
Abstract
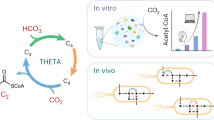
The capture of CO2 by carboxylases is key to sustainable biocatalysis and a carbon-neutral bio-economy, yet currently limited to few naturally existing enzymes. Here, we developed glycolyl-CoA carboxylase (GCC), a new-to-nature enzyme, by combining rational design, high-throughput microfluidics and microplate screens. During this process, GCC’s catalytic efficiency improved by three orders of magnitude to match the properties of natural CO2-fixing enzymes. We verified our active-site redesign with an atomic-resolution, 1.96-Å cryo-electron microscopy structure and engineered two more enzymes that, together with GCC, form a carboxylation module for the conversion of glycolate (C2) to glycerate (C3). We demonstrate how this module can be interfaced with natural photorespiration, ethylene glycol conversion and synthetic CO2 fixation. Based on stoichiometrical calculations, GCC is predicted to increase the carbon efficiency of all of these processes by up to 150% while reducing their theoretical energy demand, showcasing how expanding the solution space of natural metabolism provides new opportunities for biotechnology and agriculture.

Similar content being viewed by others

Main
The conversion of the C2 compounds glycolate and glyoxylate into C3 metabolites plays a central role in many carbon metabolic processes, such as photorespiration and fatty acid assimilation, as well as formato- and methylotrophy. Moreover, glyoxylate is the product of several synthetic CO2 fixation pathways1,2, where it serves as a hub metabolite that connects to central carbon metabolism.
However, there are only very few natural metabolic routes that allow the direct conversion of these C2 intermediates into C3 metabolites, and all of them result in the loss of carbon. The glyoxylate cycle3 and the recently described β-hydroxyaspartate cycle4 arrive at C4 compounds, which need to be decarboxylated to generate C3 metabolites. Similarly, photorespiration and the glycerate pathway convert two glyoxylate molecules into glycerate through the release of CO25,6. The inevitable loss of CO2 in all of these pathways strongly limits their carbon efficiency, which is especially apparent for photorespiration. It was estimated that in hot and dry climates, agricultural crop yield is diminished by up to 50% due to photorespiratory carbon losses7. Thus, circumventing energy and carbon loss during glycolate assimilation through synthetic pathways is expected to enhance productivity substantially.
Recently, the tartronyl-CoA (TaCo) pathway was proposed as a direct route for the assimilation of glycolate into central carbon metabolism8. This hypothetical pathway was designed to fix CO2 instead of releasing it, and is expected to outperform all naturally evolved glycolate assimilation routes (Fig. 1). However, the pathway remained theoretical until now, as neither tartronyl-CoA nor the individual enzymatic reactions of the TaCo pathway are known to occur in nature. This presented a challenge for its realization, with the engineering of glycolyl-CoA carboxylase (GCC)—the key enzyme of the TaCo pathway—as the biggest obstacle.

a, In the TaCo pathway (blue box), glycolate is activated by GCS to glycolyl-CoA, which is carboxylated by the key enzyme GCC to tatronyl-CoA. Tartronyl-CoA is reduced to glycerate by TCR. The CBB (green) and CETCH cycles (yellow) can be interfaced with the TaCo pathway on the level of glycolate. Ethylene glycol (EG) assimilation (red) can be interfaced via glycolyl-CoA. GA, glycolaldehyde; Pgp, 2-PG phosphatase; Pi, inorganic phosphate; PPi, inorganic pyrophosphate; TCA, tricarboxylic acid. b–d, Stepwise reconstitution of the TaCo pathway in vitro, starting from glycolate and 13C-bicarbonate. Shown are extracted-ion count chromatograms for glycolyl-CoA ([M + H]+ at 826.1 m/z) (b), 13C-labelled tartronyl-CoA ([M + H]+ at 871.1 m/z) (c) and 13C-labelled, 3-NPH-derivatized glycerate ([M − H]− at 241.1 m/z) (d) at t0 (no enzyme), after the addition of GCS, after the addition of GCC and after the addition of TCR. e–g, Performance of the TaCo pathway for different applications. The data are shown for two independent experiments each. e, Demonstration of the TaCo pathway as synthetic photorespiratory bypass using malate as a read-out (Fig. 4). The malate yield was increased in the presence of the TaCo pathway (inset). f, Ethylene glycol assimilation via the ethylene glycol module and the TaCo pathway. Shown is glycerate formation from ethylene glycol in an optimized experimental set-up. g, The TaCo pathway as an additional carbon-fixing module for the CETCH cycle. Shown is glycerate formation from propionyl-CoA and CO2 as starting substrates under optimized experimental conditions.
