue-specific fibroblasts and smooth muscle lineages (displayed as purple) by virtue of EMT. For further details, see the main body of text
Liver development
In mice, the liver is initially formed as an out pocket of the foregut endoderm, but subsequently invades the surrounding septum transversum (which also contributes to heart development) and forms a hepatic cord to complete its development. Cells of the septum transversum proliferate actively to cover the hepatic lobes and migrate inward to generate mesenchymal lineages33. Lineage-tracing studies with MslnCreERT2 (ref. 6), Wt1CreERT2 (refs. 34,35) and Gata4Cre (ref. 36), combined with reporter lines have shown that coelomic epithelium progenitors give rise to hepatic stellate cells, smooth muscle cells and fibroblasts that incorporate into the sinusoidal walls of the liver lobes (Fig. 1). The same molecular programmes that induce the epicardium to undergo EMT and to contribute cellular derivatives during heart development are observed in the developing liver. Consequently, many of the transgenic lines where EMT genes are knocked out show liver phenotypes in addition to heart phenotypes. For example, Wt1 and Rxra knockout mice fail to properly generate mesenchymal cell lineages, resulting in liver defects through a reduction in liver size and lobe abnormalities37. Similarly, coelomic epithelium from the septum transversum requires Wt1-driven WNT/β-catenin38, and loss of β-catenin in the coelomic epithelium and stellate cells expressing TWIST2 causes abnormal activation of stellate cells in embryonic livers39. In Gata4-deficient mice, the liver bud is able to form a pseudo-stratified epithelial liver, but fails to expand27,36,40. Both Gata4 (ref. 36) and Wt1 (ref. 37) knockouts have an increased activation state of hepatic stellate cells, with abnormally high expression of α-SMA and deposition of ECM, reminiscent of myofibroblastic differentiation. This suggests that next to EMT, lineage-specific programmes may be regulated through GATA4 and WT1 (ref. 41).
Development of the reproductive (gonadal) system
The adrenal cortex and gonads share a common developmental origin, the adrenogonadal primordium. In mice, the adrenogonadal primordium can be first detected as a thickening of the coelomic epithelium at approximately E9.5 (refs. 42,43). By E10.5, the adrenogonadal primordium splits into the adrenal and gonadal primordia that will continue to differentiate separately. The gonadal primordium, also known as the genital ridge, is initially bi-potential, and begins development through the increased proliferation of the coelomic epithelium (Fig. 1). This increased proliferation leads to the transformation of the monolayer epithelium into a dense and pseudo-stratified layer. Lineage-tracing studies using cell-permeable dyes44 or genetic Cre lines (Wt1 (refs. 45,46), Tbx18 (refs. 47,48), or steroidogenic factor 1 (Sf1)49) have shown that following these events, the basement membrane underneath the coelomic epithelium disintegrates, which allows the proliferating cells of the coelomic epithelium to undergo EMT, and to ingress and form a cluster of SF1+ gonadal somatic precursor cells50,51, which later give rise to the supporting Sertoli cells of the testis cords and seminiferous tubules52,53, or the follicular (or granulosa) cells of the ovarian follicles54,55. The supporting cells are not the only cells derived from the coelomic epithelium in the primitive gonads of mice. Cell-tracking experiments have shown that the SF1+ gonadal precursor cells generate the testicular peritubular myoid cells (PMCs, the smooth muscle cells surrounding the seminiferous tubules in the testis) through EMT and the steroidogenic Leydig cells and Theca cells in testis and ovaries, respectively44,46,47,48,53,56,57,58. The molecular mechanisms driving coelomic epithelium transformation into the genital ridge have not been laid out as comprehensively as for the embryonic heart or liver, but a similar gene signature in gonadal tissues can be observed around this time. Like the heart and liver, in the developing gonads Wt1, Gata4, Tcf21 and Tbx18 are one of the earliest activated genes, present throughout the whole urogenital ridge. Their expression is quickly followed by Sf1, which remains restricted to the genital ridge59. Tbx18 null mice appear to have defective EMT in that they develop ectopic patches of fibroblasts along the developing gonads, and Tcf21 knockouts lack a distinct mesenchymal compartment60,61. GATA4 is thought to be most important in the undifferentiated precursor population giving rise to the genital ridge. Gata4 null mice do not develop a genital ridge at all, and the expression of SF1, but not WT1, is significantly decreased59. Similarly, Fog2 mutants have abnormal gonads through impaired thickening of the genital ridge62. Mice lacking functional WT1 initiate gonad formation by thickening of the coelomic epithelium through active division, but the genital ridge quickly regresses before it is completely formed15,45. There is further evidence to suggest that WT1 regulates SF1 in the undifferentiated genital ridge63. Consequently, disruption of SF1 expression results in delayed and decreased EMT, disrupted cell ingression during the formation of the genital ridges, and consequently fewer SF1+ gonadal precursor cells and gonad size64. A microarray screen for WT1 wildtype and knockout E11.0 embryos highlighted Muc16 as the top gene with regard to the coelomic epithelium, which was completely absent in knockout tissues65. Mucin 16 is highly expressed in mesothelial cancers, and has been shown to promote EMT, in part through interaction with MSLN66,67. Thus, similar to heart and liver development, WT1 appears to regulate EMT in the developing gonads, in part through SF1, while GATA4 closely cooperates alongside WT1 to drive initial formation of the genital ridge. In XY mice, knocking out Wt1 after sex determination results in disruption of developing seminiferous tubules through the aberrant differentiation and development of Sertoli cells68,69. Sertoli-specific Wt1 mutants also show abnormal PMC and Leydig development70. Interestingly, the precursors of Sertoli cells express only very low amounts of SF1, despite a high abundance of WT1, and it has been shown that after sex determination, SF1 is repressed by WT1 through direct binding to the promoter region71. In the absence of WT1, high SF1 expression leads undifferentiated somatic cells to develop into steroidogenic cells of the gonads (Leydig and Theca cells in the testis and ovaries, respectively)69,71,72, leading to structural impairments. These findings are consistent with the natural progression of SF1 expression throughout the developing gonads. In XY mice, SF1 expression starts to cease in the coelomic epithelium covering the genital ridge, but remains expressed in gonadal precursor-derivatives (pre-Sertoli cells and Leydig cells) around E12.5, and later persists only in Leydig cells73. Thus, similar to the Stellate cell of the liver, after sex determination WT1 acts to maintain lineage specification in the gonads. This may explain why WT1 expression is maintained in ovarian granulosa cells and testicular Sertoli cells of adult mice15; however, the origin of gonadal cell fates and the mechanisms of their differentiation still require further studies.
Development of the lungs
In mouse embryos, the lung primordium emerges from the ventral foregut around E9.5, consisting of a simple tube of endodermal epithelium surrounded by splanchic mesodermal mesenchyme that is lined by a coelomic epithelium layer. This tube then branches to form the pulmonary sacs74. Anterior to the developing lungs are bilateral folds of tissue which extend from the body wall, known as the pleuropericardial membranes, which grow towards the midline, each encapsulating a common cardinal vein and phrenic nerve. They are further stretched when the initially straight embryonic heart tube is transformed into a helical loop, to finally fuse with the root of the lungs to close off the remaining communication between the pericardial and pleural spaces around E13.5 (ref. 75) (Fig. 1). Because of the contiguous nature of the heart and pleura, many gene pathways are shared between these organs. WT1, TBX18, RALDH2, β-catenin, Notch1, and GATA4, at some stage in lung development, are all selectively expressed in coelomic epithelium cells surrounding the lung buds or pleuropericardial folds76,77,78,79,80,81. Lineage-tracing studies have confirmed that by E11.5, WT1+78,79,82,83,84, MSLN+6 and Notch1+79 coelomic epithelium cells covering the lungs migrate inward to give rise to α-SMA+ fibroblast and smooth muscle cells that make up both arteries and veins in the vascular wall, or those positioned circumferentially around the airways. These results have been extended towards the postnatal lung using inducible Cre lines, indicating that the coelomic epithelium contributes to these lineages even after birth82. Equally, WT1+ descendants from the pleuropericardial folds generate the mesenchyme surrounding the cardinal veins adjacent to the growing lungs85. Knockout studies have corroborated these findings. Defects in Gata4 or its co-factor Fog2 cause lung lobulation abnormalities, which can be attributed to growth delay and abnormal branching morphogenesis81,86. Internally, Gata4 mutants show dilated distal airways and patches of thickened mesenchyme, characterized as ectopic expression of α-SMA in the mesothelial and submesothelial layer87. Similar observations have been made in the pleura of mice harbouring a mutant of P300, another known co-factor for GATA4 (ref. 88). These results suggest defects in GATA4 expression perturb coelomic epithelium-driven EMT during lung morphogenesis, presumably through impaired signalling from the mesothelium to the underlying endoderm. Similarly, coelomic epithelium-selective (Dermo1+) β-catenin mutants show impaired lung growth as early as E12.5, primarily through reduced mesenchymal expansion89,90, and defective Notch signalling in Dermo1+ RBPjκ mutants results in impaired vascular, but not bronchial smooth muscle differentiation79. Systemic Wt1 knockout embryos show severe growth defects in the lungs, displaying an irregular rounded-shape and abnormally fused lobes15,78. Raldh2 mutant mice91,92 or lung explants treated with the pan-RXR inverse agonist BMS493 have perturbed lung bud formation93. Wt1 and Raldh2 mutants initially form a pleuropericardial fold, but do not fuse with the coelomic epithelial lining of the lung hilus75,85, and Tbx18 knockout mice fail to develop the pleuropericardial membranes at all75. In this regard, drivers of EMT have been shown to be essential in the underlying pathology. Wt1 knockouts maintain an intact basal lamina under the pulmonary coelomic epithelium, and show strongly reduced vimentin immunoreactive cells in the lung mesenchyme, indicative of impaired mesothelial EMT78. Several downstream targets have been shown elementary in this process. Embryonic lung explants exposed to CCSFE to label pleural coelomic epithelium show extensive EMT after 48 h, which can be further controlled through TGF-β or Notch signalling modulation79. Another putative regulator of mesothelial EMT is Enhancer of zeste homologue 2 (EZH2), a histone methyltransferase that in mesothelial cancers is known to repress EMT94. Wt1-dependent EZH2 knockouts display uncontrolled EMT in the coelomic epithelium, and develop multiple heart and lung abnormalities by generating ectopic patches of smooth muscle around E14.5 (ref. 95), originating at the proximal end of lung lobes, and then spreading out distally around E18.5. These patches co-express smooth muscle protein 22 (SM-22)α and α-SMA, indicative of a smooth muscle phenotype, and are closely associated with WT1+ cells at the periphery of the lung.