Here, we show the successful reconstitution and in vitro implementation of the complete TaCo pathway, applying rational design and high-throughput evolution of enzymes. Furthermore, we interface the TaCo pathway with several biotechnologically and agriculturally relevant processes (in particular, photorespiration, ethylene glycol conversion and synthetic CO2 fixation) as proof of principle.
Results
Identifying the enzymatic framework for the TaCo pathway
To establish a biosynthetic route to the non-native carboxylation substrate glycolyl-CoA, we investigated two possible options: a coenzyme A (CoA) transfer from another acyl-CoA donor by CoA transferases, as well as the direct ligation of glycolate to CoA by acyl-CoA synthetases. We screened 11 different native and engineered enzymes (Supplementary Table 1). All of the tested enzymes catalysed the activation of glycolate. The best transferase from Clostridium aminobutyricum (AbfT) showed a catalytic efficiency of 120 M−1 s−1 for the activation of glycolate with acetyl-CoA as the CoA donor (Supplementary Table 1).
The best CoA synthetase was an acetyl-CoA synthetase (ACS) homologue from Erythrobacter sp. NAP1 (EryACS1), which showed a catalytic efficiency of 20 M−1 s−1 with glycolate. It is well known that ACSs are post-translationally regulated through lysine acetylation in vivo9,10. To suppress post-translational inactivation of EryACS1 during protein production, we changed surface loop residue Leu641 into a proline, which has been reported to prevent acetylation of ACS from Salmonella enterica11. Indeed, EryACS1 Leu641Pro showed a twofold higher specific activity, but also a tenfold increased apparent Km for glycolate (Supplementary Table 1). Therefore, we decided to create a lysine acetylase knockout strain for protein expression (Escherichia coli BL21 (DE3) AI ΔpatZ). When we produced EryACS1 in this knockout strain, the catalytic efficiency of the enzyme was increased almost 30-fold to 540 M−1 s−1 (Supplementary Table 1), which is sevenfold higher than the catalytic efficiency of a previously reported engineered E. coli ACS for the activation of glycolate (82 M−1 s−1; deacetylated enzyme)8. Based on a homology model created with ACS of S. enterica (Protein Data Bank (PDB) ID: 2P2B; 62% sequence identity)12,13, we identified Val379 as a target for directed mutagenesis to open up the active site for accommodation of the slightly larger glycolate (Supplementary Fig. 1). A substitution of Val379 by alanine (Val379Ala) had previously been reported to enhance the activity of the enzyme with the slightly larger propionate in S. enterica ACS12. We tested three different Val379 substitutions (Supplementary Table 1), isolating the variant Val379Ala (glycolyl-CoA synthetase (GCS)), which showed an improved apparent Km for glycolate (13 ± 3 mM) and a catalytic efficiency of 853 M−1 s−1 (Table 1). Based on the favourable kinetic parameters and thermodynamic considerations (that is, the irreversibility of the reaction because of immediate hydrolysis of inorganic pyrophosphate in vivo), as well as the independence from other acyl-CoA pools in vivo compared with transferases, we decided to further rely on the engineered GCS for glycolyl-CoA synthesis (Table 1 and Supplementary Fig. 1).
In the TaCo pathway, the hypothetical product of glycolyl-CoA carboxylation, tartronyl-CoA, is reduced to glycerate via tartronic semialdehyde. We tested different reductases and found that the bifunctional malonyl-CoA reductase from Chloroflexus aurantiacus14 (CaMCR; tartronyl-CoA reductase (TCR)) was able to convert tartronyl-CoA directly to glycerate in two steps with a kcat of 1.4 s−1 and a very favourable apparent Km of 26 µM for tartronyl-CoA (Table 1 and Supplementary Table 1).