Injury and disease in adult life: recapitulation of coelomic epithelial genes
Pathology of the heart
The mesothelium covering the heart (epicardium) is directly involved in several types of heart diseases, an example being myocardial infarction. Induction of myocardial infarction in mice results in re-activation of the epicardium and the re-expression of several embryonic epicardial genes, such as Raldh2, Wt1, Gata4, and Msln96,97,98,99. The re-expression of embryonic genes is typically confined to the lesion site, where the epicardium progressively expands and undergoes EMT to generate cardiac fibroblasts within the vicinity of the infarct96,100,101,102. These areas are rich in expression of mesenchymal cell type markers, including fibroblast-specific protein 1, pro-collagen I, collagen III, fibronectin, α-SMA, SM-22α, and smooth muscle myosin heavy chain96, indicative of fibroblasts that are actively depositing extracellular matrix proteins. Epicardium-specific inhibition of these coelomic epithelial genes significantly diminishes cardiac fibrosis. For example, Wt1Cre;Ctnnb1flox mice are null for β-catenin expression in the epicardium, and show minimal signs of epicardial expansion and EMT when subjected to ischaemia reperfusion injury101. In mammals, expression of a dominant-negative CCAAT/enhancer binding protein, which in essence is upstream of and controls the transcription of Raldh2 and Wt1 in adult mouse epicardium, can instil a cardioprotective effect and partially prevent myocardial fibrosis after ischaemia reperfusion97. In spite of the fibrotic potential of injured mesothelium of the heart, its presence is probably required for normal wound repair. In adult zebrafish, a species with robust regenerative potential after injury, genetic depletion of the epicardium after experimentally resecting parts of the heart ventricle inhibits cardiomyocyte proliferation and delays muscle regeneration103,104. Similarly, disruption of thymosin β4-mediated paracrine signalling towards the epicardium impairs smooth muscle cell development in mice, and its expression facilitates vascular repair following myocardial infarction105,106. It is worth mentioning that not every cardiac injury model shows evidence for epicardial EMT, for example, in the pressure overload model through constriction of the ascending aortus107,108. However, this model primarily results in perivascular fibrosis, with no signs of elevated WT1 or TBX18 expression in the epicardium109, and thus likely originates from a different, possibly endothelial source107,108.
Pathology of the liver
The mesothelium covering the liver is comprehensively involved in liver pathologies. Different hepatic injury models that exhibit varying degrees of fibrosis show varied involvements of the hepatic surface mesothelium, or hepatic stellate cells (HSCs), which are derived from mesothelium. In fact, different injury models show diverged severities of fibrosis that stem from the level of activation of the mesothelium and HSCs. Lineage-tracing studies have confirmed an active role of the surface mesothelium as the cell-of-origin of HSCs and myofibroblasts in response to carbon tetrachloride, which can be inhibited through antagonism of TGF-β signalling110. Similarly, activated portal fibroblast (the predominant source in cholestatic liver injury induced by bile duct ligation) exhibit a high expression of MSLN upon bile duct injury, further suggesting that these cells originate from a mesothelial source34,111. Consequently, MSLN-deficient mice are less susceptible to liver fibrosis compared to wild-type mice112, and conditional ablation of mesothelial-derived MSLN+ portal fibroblasts in bile duct-injured mice show significantly reduced liver fibrosis113. The mechanism by which MSLN promotes fibrosis presumably acts through its antagonistic actions on a TGFβR1 inhibitory complex called thymocyte antigen 1 (ref. 113). Like the heart, the mesothelium also supports liver regeneration upon injury. The mesothelium-derived paracrine factors MDK and PTN facilitate hepatocyte growth following hepatectomy114,115, analogous to the mesothelium’s stimulatory and regenerative role during intestinal crypt116 and embryonic heart regeneration117. Conditional Gata4 knockout mice have a high degree of liver fibrosis when treated with subcutaneous injections of carbon tetrachloride, inducing hepatoxic liver injury36. Chronic exposure in GATA4-deficient mice results in increased liver hydroxyproline content, infiltrating CD45+ cells, and serum alanine aminotransferase and aspartate aminotransferase levels compared to control livers36, all indicative of fibrosis associated with progressive liver failure. These findings likely indicate that mesothelial expression of GATA4 inhibits liver fibrosis through its effects on lineage maintenance in Stellate cells.
Repair of the (female) reproductive system
The ovaries regenerate their surface mesothelium (also called ovarian surface epithelium, OSE) throughout postnatal life. Rupturing of the follicles that follows each ovulatory cycle is followed by complete regeneration of the mesothelium without fibrosis. In spite of this, ovulatory wound repair is nonetheless associated with mesothelial EMT that temporarily permits displaced OSE to assume a mesenchymal phenotype within the ovarian cortex and migrate actively towards the ovulatory wound118,119,120,121. Additionally, the EMT regulator TGF-β1 is highly expressed in the serosal fluid around the ovaries in response to ovulation, and significantly increases the sphere-forming efficiency of OSE progenitor cells, further suggesting EMT is a requisite for the maintenance of OSE integrity122. It has been proposed that discrete regions within the OSE may serve as cell sources that replenish or close the ovulatory wounds123, and several markers have been identified124,125, including lymphocyte antigen 6 complex, locus A (LY6A), leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5), and RALDH (WNT target genes). For example, a subset of the OSE expresses LGR5 that serves to maintain OSE homoeostasis and act as a cell-of-origin for new OSE during the ovulatory cycle121,126. The exact location of LGR5+ cells is still under debate, as to whether they are restricted to the ovary hilum121, or are more widespread, and respond locally to rupturing follicles by actively proliferating126. Nonetheless, these findings suggest that the activity of LGR5+ mesothelial cells are most likely stem cells that are tailored to meet the specific growth requirement of the local OSE. The integrity of LGR5+ cells is likely maintained by active WNT signalling, as these cells secrete WNT4 and express several WNT target genes such as TROY and inhibitor of DNA binding 2 (ref. 126). High RALDH expression has also been identified in the OSE transitional zone, which constitutes approximately 5% of the total OSE population. In vitro studies have shown this cell subset has higher capacities to generate cell clones as compared to RALDH- cells, and similar to LGR5+ cells, these cells exhibit a greater degree of proliferation after ovulation121. It is unclear at this point whether these cells represent the same cell population, and importantly, whether these genes are involved in regulating mesothelial EMT to drive ovulatory wound repair.
Pathology of the lungs
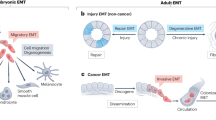
There are two scarring pathologies of the lungs in which pleural mesothelium is known to be involved: pleural fibrosis (PF), and interstitial lung disease. PF manifests either as discrete localized fibrotic lesions, or more diffusely organized pleural fibrotic patches, which in some cases result in fusion of the pleural membranes (Fig. 2)127. Both parietal and visceral layers can be affected, but is generally only clinically significant when it involves the viscera128. Various etiologies can account for PF, from pleural effusions (e.g. rheumatoid, tuberculous, or uraemic pleurisy, bacterial empyema, retained hemothorax), to particle or compound exposures (e.g. asbestos, carbon nanoparticles, medications) or secondary malignancies128. Experimental models to study PF have relied mostly on variations of the bleomycin mouse model. In mice receiving intraperitoneal or intratracheal bleomycin, increased expression of the mesothelial markers calretinin and WT1 combined with TGF-β1-Smad2/3 is observed in the pleura and underlying parenchyma of mice129,130. Consequently, mice treated with the TGF-βRII targeting miRNA miR-18a-5p have reduced (sub-)PF after bleomycin administration131. Strikingly, intratracheal bleomycin combined with carbon nanoparticles result in a more persistent and severe pulmonary response, including adhesions and severe and progressive (sub-)PF, preceded by pleural thickening and inflammation132. The engulfment of carbon nanoparticles by the mesothelium contributes to the severity of this model, driving rapid upregulation of α-SMA and release of MMP-9 and TGF-β1 in the pleural fluids132,133. A role for β-catenin signalling has been described in a model for pleural effusion, a known risk factor for PF. Mice subjected to Streptococcus pneumoniae develop empyema accompanied by α-SMA+ expression in the pleura, pleural thickening and reduced lung function, which can inhibited by GSK-3β antagonist 9ING41 (ref. 134).