Identification and engineering of GCC
With both GCS and TCR in hand, we focused our efforts on identifying a suitable enzyme candidate for the key reaction of the TaCo pathway, the carboxylation of glycoyl-CoA to tartronyl-CoA. We screened for promiscuity in four different biotin-dependent propionyl-CoA carboxylases (PCCs), because of the structural similarity between glycolyl-CoA and propionyl-CoA (Supplementary Table 1). Of the tested enzymes in this study, only PCC from Methylorubrum extorquens (MePCC) had minuscule activity with glycolyl-CoA. Co-expression of the cognate biotin ligase gene of M. extorquens resulted in very low but measurable activity (kcat = 0.01 s−1; Fig. 2 and Supplementary Table 1) accompanied by a high ratio of futile ATP hydrolysis compared with tartronyl-CoA formation (~100:1).

a, Glycolyl-CoA carboxylation activities (kcat) of GCC variants. The data represent means ± s.d., as determined from n = 18 independent measurements using nonlinear regression. b, Catalytic efficiencies of GCC variants. The data represent means calculated from the results of nonlinear regression analysis. c, Ratio of ATP hydrolysed per carboxylation reaction for the MePCC wild type (WT) and engineered GCC variants. The data represent the means of n = 2, n = 3 or n = 4 independent measurements. Individual data points are shown as dots. d, 1.96-Å-resolution cryo-EM structure of the β core of GCC M5. e, Variants of GCC compared with the MePCC wild type. M2 and M3 were obtained by rational design, whereas M4 and M5 were obtained from subsequent rounds of random mutagenesis. The structures of the active sites of the MePCC wild type (3.48 Å; PDB ID 6YBP) and engineered GCC M5 (1.96 Å, PDB ID 6YBQ) were obtained from cryo-EM. The GCC M3 structure was modelled on the structure of GCC M5.
Next, we sought to alter the substrate preference of MePCC. Such engineering of biotin-dependent acyl-CoA carboxylases had only been attempted in a few studies15,16,17. For structure-guided rational design, we built a structural model of MePCC, for which we obtained a 3.48-Å cryo-electron microscopy (cryo-EM) dataset (Fig. 2, Supplementary Fig. 2 and Supplementary Table 2). We then targeted several residues in the first and second shell of the active site of the carboxyltransferase subunit, which form the substrate binding pocket. The selected residues were assumed to be involved in direct or indirect binding of the natural substrate propionyl-CoA, which differs in the Cα position from glycolyl-CoA by a hydroxyl group (Fig. 2, Supplementary Fig. 3 and Supplementary Table 1). To accommodate this hydroxyl group of glycolyl-CoA in the active site, we created variant GCC M2, in which we introduced a Tyr143His substitution to enable direct hydrogen bonding between enzyme and substrate, and further directed the hydroxyl group towards His143 via a Asp407Ile substitution, which had been reported to enhance substrate promiscuity in a PCC from Streptomyces coelicolor15. We introduced a third substitution in the active site’s second shell (Leu100Ser), to facilitate the formation of a hydrogen-bonding network and further strengthen the interaction of His143 with glycolyl-CoA. Compared with the wild type, the corresponding triple mutant (GCC M3) exhibited a more than 50-fold increased catalytic efficiency and a more than 15-fold decreased futile ATP hydrolysis (Fig. 2a–c and Supplementary Table 3).
Next, we applied directed evolution using high-throughput approaches to screen for mutants with further decreased futile ATP hydrolysis. We diversified the carboxyltransferase subunit of GCC M3 using error-prone PCR. To assess whether the libraries contained enzymes with decreased futile ATP hydrolysis, we established a high-throughput microfluidics screen (Supplementary Fig. 4). In this screen, the formation of tartronyl-CoA was monitored in a TCR-coupled assay under ATP-limiting conditions within picolitre droplets. We used previously described microfluidic workflows18 to develop short- and long-term multiplexed endpoint assays with single E. coli cells expressing individual GCC variants (Supplementary Figs. 5–7). Analysis of the pooled libraries showed that 4.7% of the variants exhibited more favourable ATP hydrolysis ratios compared with GCC M3 (Supplementary Fig. 8). This fraction of positive hits made it feasible to switch to microplate screens, which had the additional benefit that time-dependent kinetics of single variants could be directly obtained. We assessed each library separately to identify and characterize potentially improved enzyme variants. The best variant obtained from two rounds of subsequent random mutagenesis (GCC M5) possessed two additional substitutions (Ile450Val and Trp502Arg; Fig. 2e), a 560-fold increased kcat of 5.6 s−1 and a more than 25-fold lower ATP hydrolysis compared with the MePCC wild type (Table 1 and Fig. 2a–c). Overall, GCC M5 had a catalytic efficiency of 3.6 × 104 M−1 s−1 for glycolyl-CoA carboxylation, which is three orders of magnitude higher compared with the wild type (Supplementary Table 3) and similar to the catalytic efficiencies of naturally occurring biotin-dependent acyl-CoA carboxylases (Supplementary Fig. 9).