Fibroblastic lineage commitment is a common mechanism driving mesothelial pathologies. Many, if not all mesothelial pathologies ultimately converge on tissue fibrosis through the mesothelium’s ability to undergo EMT and generate fibroblastic lineages: (1) Epithelioid mesothelial cancers invade through their ability to influence resident mesothelium, in part by secreting exosomes, which then undergoes EMT and forms a cancer niche. (2) Sarcomatoid mesothelial cancers undergo EMT themselves to ingress in the parenchyma. (3) Mesothelial adhesions are similarly derived from EMT through various insults, which generates surface scarring that can spread to apposing organs and glues them together. (4) Damage to the surface mesothelium does not always result in adhesions, primarily when fibrosis progresses into the interiors of the (in most cases visceral) organs
Interstitial lung diseases represent another collection of heterogeneous fibrotic parenchymal disorders, of which idiopathic pulmonary fibrosis (IPF) is a more clearly defined subset. In line with the mesothelium as the site of origin, clonal analyses in IPF lungs have revealed the presence of a polyclonal, rather than a monoclonal population of fibroblasts. This suggests fibrotic foci are due to a reactive process responsive to local environmental stimuli, rather than a single malignant neoplasm growing through the lung. These foci are part of a continuous fibrotic reticulum that extends from the mesothelial surface into the lung parenchyma, identifying the mesothelium as the most likely source of origin (Fig. 2)135. Further evidence has been derived from mathematical models, in which increased stiffness of the visceral pleura, e.g., following EMT and matrix synthesis, is predicted to be key in driving mechanotransduction pathways to propel a fibroblast activation chain towards the parenchyma136. In support of this, the pleura and underlying parenchyma of human IPF patients show strong immuno-labelling for MSLN, WT1 or calretinin130,137,138, and when isolated and cultured display increased contractility and movement compared to healthy mesothelium130. The extend of labelling correlates positively with the degree of parenchymal fibrosis in patients137. Importantly, these markers are absent in other fibrotic diseases of the lung, such as chronic obstructive pulmonary disease or cystic fibrosis, in which the mesothelium plays no significant role. In vitro, pleural mesothelial EMT is controlled by a WT1–TGF-β1–Smad-2 axis138, which is further supported by mouse models. Adenoviral transmission of intrapleural TGF-β1 induces progressive PF of the viscera and underlying parenchyma, with strong cytokeratin and α-SMA in both tissue compartments139. Lineage tracing of these cells with WT1GFP-Cre mice or GFP-labelled pleural mesothelium injected in the pleural space, combined with intratracheal recombinant TGF-β1 show strong GFP and α-SMA co-labelling in the lung parenchyma underneath the pleura130,137, indicating active migration of the mesothelium.
Pathology of the abdominal wall (peritoneum)
Damage to mesothelium covering the three cavity walls or the internal organs induces the development of fibrosis on their surfaces that can attach to neighbouring healthy tissues, termed adhesions140 (Fig. 2). Adhesions are the most common side-effect of abdominal surgery that stems from mechanical trauma due to surgical tissue handling, ischaemia at incision sites, foreign bodies or tissue desiccation141,142. As well as after surgery, adhesions can result from inflammatory processes, from infections, or as an adverse response to dialysis143. Kidney dialysis takes advantage of the peritoneal surface mesothelium as a means of extravasation of plasma fluids from the blood. The dialysate solution, which contains solvents that are hyperosmotic, hyperglycaemic, and of low pH, gradually damages the exposed surface mesothelium and leads to its activation144,145. Exposure of the mesothelium to dialysis solution increases mesothelial cell death146, but also eventually leads to thickening of the mesothelium and generation of myofibroblasts and fibrous collagen bands at the injured surface144,147. Once fibrous bands initiate at the surface, the mesothelium never reaches complete regeneration, even months after cessation of dialysis146. More severe cases involve the development of encapsulating peritoneal sclerosis on the surface mesothelium that follows prolonged exposure to dialysis, chronic inflammation, or several other secondary acute intra-abdominal events. Similar to other mesothelial pathologies, the pathomechanism that leads to encapsulation is poorly understood. Organ encapsulation stems from thickening and fibrosis of the surface mesothelium and leads to a range of ailments, including obstruction and shortening of the bowels, abdominal pain, weight loss, malnutrition, sepsis, or death148. The level of encapsulation and thickening directly correlates with the level of mesothelial activation149, suggestive of a pathomechanism wherein mesothelial cells undergo varied extents of EMT in response to durations of dialysis150,151,152. Human peritoneal–intestinal adhesion tissues resulting from abdominal surgery show strong immuno-labelling for cytokeratin, calretinin, WT1, and α-SMA in the mesothelium and surrounding fibroblasts, suggesting these fibroblasts derive from mesothelial cells that have undergone EMT153. The advent of lineage tracing has confirmed these observations. Although WT1+ mesothelial cells have been reported to proliferate and actively repair the peritoneal surface upon injury154, various models of peritoneal fibrosis (i.e. hypochlorite, daily intraperitoneal injection of dialysate or mouse TGF-β adenoviral administration) have reported a mesothelial and submesothelial origin for myofibroblasts in peritoneal fibrosis154,155. Mechanistic studies have suggested TGF-β to be an active player in peritoneal EMT156,157,158. Conditional deletion of the Tgfbr2 gene in WT1+ mesothelial cells, treatment with the TGF-β1-blocking peptide p144 or p17, or use of mutant mice deficient in Smad3 all significantly attenuate the fibrotic response in (both) visceral and parietal mesothelium in several peritoneal fibrosis models153,155,159,160. Accordingly, these events are parralelled by downregulation of EMT markers, such as Snail153. We have recently established a mouse model of abdominal adhesions, wherein ligating small areas within the peritoneum, combined with gentle localized abrasion generates adhesions between the body wall and intestine161. In this model, MSLN and WT1 are rapidly upregulated in the mesothelial layer, followed by active proliferation. Within 1 h mesothelial cells show early morphologic signs of EMT, extending cilial protrusions into the peritoneal space and subsequently detaching from the basement membrane and neighbouring cells (Fig. 2). Cell tracking studies confirmed a mesothelial origin for peritoneal adhesions. Remarkably, these changes are associated with a massive genome-wide transcriptional rearrangement (more than 8000 genes) within 24 h after injury. Early gene programmes involved upregulation of angiogenesis and hypoxia, followed by inflammatory responses, as well as cell cycle markers and the downregulation of adhesion molecules. Many markers that are highly expressed by the peritoneal mesothelium during embryonic development and with very low expression during normal homoeostasis of adult mice, e.g. MSLN, Uroplakin-1B, and WT1, all peaked at 24 h after injury. Genes associated with fibroblasts (fibroblast-specific protein 1 and α-SMA) were also highly upregulated, paralleled by spindle-shaped mesothelial cells, further suggesting active EMT.
Pathology of mesothelium-derived cancers and cancer-associated fibroblasts
Mesothelial EMT is also linked with the progression of various tumours. The most well-known are malignant mesothelioma, a rare kind of cancer that originates from the visceral or parietal mesothelium of the thoracic, pericardial, or trunk cavities162,163, and ovarian cancer, particularly type II high-grade serous carcinoma. Mesothelioma most often originates in the pleura164, and is usually triggered by asbestos exposure165, or inherent germline mutations166, which can act synergistically167,168. It is broadly divided into three histological subtypes: epithelioid, biphasic (or mixed) and sarcomatoid, the latter occurring in approximately 10% of patients169. The prognosis for patients with mesothelioma is dismal, and patients with sarcomatoid mesothelioma have particularly poor outcomes compared to patients with epithelioid histology169. Ovarian cancer was initially thought to originate exclusively in the ovaries170, but the discovery of a possible precursor lesion in the fallopian tube fimbria provided evidence for possible multiple sites of origin171,172,173. Ovarian cancer, but also mesothelioma, spreads by disseminating from the primary site174,175, where cells enter the serosal fluid to be carried to secondary sites176. Both mesothelioma and ovarian cancer are highly aggressive, evolve rapidly, and are usually diagnosed in an advanced stage where curative surgery is no longer an option. Both OSE and fallopian tube fimbria-derived human tumours, as well as mesotheliomas, are characterized by high expression of Notch1 and MSLN or its derivative, serum soluble mesothelin-related peptide175,177,178, and cell culture studies have indicated MSLN is involved in driving cancer EMT179. Mesothelial-derived cancers are remarkably heterogeneous. Most metastasizing solid tumours must first invade the tumour stroma and access the vasculature. Therefore, an early EMT is necessary in order to adopt a motile, invasive phenotype. However, the unique trans-coelomic route of mesothelial-derived cancer cells creates an exceptional microenvironment. In contrast to most solid tumours, mesothelium-derived cancer cells exfoliate directly into the peritoneal cavity. Thus, early events in metastatic dissemination do not necessarily require a mesenchymal phenotype. As a result, both pleural and ovarian mesothelial cancer cells can display either epithelial or mesenchymal morphologies, or exist as an intermediate state. For example, sarcomatoid mesotheliomas resemble spindle-shaped cells that mimics the morphology of mesenchymal tumours, whereas epithelioid mesotheliomas represent a more epithelial-like morphology169. In sarcomatoid histology, expression of MSLN or WT1 is generally absent, concomitant with high activation of mesenchymal genes, such as Loxl2 or Vim180. Conversely, epithelioid mesotheliomas retain epithelial features, such as expression of the junctional protein E-cadherin181,182, and express high levels of mesothelial markers, among which UPK3B, R-spondin 1, WT1, and MSLN. Genes necessary for EMT are lowly transcribed, such as Cldn15 (refs. 180,181). Importantly, different EMT states can predict survival outcome in ovarian cancer patients, where highly mesenchymal cancers correlate with poorer disease-free survival, and epithelial cancers show better overall survival183. The ability of epithelioid mesothelioma to metastasize, presumably without requiring EMT, may represent a common behaviour of epithelial cancers184,185. It has been proposed that epithelial cancer cells can exploit non-malignant stromal cell types to develop cooperative invasion strategies. Cancer-associated fibroblasts are ideal stromal partners to enable collective cancer cell invasion186,187,188. A body of literature has suggested cancer-associated fibroblasts to originate from a local mesothelial source189,190,191,192, which has been confirmed with more recent lineage tracing studies. In a model where gastric cancer cells were implanted on the stomach surface, both resident mesothelium and implanted cancer cells mutually migrate towards each other after a rapid proliferative response by the mesothelial layer193. It was shown that the release of CD63+ exosomes by cancer cells largely dictated this response, where exosomes can always be found ahead of invading cancer cells and are actively incorporated into the infiltrated mesothelium (Fig. 2). Exosomes may contain growth factors used to directly modify the mesothelium, or matrixmetalloproteases (MMPs) to disrupt the basement membrane, and is not restricted to gastric cancer: CD63+, CD44+194, MSLN+195, or MMP1+196 exosomes have been found in ovarian cancer as well. Isolated exosomes derived from cancer cells can induce proliferation197 and subsequent EMT198 in the peritoneum of mice when injected, promoting tumour survival. WNT/β-catenin and RALDH1 signalling appear elementary in these events, as they are highly upregulated in both gastric cancer and infiltrated mesothelium. Consequently, knockdown of these pathways effectively inhibits cancer invasion. A similar role has been shown for the peritoneum of mice. Gastric cancer cells injected into the peritoneal space quickly home to the peritoneum and start actively proliferating. In this study, like the stomach, invasion of cancer cells into the underlying stroma was always preceded by the rapid invasion of infiltrating mesothelial cells into the muscle layer. Infiltrated mesothelial cells covered a large area of the muscle layer, and were found abundantly within tumour nodules192. Lineage tracing confirmed that more than 90% of α-SMA expressing fibroblasts derived from mesothelium, indicating the occurrence of active EMT and a mesothelial origin of cancer-associated fibroblasts. These observations suggest that the mesothelium promotes cancer development by creating a niche that favours cancer cell invasion. Elevated expression of Tks5 (a gene involved in the formation of actin-rich protrusions called invadopodia) was detected in mesothelial cells located on the invading edge of gastric cancer cells, which was abolished by neutralizing peptides192.
Concluding remarks
Here we have presented evidence from various organ systems indicating that some of the most common trunk organ pathologies, such as infarctions, ischaemia, fibrosis, adhesions, and cancer, involve re-activation of genetic programmes that are normally expressed during the mesothelium’s embryonic EMT. These programmes initiate and regulate organ disease, rather than being a consequence of it. Moreover, the similarities in gene expression programmes across diverse organ systems and disease states suggest common roles of the mesothelium in driving organ pathophysiology. A deeper understanding of trunk organ diseases could thus emerge from studying the mechanisms underlying embryonic mesothelial EMT. Such a developmental focus might also identify much-needed new therapeutic avenues for trunk organ disease. For further research, three main aspects in mesothelium’s biology are particularly relevant: (1) intermediate stages of EMT, (2) common and organ-unique mechanisms of EMT, and (3) mesothelium’s cell type heterogeneity and their relation with EMT.
Intermediate stages of EMT
Coelomic epithelium cells of the proepicardial organ need to go through a series of highly orchestrated steps to populate the embryonic heart primordium: (1) directed migration towards the heart surface, (2) controlled proliferation to increase cell numbers, (3) migration towards the inner compartments of the heart, and (4) gain of mesenchymal traits to adopt fibroblastic and smooth muscle characteristics. Not only do these stepwise events require alternating commitments to EMT and MET to sustain epicardial identity while also providing the mesenchymal lineages of the heart, they also require intermediate stages within the EMT spectrum. The plethora of genes described here that regulates these events, and the disparity between knockout lines with regard to their effects on EMT, highlights the complexity of (different stages of) EMT and the gene regulatory networks that are at play. Hence, it is important to consider that during scarring and wound healing in adult life, the same extent of heterogeneity may be expected, where cells may linger in intermediary stages between a full epithelial and mesenchymal state to promote wound healing or scarring. For example, studies on mesothelial ovarian cancers revealed they could be grouped in different compositions along the EMT spectrum178,199,200, which may reflect differently on their ability to migrate, adhere, invade or survive201. This stresses the importance to focus on the entire range of EMT to allow for a more dynamic interpretation of the fluidity and plasticity of this cell state. It may also provide more insight into the different scarring signatures recognized among mesothelial pathologies, e.g. why is the viscera sometimes more affected than the parietal surface, and why do some pathologies involve fibrous scars to exclusively develop on organ surfaces, whereas others extend into the parenchyma?
Mechanisms of known EMT drivers
At present there is an underestimation of the complexity of WT1 signal regulation, and the networks in which it functions. This complexity includes alternative start codons, splice sites, and RNA editing that can theoretically give rise to 36 different proteins and many more potential dimers202. The creation of mice ablated for specific WT1 isoforms has provided direct evidence that Wt1 splicing variants perform distinct functions in embryonic development45,203. Moreover, there may be gene elements that can influence the effects of WT1 isoforms. For example, WT1 drives EMT in the coelomic epithelium covering the heart, liver, and lung primordium through close cooperation with GATA4 under control of the G2 enhancer domain204. Contrary, in kidney development WT1 maintains epithelial integrity, where the Gata4 gene is not driven by the G2 enhancer205. Thus, much work remains on the exact mechanisms that control mesothelial EMT, which likely requires a more systems-based approach. Establishing such gene regulatory networks and feedback loops will further our chance of developing successful therapies to reduce scarring and prevent fibrosis.
Cell heterogeneity within the mesothelium
Single-cell transcriptomic studies have identified at least three epicardial cell subsets in the healthy adult heart of zebrafish104, and a sub-population of c-kit+ epicardials cells exists in the adult mouse epicardium98,206. However, although cultured epicardial cells from human and mouse adults are able to recapitulate at least part of the differentiation potential of their embryonic counterparts117,207, isolated adult epicardial cells of mice are not able to generate colony-forming units ex vivo208. As such, if the adult mesothelium of mice and humans contains a sub-population of cells with stem-like properties, they may exist in a dormant state209. A study in mice has recently revealed the presence of epicardial cell heterogeneity after subjecting hearts to models of myocardial infarction210. These cells expressed the progenitor markers CD90+, CD44+, CD105+, c-kit+, but were distinct from their embryonic counterparts99,210. Rather than an existing dormant progenitor state, stemness may be conferred to differentiated cells by changes in the local microenvironment in response to tissue injury. The idea that cell reprogramming can generate facultative stem cells emerged independently from several experimental models. For example, pancreatic ductal ligation causes broad degeneration of acinar cells211. However, the resulting survivor population is reprogrammed and exhibits embryonic multi-potency factors capable of partial tissue regeneration212. Whether this applies to adult mesothelial tissues remains to be explored. Further insight into the heterogeneity, the factors necessary to activate dormant or reprogrammable progenitor states, and how each of these populations contribute to EMT, scarring, and wound healing, will be future areas of research.
In recent years there has been a proliferation of studies on drugs targeting WT1 and MSLN, many of which are in advanced stages of clinical testing. Drugs include recombinant immunotoxin proteins, vaccines, monoclonal antibodies, and antibody-drug conjugates213,214,215. One example is Amatuximab, a chimeric high-affinity monoclonal IgG1/k antibody targeting MSLN, which elicits cellular toxicity against MSLN-expressing tumour cells, and reduces invasive capacity and tumour sphere formation216. Amatuximab improved median overall survival and was well tolerated in phase II clinical trials of patients with advanced pleural mesothelioma217. It would be important to determine whether patients receiving therapies targeting MSLN and WT1 have equally reduced risks of developing additional diseases such as ischaemia, chronic fibrosis, and adhesions. In this regard, other embryonic markers, such as WNT effectors, β-catenin, Notch1, and RALDH2, some of which are highly expressed in mesothelial cancers similarly warrant further investigation218,219,220. Clinical studies that target mesothelial cancers may help clarify the extent of co-morbidity with other mesothelial pathologies where EMT plays an important role, opening new clinical avenues through which our current understanding of the EMT programme and transitional events can be exploited and organ disease can be curtailed.