To exclude incomplete biotinylation of our GCC variants during protein production, we performed avidin-gel shift assays, which confirmed full biotinylation of all characterized enzymes (Supplementary Fig. 10). Furthermore, we assessed the thermostability of all enzymes by circular dichroism spectroscopy. All GCC variants exhibited melting temperatures similar to the wild type, indicating that enzyme stability was not affected through our mutations (Supplementary Fig. 11). Finally, we determined the catalytic efficiency of the enzymes with the natural substrate, propionyl-CoA. The catalytic efficiency varied among the different variants (Supplementary Table 3) and was decreased about twofold in GCC M5 compared with the wild type. Taken together, all of these results show that we successfully created a GCC that behaves like a naturally evolved enzyme.
Atomic-resolution cryo-EM structure of GCC
To investigate the structural changes in GCC M5, we obtained a detailed 1.96-Å cryo-EM structure of the enzyme that validated our (rational) design strategy at atomic resolution (Fig. 2d,e, Supplementary Figs. 3 and 12 and Supplementary Table 2). For the cryo-EM structures of both MePCC and GCC M5, the central core comprised six β subunits, was well resolved and could be modelled without any gaps. In contrast, only the biotin carboxyl carrier protein domain and an anchoring domain could be modelled for the α subunits. Both of these domains are located right on top of their respective neighbouring β subunits and comprise about 200 amino acids of the C-termini. The actual catalytic domain of the α subunits, the biotin carboxylase domain, could not be modelled due to a much lower resolution and/or mostly disconnected weak electron densities (Supplementary Fig. 12e). The strong discrepancy in resolution and electron density between the β subunit core and the α subunits may hint at extreme flexibility within α subunits. It is therefore possible that, during catalysis, it is not just the biotin carboxyl carrier protein domain that undergoes conformational changes to bridge the distance between the active sites of the biotin carboxylase and the carboxyltransferase, as proposed by the swinging domain model19,20. Instead, also, the biotin carboxylase domains themselves may move or bend towards the β subunit core. In both structures, we also observed the biotin cofactor—interestingly, in a position analogous to what was reported for the crystal structure of a chimeric PCC holoenzyme19. The biotin is notably not located in a catalytically relevant position, as the nitrogen atom that accepts the carboxyl group is in close coordination contact with a backbone carbonyl oxygen (Supplementary Fig. 13d). Moreover, although the biotin is inserted into the neighbouring β subunit, it is still about 10 Å away from the site of the carboxyl transfer to the CoA thioester substrate. The fact that this exact position of the biotin was observed in our cryo-EM structures, as well as in the crystal structure of a homologous enzyme from another organism, suggests that this location is a possible parking position for the cofactor. Unfortunately, we were not able to observe the supplied substrate glycolyl-CoA in the active site of the carboxyltransferase β subunits, but only free CoA (Supplementary Fig. 13b,c), probably due to spontaneous hydrolysis of the CoA thioester bond. Nevertheless, the resolution of the β subunit core of GCC M5 was high enough to model almost 900 ordered solvent water molecules with very low B factors. This allowed us to determine the actual distances between important active-site residues, which indicated that the Leu100Ser substitution did not directly hydrogen bond to His143 as initially assumed (Fig. 2e), but probably provided space for a new hydrophilic interaction between His143 and Asp171 in GCC M5. The Tyr143His substitution that was introduced to accommodate the hydroxyl group of glycolyl-CoA was actually held in a more favourable rotamer conformation by Asp171 through an almost perfect H bond of 2.8 Å (Fig. 2e and Supplementary Fig. 3). During random mutagenesis, two additional substitutions were introduced: Ile450Val and Trp502Arg. The Ile450Val substitution is located close to the active site in an α helix (Fig. 2e and Supplementary Fig. 3). Compared with the wild type, the Ile450Val substitution may slightly influence interatomic distances during catalysis. In contrast, the Trp502Arg substitution is far away from the active site, close to the rotational symmetry centre of the β subunit core, where it only affects the position of the loop region between Lys496 and Lys503. Its impact on enzyme activity cannot be simply rationalized by our structures, which represent a single non-catalytic state of the enzyme.
In vitro reconstitution of the TaCo pathway
Having established and engineered all of the enzymes, we next calculated the thermodynamic profile of the TaCo pathway using component contribution21. Overall, the max–min driving force (MDF)—representing the minimum thermodynamic driving force via the pathway reactions after optimizing metabolite concentrations within a physiological range22—was above 7 kJ mol−1 for the TaCo pathway (Methods and Supplementary Fig. 14). This high MDF predicts that all pathway reactions could work with minimal backward flux (that is, <20% of the total flux)22 and hence close to the maximum rate23. In fact, the tartronyl-CoA module seems to be almost as thermodynamically favourable as the Calvin–Benson–Bassham (CBB) cycle itself (MDF approaching 8 kJ mol−1; Supplementary Fig. 14) and more thermodynamically favourable than most central metabolism pathways, including Embden–Meyerhof–Parnas glycolysis (MDF < 2 kJ mol−1)22.
Next, we aimed to confirm the functionality of the full TaCo pathway by sequentially reconstituting its reaction sequence and isotopic labelling with 13C-bicarbonate (Fig. 1b–d). The reconstituted TaCo pathway converted glycolate into glycerate at a rate of 27 ± 1 nmol min−1 mg−1 total protein (Supplementary Fig. 15). To continuously operate and optimize the TaCo pathway, we coupled it to different ATP regeneration modules2,24,25. While the use of a polyphosphate kinase-based system was limited due to the precipitation of polyphosphate at concentrations above 20 mM, a phosphocreatine-based system proved three times more effective compared with the polyphosphate system (Supplementary Fig. 16). With these optimized conditions, we next aimed to test the TaCo pathway for three different potential applications.
The TaCo pathway as photorespiratory bypass
As proof of concept, we first interfaced the TaCo pathway with photorespiration. Natural photorespiration yields 2-phosphoglycolate (2-PG), which is recycled back into the CBB cycle through a complicated reaction sequence of 11 enzymes releasing NH3 and CO2 (Supplementary Fig. 17a). According to theoretical calculations, replacing natural photorespiration with the TaCo pathway would allow the direct conversion of 2-PG into the CBB cycle intermediate 3-phosphoglycerate (3-PGA) with only five enzymes, while fixing an additional carbon, instead of releasing it, thereby increasing the carbon efficiency from 75% to 150% (Supplementary Table 4). Furthermore, flux balance analysis showed that the combination of the CBB cycle and the TaCo pathway requires 21% less ATP and 29% less reducing equivalents for the net formation of one 3-PGA molecule from three CO2 molecules, compared with the CBB cycle coupled with natural photorespiration (Fig. 3 and Supplementary Table 4).

a, The numbers for ATP and reducing equivalents are derived from flux balance analysis and refer to the net fixation of three CO2 molecules and the formation of one 3-PGA molecule via the CBB cycle in combination with the specified photorespiratory pathway. For specific values, see Supplementary Table 4. b, Number of photorespiratory enzymes needed for the specified pathways (excluding CBB cycle enzymes). 3OHP, 3-hydroxypropionate bypass; A5P, arabinose-5-phosphate shunt; GLC, glycerate bypass; NPR, natural photorespiration; OX, glycolate oxidation pathway; TaCo*, TaCo pathway including futile ATP hydrolysis.
Notably, TaCo-based photorespiration excels not only natural photorespiration regarding carbon and energy efficiencies (that is, cofactor requirements), but also other synthetic bypasses8,26,27,28,29,30 that were recently proposed and/or realized (Fig. 3, Supplementary Fig. 17 and Supplementary Table 4).
Starting from the photorespiration product 2-PG, we tested the TaCo pathway together with 2-PG phosphatase and glycerate kinase (GlxK) using 13C-labelled bicarbonate. Our synthetic pathway produced (R)-glycerate, which was further converted into labelled phosphoglycerate at a rate of 12.3 nmol min−1 mg−1, showing that the TaCo pathway can be successfully interfaced with photorespiration (Supplementary Fig. 18).