References
Wang, N. S. The regional difference of pleural mesothelial cells in rabbits. Am. Rev. Respir. Dis. 110, 623–633 (1974).
Ishihara, T. et al. Histologic and ultrastructural features of normal human parietal pericardium. Am. J. Cardiol. 46, 744–753 (1980).
Albertine, K. H., Wiener-Kronish, J. P., Roos, P. J. & Staub, N. C. Structure, blood supply, and lymphatic vessels of the sheep’s visceral pleura. Am. J. Anat. 165, 277–294 (1982).
Mutsaers, S. E. Mesothelial cells: their structure, function and role in serosal repair. Respirology. 7, 171–191 (2002).
Moore, K., Persaud, T. & Torchia, M. Before We Are Born. Essentials of Embryology and Birth Defects (Elsevier, Oxford, 2015).
Rinkevich, Y. et al. Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat. Cell Biol. 14, 1251–1260 (2012).
Gittenberger-de Groot, A. C., Vrancken Peeters, M. P., Mentink, M. M., Gourdie, R. G. & Poelmann, R. E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 82, 1043–1052 (1998).
Pérez-Pomares, J. M. et al. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 247, 307–326 (2002).
Wilm, B., Ipenberg, A., Hastie, N. D., Burch, J. B. E. & Bader, D. M. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 132, 5317–5328 (2005).
Cai, C.-L. et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108 (2008).
Zhou, B. et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113 (2008).
Martínez-Estrada, O. M. et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 42, 89–93 (2010).
Wessels, A. et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 366, 111–124 (2012).
Takeichi, M., Nimura, K., Mori, M., Nakagami, H. & Kaneda, Y. The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of Slug in murine primary epicardial cells. PLoS ONE 8, e57829 (2013).
Kreidberg, J. A. et al. WT-1 is required for early kidney development. Cell 74, 679–691 (1993).
von Gise, A. et al. WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev. Biol. 356, 421–431 (2011).
Zhou, B., von Gise, A., Ma, Q., Hu, Y. W. & Pu, W. T. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Biol. 338, 251–261 (2010).
Zamora, M., Männer, J. & Ruiz-Lozano, P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc. Natl Acad. Sci. USA 104, 18109–18114 (2007).
Acharya, A. et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139, 2139–2149 (2012).
Braitsch, C. M., Combs, M. D., Quaggin, S. E. & Yutzey, K. E. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 368, 345–357 (2012).
Fraidenraich, D. et al. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science 306, 247–252 (2004).
Niederreither, K. et al. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 128, 1019–1031 (2001).
Guadix, J. A. et al. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 138, 1093–1097 (2011).
Azambuja, A. P. et al. Retinoic acid and VEGF delay smooth muscle relative to endothelial differentiation to coordinate inner and outer coronary vessel wall morphogenesis. Circ. Res. 107, 204–216 (2010).
Merki, E. et al. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl Acad. Sci. USA 102, 18455–18460 (2005).
Watt, A. J., Battle, M. A., Li, J. & Duncan, S. A. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc. Natl Acad. Sci. USA 101, 12573–12578 (2004).
Borok, M. J., Papaioannou, V. E. & Sussel, L. Unique functions of Gata4 in mouse liver induction and heart development. Dev. Biol. 410, 213–222 (2016).
Crispino, J. D. et al. Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev. 15, 839–844 (2001).
Zhou, B. et al. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J. Clin. Invest. 119, 1462–1476 (2009).
Xin, M. et al. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc. Natl Acad. Sci. USA 103, 11189–11194 (2006).
Kolander, K. D., Holtz, M. L., Cossette, S. M., Duncan, S. A. & Misra, R. P. Epicardial GATA factors regulate early coronary vascular plexus formation. Dev. Biol. 386, 204–215 (2014).
del Monte, G. et al. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ. Res. 108, 824–836 (2011).
Pérez-Pomares, J. M. et al. Contribution of mesothelium-derived cells to liver sinusoids in avian embryos. Dev. Dyn. 229, 465–474 (2004).
Asahina, K., Zhou, B., Pu, W. T. & Tsukamoto, H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53, 983–995 (2011).
Lua, I., James, D., Wang, J., Wang, K. S. & Asahina, K. Mesodermal mesenchymal cells give rise to myofibroblasts, but not epithelial cells, in mouse liver injury. Hepatology 60, 311–322 (2014).
Delgado, I. et al. GATA4 loss in the septum transversum mesenchyme promotes liver fibrosis in mice. Hepatology 59, 2358–2370 (2014).
Ijpenberg, A. et al. Wt1 and retinoic acid signaling are essential for stellate cell development and liver morphogenesis. Dev. Biol. 312, 157–170 (2007).
Paris, N. D., Coles, G. L. & Ackerman, K. G. Wt1 and β-catenin cooperatively regulate diaphragm development in the mouse. Dev. Biol. 407, 40–56 (2015).
Berg, T. et al. β-catenin regulates mesenchymal progenitor cell differentiation during hepatogenesis. J. Surg. Res. 164, 276–285 (2010).
Watt, A. J., Zhao, R., Li, J. & Duncan, S. A. Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev. Biol. 7, 37 (2007).
Miyamoto, Y., Taniguchi, H., Hamel, F., Silversides, D. W. & Viger, R. S. A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol. Biol. 9, 44 (2008).
Ikeda, Y., Shen, W. H., Ingraham, H. A. & Parker, K. L. Developmental expression of mouse steroidogenic factor-1, an essential regulator of the steroid hydroxylases . Mol. Endocrinol. 8, 654–662 (1994).
Hatano, O., Takakusu, A., Nomura, M. & Morohashi, K. Identical origin of adrenal cortex and gonad revealed by expression profiles of Ad4BP/SF-1. Genes Cells 1, 663–671 (1996).
Karl, J. & Capel, B. Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev. Biol. 203, 323–333 (1998).
Hammes, A. et al. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell 106, 319–329 (2001).
Liu, C., Peng, J., Matzuk, M. M. & Yao, H. H.-C. Lineage specification of ovarian theca cells requires multicellular interactions via oocyte and granulosa cells. Nat. Commun. 6, 6934 (2015).
Bohnenpoll, T. et al. Tbx18 expression demarcates multipotent precursor populations in the developing urogenital system but is exclusively required within the ureteric mesenchymal lineage to suppress a renal stromal fate. Dev. Biol. 380, 25–36 (2013).
Häfner, R., Bohnenpoll, T., Rudat, C., Schultheiss, T. M. & Kispert, A. Fgfr2 is required for the expansion of the early adrenocortical primordium. Mol. Cell Endocrinol. 413, 168–177 (2015).
Bingham, N. C., Verma-Kurvari, S., Parada, L. F. & Parker, K. L. Development of a steroidogenic factor 1/Cre transgenic mouse line. Genesis 44, 419–424 (2006).
Kusaka, M. et al. Abnormal epithelial cell polarity and ectopic epidermal growth factor receptor (EGFR) expression induced in Emx2 KO embryonic gonads. Endocrinology 151, 5893–5904 (2010).
Hummitzsch, K. et al. A new model of development of the mammalian ovary and follicles. PLoS ONE 8, e55578 (2013).
Albrecht, K. H. & Eicher, E. M. Evidence that Sry is expressed in pre-Sertoli cells and Sertoli and granulosa cells have a common precursor. Dev. Biol. 240, 92–107 (2001).
Combes, A. N. et al. Endothelial cell migration directs testis cord formation. Dev. Biol. 326, 112–120 (2009).
Chassot, A.-A. et al. Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum. Mol. Genet. 17, 1264–1277 (2008).
Piprek, R. P. Molecular mechanisms underlying female sex determination—antagonism between female and male pathway. Folia Biol. (Praha) 57, 105–113 (2009).
Brennan, J., Tilmann, C. & Capel, B. Pdgfr-alpha mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev. 17, 800–810 (2003).
Cool, J., Carmona, F. D., Szucsik, J. C. & Capel, B. Peritubular myoid cells are not the migrating population required for testis cord formation in the XY gonad. Sex. Dev. 2, 128–133 (2008).
Barsoum, I. B., Kaur, J., Ge, R. S., Cooke, P. S. & Yao, H. H.-C. Dynamic changes in fetal Leydig cell populations influence adult Leydig cell populations in mice.FASEB J. 27, 2657–2666 (2013).
Hu, Y.-C., Okumura, L. M. & Page, D. C. Gata4 is required for formation of the genital ridge in mice. PLoS Genet. 9, e1003629 (2013).
Airik, R., Bussen, M., Singh, M. K., Petry, M. & Kispert, A. Tbx18 regulates the development of the ureteral mesenchyme. J. Clin. Invest. 116, 663–674 (2006).
Cui, S. et al. Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development 131, 4095–4105 (2004).
Tevosian, S. G. et al. Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2. Development 129, 4627–4634 (2002).
Wilhelm, D. & Englert, C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. 16, 1839–1851 (2002).
Fujimoto, Y. et al. Homeoproteins Six1 and Six4 regulate male sex determination and mouse gonadal development. Dev. Cell 26, 416–430 (2013).
Klattig, J., Sierig, R., Kruspe, D., Makki, M. S. & Englert, C. WT1-mediated gene regulation in early urogenital ridge development. Sex. Dev. 1, 238–254 (2007).
Ponnusamy, M. P. et al. Emerging role of mucins in epithelial to mesenchymal transition. Curr. Cancer Drug Targets 13, 945–956 (2013).
Chen, S.-H., Hung, W.-C., Wang, P., Paul, C. & Konstantopoulos, K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci. Rep. 3, 1870 (2013).
Lécureuil, C., Fontaine, I., Crepieux, P. & Guillou, F. Sertoli and granulosa cell-specific Cre recombinase activity in transgenic mice. Genesis 33, 114–118 (2002).
Gao, F. et al. The Wilms tumor gene, Wt1, is required for Sox9 expression and maintenance of tubular architecture in the developing testis. Proc. Natl Acad. Sci. USA 103, 11987–11992 (2006).
Wen, Q., Wang, Y., Tang, J., Cheng, C. Y. & Liu, Y.-X. Sertoli cell Wt1 regulates peritubular myoid cell and fetal Leydig cell differentiation during fetal testis development. PLoS ONE 11, e0167920 (2016).
Chen, M. et al. Wt1 directs the lineage specification of sertoli and granulosa cells by repressing Sf1 expression. Development 144, 44–53 (2017).
Zhang, L. et al. Reprogramming of Sertoli cells to fetal-like Leydig cells by Wt1 ablation. Proc. Natl Acad. Sci. USA 112, 4003–4008 (2015).
Bandiera, R. et al. WT1 maintains adrenal-gonadal primordium identity and marks a population of AGP-like progenitors within the adrenal gland. Dev. Cell 27, 5–18 (2013).
Hogan, B. L. Morphogenesis. Cell 96, 225–233 (1999).
Norden, J., Grieskamp, T., Christoffels, V. M., Moorman, A. F. M. & Kispert, A. Partial absence of pleuropericardial membranes in Tbx18- and Wt1-deficient mice. PLoS ONE 7, e45100 (2012).
Malpel, S., Mendelsohn, C. & Cardoso, W. V. Regulation of retinoic acid signaling during lung morphogenesis. Development 127, 3057–3067 (2000).
Niederreither, K., Fraulob, V., Garnier, J.-M., Chambon, P. & Dollé, P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech. Dev. 110, 165–171 (2002).
Cano, E., Carmona, R. & Muñoz-Chápuli, R. Wt1-expressing progenitors contribute to multiple tissues in the developing lung. Am. J. Physiol. Lung Cell Mol. Physiol. 305, L322–L332 (2013).
Morimoto, M. et al. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J. Cell Sci. 123, 213–224 (2010).
Kaarteenaho, R., Lappi-Blanco, E. & Lehtonen, S. Epithelial N-cadherin and nuclear β-catenin are up-regulated during early development of human lung. BMC Dev. Biol. 10, 113 (2010).
Ackerman, K. G. et al. Gata4 is necessary for normal pulmonary lobar development. Am. J. Respir. Cell Mol. Biol. 36, 391–397 (2007).
Dixit, R., Ai, X. & Fine, A. Derivation of lung mesenchymal lineages from the fetal mesothelium requires hedgehog signaling for mesothelial cell entry. Development 140, 4398–4406 (2013).
Que, J. et al. Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc. Natl Acad. Sci. USA 105, 16626–16630 (2008).
von Gise, A. et al. Contribution of fetal, but not adult, pulmonary mesothelium to mesenchymal lineages in lung homeostasis and fibrosis. Am. J. Respir. Cell Mol. Biol. 54, 222–230 (2016).
Norden, J. et al. Wt1 and retinoic acid signaling in the subcoelomic mesenchyme control the development of the pleuropericardial membranes and the sinus horns. Circ. Res. 106, 1212–1220 (2010).
Ackerman, K. G. et al. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 1, 58–65 (2005).
Jay, P. Y. et al. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev. Biol. 301, 602–614 (2007).
Shikama, N. et al. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 22, 5175–5185 (2003).
Yin, Y. et al. An FGF-WNT gene regulatory network controls lung mesenchyme development. Dev. Biol. 319, 426–436 (2008).
De Langhe, S. P. et al. Formation and differentiation of multiple mesenchymal lineages during lung development is regulated by beta-catenin signaling. PLoS ONE 3, e1516 (2008).
Desai, T. J. et al. Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev. Biol. 291, 12–24 (2006).
Wang, Z., Dollé, P., Cardoso, W. V. & Niederreither, K. Retinoic acid regulates morphogenesis and patterning of posterior foregut derivatives. Dev. Biol. 297, 433–445 (2006).
Chazaud, C., Dollé, P., Rossant, J. & Mollard, R. Retinoic acid signaling regulates murine bronchial tubule formation. Mech. Dev. 120, 691–700 (2003).
Cardenas, H., Zhao, J., Vieth, E., Nephew, K. P. & Matei, D. EZH2 inhibition promotes epithelial-to-mesenchymal transition in ovarian cancer cells. Oncotarget 7, 84453–84467 (2016).
Snitow, M., Lu, M., Cheng, L., Zhou, S. & Morrisey, E. E. Ezh2 restricts the smooth muscle lineage during mouse lung mesothelial development. Development 143, 3733–3741 (2016).
Zhou, B. et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Invest. 121, 1894–1904 (2011).
Huang, G. N. et al. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science 338, 1599–1603 (2012).
Limana, F. et al. Identification of myocardial and vascular precursor cells in human and mouse epicardium. Circ. Res. 101, 1255–1265 (2007).
Limana, F. et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit(+) cells: role of the pericardial fluid. J. Mol. Cell Cardiol. 48, 609–618 (2010).
Kikuchi, K. et al. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 20, 397–404 (2011).
Duan, J. et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 31, 429–442 (2012).
Ruiz-Villalba, A. et al. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J. Am. Coll. Cardiol. 65, 2057–2066 (2015).
Wang, J., Cao, J., Dickson, A. L. & Poss, K. D. Epicardial regeneration is guided by cardiac outflow tract and Hedgehog signalling. Nature 522, 226–230 (2015).
Cao, J. et al. Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 143, 232–243 (2016).
Bock-Marquette, I. et al. Thymosin beta4 mediated PKC activation is essential to initiate the embryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J. Mol. Cell Cardiol. 46, 728–738 (2009).
Smart, N. et al. Thymosin beta4 facilitates epicardial neovascularization of the injured adult heart. Ann. NY Acad. Sci. 1194, 97–104 (2010).
Moore-Morris, T. et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 124, 2921–2934 (2014).
Ali, S. R. et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 115, 625–635 (2014).
Braitsch, C. M., Kanisicak, O., van Berlo, J. H., Molkentin, J. D. & Yutzey, K. E. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J. Mol. Cell Cardiol. 65, 108–119 (2013).
Li, Y., Wang, J. & Asahina, K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc. Natl Acad. Sci. USA 110, 2324–2329 (2013).
Asahina, K. et al. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 49, 998–1011 (2009).
Iwaisako, K. et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl Acad. Sci. USA 111, E3297–E3305 (2014).
Koyama, Y. et al. Mesothelin/mucin 16 signaling in activated portal fibroblasts regulates cholestatic liver fibrosis. J. Clin. Invest. 127, 1254–1270 (2017).
Ochiai, K., Muramatsu, H., Yamamoto, S., Ando, H. & Muramatsu, T. The role of midkine and pleiotrophin in liver regeneration. Liver Int. 24, 484–491 (2004).
Inagaki, N. F., Inagaki, F. F., Kokudo, N. & Miyajima, A. Use of mouse liver mesothelial cells to prevent postoperative adhesion and promote liver regeneration after hepatectomy. J. Hepatol. 62, 1141–1147 (2015).
Miyoshi, H., Ajima, R., Luo, C. T., Yamaguchi, T. P. & Stappenbeck, T. S. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science 338, 108–113 (2012).
Smart, N. et al. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445, 177–182 (2007).
Auersperg, N., Wong, A. S., Choi, K. C., Kang, S. K. & Leung, P. C. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr. Rev. 22, 255–288 (2001).
Ahmed, N. et al. Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am. J. Physiol. Cell Physiol. 290, C1532–C1542 (2006).
Zhu, Y., Nilsson, M. & Sundfeldt, K. Phenotypic plasticity of the ovarian surface epithelium: TGF-beta 1 induction of epithelial to mesenchymal transition (EMT) in vitro. Endocrinology 151, 5497–5505 (2010).
Flesken-Nikitin, A. et al. Ovarian surface epithelium at the junction area contains a cancer-prone stem cell niche. Nature 495, 241–245 (2013).
Gamwell, L. F., Collins, O. & Vanderhyden, B. C. The mouse ovarian surface epithelium contains a population of LY6A (SCA-1) expressing progenitor cells that are regulated by ovulation-associated factors. Biol. Reprod. 87, 80 (2012).
Szotek, P. P. et al. Normal ovarian surface epithelial label-retaining cells exhibit stem/progenitor cell characteristics. Proc. Natl Acad. Sci. USA 105, 12469–12473 (2008).
Beller, U., Haimovitch, R. & Ben-Sasson, S. Periovulatory multifocal mesothelial proliferation: a possible association with malignant transformation. Int. J. Gynecol. Cancer 5, 306–309 (1995).