We further aimed at testing the TaCo pathway under conditions mimicking 100% RuBisCO oxygenation by the addition of equimolar amounts of 3-PGA and 2-PG. For quantification, we developed a malate read-out module based on isotopic labelling. The read-out module converts 3-PGA into malate, while 2-PG is only converted to malate when the TaCo pathway is active (Fig. 4a). Isotopic labelling with 13C-bicarbonate allowed us to distinguish the fraction of malate derived from 3-PGA from that derived through the TaCo pathway from 2-PG. 3-PGA is converted into malate via one carboxylation step (that is, the carboxylation of phosphoenolpyruvate (PEP) to oxaloacetate by PEP carboxylase; Fig. 4). This carboxylation introduces one 13C label, which leads to a single-labelled malate. The conversion of 2-PG into malate only takes place in the presence of the TaCo pathway. This requires two carboxylation reactions (and therefore double incorporation of H13CO3−): first, the carboxylation of glycolyl-CoA to tartronyl-CoA by GCC; and second, the carboxylation of PEP to oxaloacetate by PEP carboxylase (Fig. 4). Thus, the malate formed via the TaCo pathway is double labelled while malate formed from 3-PGA is only single labelled. Malate formation increased in the presence of the TaCo pathway by 130 µM, corresponding to a surplus of 33%. 13C labelling confirmed that the malate surplus was provided by the TaCo pathway (Figs. 1e and 4). In a control experiment, in which the TaCo pathway was disconnected from the read-out module by omission of GlxK, no additional malate was formed, but 139 µM glycerate accumulated (Fig. 4d).

a, Schematic of the malate read-out developed in this study. Enzymes in blue represent the TaCo pathway. Enzymes in green represent naturally occurring enzymes. Red asterisks indicate 13C incorporation into intermediates and products from labelled bicarbonate. 2-PGA, 2-phosphoglycerate; Eno, enolase; Gpm, phosphoglycerate mutase; MDH, malate dehydrogenase; PEPC, PEP carboxylase; RuBP, ribulose-1,5-bisphosphate. b, Isotopic fractions of malate produced by the TaCo pathway and the read-out module. M + 0, non-labelled malate; M + 1, malate containing one 13C atom; M + 2, malate containing two 13C atoms. The assays for the negative control (no TaCo) contained GlxK but no GCS, GCC or TCR. The assays containing the TaCo enzymes (with TaCo) contained GlxK, GCS, GCC M4 and TCR. The assays for the no GlxK control contained GCS, GCC M4 and TCR but no GlxK. All of the assays were started by the addition of 5 mM ATP, 1 mM 2-PG and 1 mM 3-PGA. EIC, extracted-ion chromatogram. c, Malate concentrations (all isotopic fractions added up). d, Glycerate concentrations (all isotopic fractions added up). In b–d, individual data points are shown for two independent experiments.
Notably, GlxK from E. coli only accepts the (R)-stereoisomer of glycerate31,32, thus confirming that the physiologically relevant stereoisomer (R)-glycerate is the product of the TaCo pathway. Overall, these results showcase the potential of the TaCo pathway as synthetic, carbon-positive photorespiratory bypass that is still able to drive CO2 fixation, notably even in a 100% RuBisCO oxygenation reaction (that is, maximum photorespiration in the CBB cycle).
Ethylene glycol conversion via the TaCo pathway
Next, we tested the TaCo pathway in the context of ethylene glycol conversion. Ethylene glycol, a constituent of polyethylene terephthalate and a defrosting agent, is an environmental pollutant that is degraded by aerobic microbes via the glycerate pathway under CO2 release33,34. We conceived of a TaCo pathway-based route for the conversion of ethylene glycol into the central metabolite glycerate that would increase the carbon efficiency of ethylene glycol assimilation from 75% to 150% (Fig. 1a). We combined GCC and TCR of the TaCo pathway with (L)-lactaldehyde dehydrogenase of E. coli (FucO35, which oxidizes ethylene glycol into glycolaldehyde) and an aldehyde dehydrogenase of Rhodopseudomonas palustris BisB18 (PduP8,36, to convert glycolaldehyde into glycolyl-CoA). A first version of the synthetic pathway produced 77 µM glycerate over 2 h with an initial rate of 0.4 nmol min−1 mg−1 in vitro (Supplementary Fig. 19). To improve the initial ethylene glycol oxidation, we replaced FucO with Gox0313 from Gluconobacter oxydans37 (Supplementary Table 1) and introduced a water-forming NADH oxidase38 to maintain a high NAD+ concentration. Together with the integration of an efficient ATP regeneration system, glycerate production was increased to 485 µM at a fivefold higher rate of 2.1 nmol min−1 mg−1, demonstrating efficient CO2-dependent conversion of the environmental pollutant and plastic component ethylene glycol into a central carbon metabolite through the TaCo pathway (Fig. 1f and Supplementary Fig. 19).