Tan, O. L. & Fleming, J. S. Proliferating cell nuclear antigen immunoreactivity in the ovarian surface epithelium of mice of varying ages and total lifetime ovulation number following ovulation. Biol. Reprod. 71, 1501–1507 (2004).
Ng, A. et al. Lgr5 marks stem/progenitor cells in ovary and tubal epithelia. Nat. Cell Biol. 16, 745–757 (2014).
Batra, H. & Antony, V. B. Pleural mesothelial cells in pleural and lung diseases. J. Thorac. Dis. 7, 964–980 (2015).
Huggins, J. T. & Sahn, S. A. Causes and management of pleural fibrosis. Respirology 9, 441–447 (2004).
Chen, L.-J. et al. Bleomycin induced epithelial-mesenchymal transition (EMT) in pleural mesothelial cells. Toxicol. Appl. Pharmacol. 283, 75–82 (2015).
Zolak, J. S. et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 182, 1239–1247 (2013).
Zhang, Q. et al. miR-18a-5p inhibits sub-pleural pulmonary fibrosis by targeting TGF-β receptor II. Mol. Ther. 25, 728–738 (2017).
Decologne, N. et al. Bleomycin induces pleural and subpleural fibrosis in the presence of carbon particles. Eur. Respir. J. 35, 176–185 (2010).
Tucker, T. A. et al. Plasminogen activator inhibitor-1 deficiency augments visceral mesothelial organization, intrapleural coagulation, and lung restriction in mice with carbon black/bleomycin-induced pleural injury. Am. J. Respir. Cell Mol. Biol. 50, 316–327 (2014).
Boren, J. et al. Inhibition of glycogen synthase kinase 3β blocks mesomesenchymal transition and attenuates Streptococcus pneumonia-mediated pleural injury in mice. Am. J. Pathol. 187, 2461–2472 (2017).
Cool, C. D. et al. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am. J. Respir. Crit. Care Med. 174, 654–658 (2006).
Wellman, T. J., Mondoñedo, J. R., Davis, G. S., Bates, J. H. T. & Suki, B. Topographic distribution of idiopathic pulmonary fibrosis: a hybrid physics- and agent-based model. Physiol. Meas. 39, 064007 (2018).
Mubarak, K. K. et al. Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur. Respir. J. 39, 133–140 (2012).
Karki, S. Wilms' tumor 1 (Wt1) regulates pleural mesothelial cell plasticity and transition into myofibroblasts in idiopathic pulmonary fibrosis. FASEB J. 28, 1122–1131 (2014).
Decologne, N. et al. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J. Immunol. 179, 6043–6051 (2007).
Suzuki, T. et al. An injured tissue affects the opposite intact peritoneum during postoperative adhesion formation. Sci. Rep. 5, 7668 (2015).
Stangel, J. J., Nisbet, J. D. & Settles, H. Formation and prevention of postoperative abdominal adhesions. J. Reprod. Med. 29, 143–156 (1984).
Rocca, A. et al. Prevention and treatment of peritoneal adhesions in patients affected by vascular diseases following surgery: a review of the literature. Open Med. Wars. Pol. 11, 106–114 (2016).
Liu, Y. et al. Transition of mesothelial cell to fibroblast in peritoneal dialysis: EMT, stem cell or bystander? Perit. Dial. Int. 35, 14–25 (2015).
Di Paolo, N. & Sacchi, G. Peritoneal vascular changes in continuous ambulatory peritoneal dialysis (CAPD): an in vivo model for the study of diabetic microangiopathy. Perit. Dial. Int. 9, 41–45 (1989).
Honda, K., Nitta, K., Horita, S., Yumura, W. & Nihei, H. Morphological changes in the peritoneal vasculature of patients on CAPD with ultrafiltration failure. Nephron 72, 171–176 (1996).
Di Paolo, N. et al. Morphology of the peritoneal membrane during continuous ambulatory peritoneal dialysis. Nephron 44, 204–211 (1986).
Mateijsen, M. A. et al. Vascular and interstitial changes in the peritoneum of CAPD patients with peritoneal sclerosis. Perit. Dial. Int. 19, 517–525 (1999).
Kawaguchi, Y., Kawanishi, H., Mujais, S., Topley, N. & Oreopoulos, D. G. Encapsulating peritoneal sclerosis: definition, etiology, diagnosis, and treatment. International Society for Peritoneal Dialysis Ad Hoc Committee on Ultrafiltration Management in Peritoneal Dialysis. Perit. Dial. Int. 20(Suppl 4), S43–S55 (2000).
Williams, J. D. et al. The natural course of peritoneal membrane biology during peritoneal dialysis. Kidney Int. Suppl. 64, S43–S49 (2003).
Whitaker, D., Manning, L., Robinson, B. & Shilkin, K. in The Pathobiology of the Mesothelium 25–68 (Hemisphere Publishing Corp., New York, 1992).
Yang, A. H., Chen, J. Y. & Lin, J. K. Myofibroblastic conversion of mesothelial cells. Kidney Int. 63, 1530–1539 (2003).
Yáñez-Mó, M. et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N. Engl. J. Med. 348, 403–413 (2003).
Sandoval, P. et al. Mesothelial-to-mesenchymal transition in the pathogenesis of post-surgical peritoneal adhesions. J. Pathol. 239, 48–59 (2016).
Chen, Y.-T. et al. Lineage tracing reveals distinctive fates for mesothelial cells and submesothelial fibroblasts during peritoneal injury. J. Am. Soc. Nephrol. 25, 2847–2858 (2014).
Lua, I., Li, Y., Pappoe, L. S. & Asahina, K. Myofibroblastic conversion and regeneration of mesothelial cells in peritoneal and liver fibrosis. Am. J. Pathol. 185, 3258–3273 (2015).
Ghellai, A. M. et al. Role of transforming growth factor beta-1 in peritonitis-induced adhesions. J. Gastrointest. Surg. 4, 316–323 (2000).
Fraser, D., Wakefield, L. & Phillips, A. Independent regulation of transforming growth factor-beta1 transcription and translation by glucose and platelet-derived growth factor. Am. J. Pathol. 161, 1039–1049 (2002).
Margetts, P. J. et al. Transient overexpression of TGF-{beta}1 induces epithelial mesenchymal transition in the rodent peritoneum. J. Am. Soc. Nephrol. 16, 425–436 (2005).
Loureiro, J. et al. Blocking TGF-β1 protects the peritoneal membrane from dialysate-induced damage. J. Am. Soc. Nephrol. 22, 1682–1695 (2011).
Patel, P. et al. Smad3-dependent and -independent pathways are involved in peritoneal membrane injury. Kidney Int. 77, 319–328 (2010).
Tsai, J. M. et al. Adhesions are derived from hypoxia responsive MSLN+ mesothelial cells and are resolved via targeted therapies. Sci. Transl. Med. (2018). In the Press.
Law, M. R., Hodson, M. E. & Heard, B. E. Malignant mesothelioma of the pleura: relation between histological type and clinical behaviour. Thorax 37, 810–815 (1982).
Rudd, R. M. Malignant mesothelioma. Br. Med. Bull. 93, 105–123 (2010).
Soeberg, M. J. et al. Incidence and survival trends for malignant pleural and peritoneal mesothelioma, Australia, 1982-2009. Occup. Environ. Med. 73, 187–194 (2016).
Baumann, F., Ambrosi, J.-P. & Carbone, M. Asbestos is not just asbestos: an unrecognised health hazard. Lancet Oncol. 14, 576–578 (2013).
Carbone, M. et al. Combined genetic and genealogic studies uncover a large BAP1 cancer syndrome kindred tracing back nine generations to a common ancestor from the 1700s. PLoS Genet. 11, e1005633 (2015).
Testa, J. R. et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 43, 1022–1025 (2011).
Napolitano, A. et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene 35, 1996–2002 (2016).
van Zandwijk, N. et al. Guidelines for the diagnosis and treatment of malignant pleural mesothelioma. J. Thorac. Dis. 5, E254–E307 (2013).
Kurman, R. J. & Shih, I.-M. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am. J. Surg. Pathol. 34, 433–443 (2010).
Piek, J. M. et al. Dysplastic changes in prophylactically removed Fallopian tubes of women predisposed to developing ovarian cancer. J. Pathol. 195, 451–456 (2001).
Callahan, M. J. et al. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J. Clin. Oncol. 25, 3985–3990 (2007).
Kindelberger, D. W. et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am. J. Surg. Pathol. 31, 161–169 (2007).
Eckert, M. A. et al. Genomics of ovarian cancer progression reveals diverse metastatic trajectories including intraepithelial metastasis to the fallopian tube. Cancer Discov. 6, 1342–1351 (2016).
Coscia, F. et al. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 7, 12645 (2016).
Zhang, X. Y. et al. Characteristics and growth patterns of human peritoneal mesothelial cells: comparison between advanced epithelial ovarian cancer and non-ovarian cancer sources. J. Soc. Gynecol. Investig. 6, 333–340 (1999).
Chang, K. & Pastan, I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl Acad. Sci. USA 93, 136–140 (1996).
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
He, X. et al. Mesothelin promotes epithelial-to-mesenchymal transition and tumorigenicity of human lung cancer and mesothelioma cells. Mol. Cancer 16, 63 (2017).
Bueno, R. et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 47, 407 (2015).
de Reyniès, A. et al. Molecular classification of malignant pleural mesothelioma: identification of a poor prognosis subgroup linked to the epithelial-to-mesenchymal transition. Clin. Cancer Res. 20, 1323–1334 (2014).
Bozzi, F. et al. Epithelioid peritoneal mesothelioma: a hybrid phenotype within a mesenchymal-epithelial/epithelial-mesenchymal transition framework. Oncotarget 7, 75503–75517 (2016).
Tan, T. Z. et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 6, 1279–1293 (2014).
Fischer, K. R. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476 (2015).
Zheng, X. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015).
Granot, D. et al. In vivo imaging of the systemic recruitment of fibroblasts to the angiogenic rim of ovarian carcinoma tumors. Cancer Res. 67, 9180–9189 (2007).
Schauer, I. G., Sood, A. K., Mok, S. & Liu, J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia 13, 393–405 (2011).
Rynne-Vidal, A. et al. Mesothelial-to-mesenchymal transition as a possible therapeutic target in peritoneal metastasis of ovarian cancer. J. Pathol. 242, 140–151 (2017).
Lv, Z.-D. et al. Mesothelial cells differentiate into fibroblast-like cells under the scirrhous gastric cancer microenvironment and promote peritoneal carcinomatosis in vitro and in vivo. Mol. Cell Biochem. 377, 177–185 (2013).
Sandoval, P. et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 231, 517–531 (2013).
Matte, I. et al. Ovarian cancer ascites enhance the migration of patient-derived peritoneal mesothelial cells via cMet pathway through HGF-dependent and -independent mechanisms. Int. J. Cancer 137, 289–298 (2015).
Satoyoshi, R., Aiba, N., Yanagihara, K., Yashiro, M. & Tanaka, M. Tks5 activation in mesothelial cells creates invasion front of peritoneal carcinomatosis. Oncogene 34, 3176–3187 (2015).
Tanaka, M. et al. Mesothelial cells create a novel tissue niche that facilitates gastric cancer invasion. Cancer Res. 77, 684–695 (2017).
Nakamura, K. et al. Exosomes promote ovarian cancer cell invasion through transfer of CD44 to peritoneal mesothelial cells. Mol. Cancer Res. 15, 78–92 (2017).
Greening, D. W. et al. Secreted primary human malignant mesothelioma exosome signature reflects oncogenic cargo. Sci. Rep. 6, 32643 (2016).
Yokoi, A. et al. Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 8, 14470 (2017).
Wei, M. et al. Malignant ascites-derived exosomes promote proliferation and induce carcinoma-associated fibroblasts transition in peritoneal mesothelial cells. Oncotarget 8, 42262–42271 (2017).
Deng, G. et al. Gastric cancer-derived exosomes promote peritoneal metastasis by destroying the mesothelial barrier. FEBS Lett. 591, 2167–2179 (2017).
Huang, R. Y.-J. et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 4, e915 (2013).
Verhaak, R. G. W. et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest. 123, 517–525 (2013).
Chu, Y.-S. et al. Prototypical type I E-cadherin and type II cadherin-7 mediate very distinct adhesiveness through their extracellular domains. J. Biol. Chem. 281, 2901–2910 (2006).
Hohenstein, P. & Hastie, N. D. The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 15(Spec No 2), R196–R201 (2006).
Bradford, S. T. et al. A cell-autonomous role for WT1 in regulating Sry in vivo. Hum. Mol. Genet. 18, 3429–3438 (2009).
Carmona, R. et al. Conditional deletion of WT1 in the septum transversum mesenchyme causes congenital diaphragmatic hernia in mice. eLife 5, e16009 (2016).
Essafi, A. et al. A wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev. Cell. 21, 559–574 (2011).
Tallini, Y. N. et al. c-kit expression identifies cardiovascular precursors in the neonatal heart. Proc. Natl Acad. Sci. USA 106, 1808–1813 (2009).
van Tuyn, J. et al. Epicardial cells of human adults can undergo an epithelial-to-mesenchymal transition and obtain characteristics of smooth muscle cells in vitro. Stem Cells 25, 271–278 (2007).
Chong, J. J. H. et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 9, 527–540 (2011).
Cabezas-Wallscheid, N. et al. Vitamin A-retinoic acid signaling regulates hematopoietic stem cell dormancy. Cell 169, 807–823.e19 (2017).
Bollini, S. et al. Re-activated adult epicardial progenitor cells are a heterogeneous population molecularly distinct from their embryonic counterparts. Stem. Cells Dev. 23, 1719–1730 (2014).
Ziv, O., Glaser, B. & Dor, Y. The plastic pancreas. Dev. Cell 26, 3–7 (2013).
Pan, F. C. et al. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 140, 751–764 (2013).
Hassan, R. et al. Anti-tumor activity of K1-LysPE38QQR, an immunotoxin targeting mesothelin, a cell-surface antigen overexpressed in ovarian cancer and malignant mesothelioma. J. Immunother. 23, 473–479 (2000).
Hung, C.-F., Calizo, R., Tsai, Y.-C., He, L. & Wu, T.-C. A DNA vaccine encoding a single-chain trimer of HLA-A2 linked to human mesothelin peptide generates anti-tumor effects against human mesothelin-expressing tumors. Vaccine 25, 127–135 (2007).
Tang, Z. et al. A human single-domain antibody elicits potent antitumor activity by targeting an epitope in mesothelin close to the cancer cell surface. Mol. Cancer Ther. 12, 416–426 (2013).
Hassan, R. et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 7, 20 (2007).
Hassan, R. et al. Phase II clinical trial of amatuximab, a chimeric antimesothelin antibody with pemetrexed and cisplatin in advanced unresectable pleural mesothelioma. Clin. Cancer Res. 20, 5927–5936 (2014).
Silva, I. A. et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 71, 3991–4001 (2011).
Cortes-Dericks, L., Froment, L., Boesch, R., Schmid, R. A. & Karoubi, G. Cisplatin-resistant cells in malignant pleural mesothelioma cell lines show ALDH(high)CD44(+) phenotype and sphere-forming capacity. BMC Cancer 14, 304 (2014).
Bodnar, L. et al. Wnt/β-catenin pathway as a potential prognostic and predictive marker in patients with advanced ovarian cancer. J. Ovarian Res. 7, 16 (2014).
Heldin, P. & Pertoft, H. Synthesis and assembly of the hyaluronan-containing coats around normal human mesothelial cells. Exp. Cell Res. 208, 422–429 (1993).
Yung, S. et al. Source of peritoneal proteoglycans. Human peritoneal mesothelial cells synthesize and secrete mainly small dermatan sulfate proteoglycans. Am. J. Pathol. 146, 520–529 (1995).
Bellingan, G. J., Caldwell, H., Howie, S. E., Dransfield, I. & Haslett, C. In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J. Immunol. 157, 2577–2585 (1996).
Bellingan, G. J. et al. Adhesion molecule-dependent mechanisms regulate the rate of macrophage clearance during the resolution of peritoneal inflammation. J. Exp. Med. 196, 1515–1521 (2002).
Gosselin, D. et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340 (2014).
Lavin, Y. et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326 (2014).
Davies, L. C. et al. Peritoneal tissue-resident macrophages are metabolically poised to engage microbes using tissue-niche fuels. Nat. Commun. 8, 2074 (2017).
Avraham-Chakim, L. et al. Fluid-flow induced wall shear stress and epithelial ovarian cancer peritoneal spreading. PLoS ONE 8, e60965 (2013).
Weber, G. F. et al. Pleural innate response activator B cells protect against pneumonia via a GM-CSF-IgM axis. J. Exp. Med. 211, 1243–1256 (2014).
Wang, J. & Kubes, P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell 165, 668–678 (2016).
Yung, S. & Chan, T. M. Intrinsic cells: mesothelial cells—central players in regulating inflammation and resolution. Perit. Dial. Int. 29(Suppl 2), S21–S27 (2009).
Rangel-Moreno, J. et al. Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity 30, 731–743 (2009).
Van Vugt, E., Van Rijthoven, E. A., Kamperdijk, E. W. & Beelen, R. H. Omental milky spots in the local immune response in the peritoneal cavity of rats. Anat. Rec. 244, 235–245 (1996).
Ansel, K. M., Harris, R. B. S. & Cyster, J. G. CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity 16, 67–76 (2002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koopmans, T., Rinkevich, Y. Mesothelial to mesenchyme transition as a major developmental and pathological player in trunk organs and their cavities. Commun Biol 1, 170 (2018). https://doi.org/10.1038/s42003-018-0180-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-018-0180-x
This article is cited by
-
Single-cell analysis of the cellular landscape of vulvar melanoma provides new insight for immunotherapy administration
BMC Cancer (2024)
-
Tumor-mesothelium HOXA11-PDGF BB/TGF β1-miR-181a-5p-Egr1 feedforward amplifier circuity propels mesothelial fibrosis and peritoneal metastasis of gastric cancer
Oncogene (2024)
-
Effect of cellular senescence on the response of human peritoneal mesothelial cells to TGF-β
Scientific Reports (2024)
-
The small and large intestine contain related mesenchymal subsets that derive from embryonic Gli1+ precursors
Nature Communications (2023)
-
Human peritoneal tight junction, transporter and channel expression in health and kidney failure, and associated solute transport
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