Synthetic CO2 fixation by the TaCo pathway
Finally, we tested the TaCo pathway in the context of the crotonyl-CoA/ethylmalonyl-CoA/hydroxybutyryl-CoA (CETCH) cycle, a recently developed synthetic CO2 fixation pathway2. The primary CO2 fixation product of the CETCH cycle is glyoxylate, which can be converted into glycolate using glyoxylate reductase (Fig. 1a). Extending the CETCH cycle with the TaCo pathway would add another CO2 fixation step and allow direct formation of the central carbon metabolite glycerate from three CO2 molecules. Thus, the use of GCC directly increases carbon efficiency of the CETCH cycle by 25% (Supplementary Table 5). For proof of principle, we coupled the CETCH cycle and TaCo pathway and optimized their interplay in several rounds. When we initially combined the 17 enzymes of CETCH (version 5.4)2 with a semialdehyde reductase for glyoxylate reduction (Gox1801 from G. oxydans) and the TaCo pathway, the TaCo pathway produced glycerate at rates of 0.1 and 0.4 nmol min−1 mg−1 in continuous and stepwise assays, respectively (Supplementary Fig. 20). Further analysis identified two potential bottlenecks of the overall system. First, the use of formate and formate dehydrogenase to regenerate NADPH led to production of the dead-end metabolite formyl-CoA due to a side reaction of GCS, trapping CoA and consuming ATP. Second, succinyl-CoA reductase of Clostridium kluyveri (CkSucD), which was used in the CETCH cycle, had a side reactivity with glycolyl-CoA of 311 ± 21 nmol min−1 mg−1. To optimize the system, we switched to glucose dehydrogenase for NADPH regeneration and replaced CkSucD with a homologue from Clostridioides difficile (CdSucD) that specifically converted glycolyl-CoA only at 53 ± 21 nmol min−1 mg−1. Furthermore, we used higher concentrations of phosphocreatine for enhanced ATP regeneration. Our final set-up produced 331 µM glycerate at an almost 50-fold higher rate of 4.8 nmol min−1 mg−1 TaCo enzymes (Fig. 1g and Supplementary Fig. 20), which is comparable to CO2 fixation rates of the CETCH cycle alone (5 nmol min−1 mg−1)2. These results demonstrate successful combination of the TaCo pathway with the synthetic CETCH cycle into a more carbon- and energy-efficient autotrophic pathway.
Conclusions
We have constructed a new-to-nature metabolic reaction sequence for the CO2-dependent assimilation of glycolate and demonstrated its use for different applications. To develop the TaCo pathway, it was necessary to identify and engineer enzymes able to catalyse reactions, which to the best of our knowledge are not known from any natural pathways. A crucial step was the engineering of a new-to-nature CO2-fixing enzyme, GCC, which shows great potential to improve photosynthetic yield in natural and synthetic carbon fixation, even under very low CO2 conditions and with 100% oxygenation of RuBisCO8.
Our engineered enzymes of the TaCo pathway show turnover numbers (1.4–11.1 s−1) that are comparable to naturally evolved enzymes, which show on average a kcat of 10 s−1 (ref. 39), while most RuBisCOs notably fall in the range of 1–10 s−1 (ref. 40). Nevertheless, to unlock the full potential of the TaCo pathway, future efforts might involve directed evolution to further improve enzyme activities, enhance flux through the pathway and potentially eliminate the unfruitful ATP hydrolysis of GCC.
Overall, our results showcase how hitherto unknown but theoretically feasible enzyme reactions can be developed on the scaffold of naturally existing proteins to extend the solution space of natural metabolism41,42,43,44,45. We expect similar future approaches to allow access to novel routes with carbon and energy efficiencies superior to naturally evolved pathways46, which may greatly impact current efforts in biotechnology and metabolic engineering.
Methods
Materials
Chemicals were obtained from Sigma–Aldrich, Carl Roth, Santa Cruz Biotechnology and Merck. NaH13CO3 was obtained from Cambridge Isotope Laboratories. Biochemicals and materials for cloning and protein expression were obtained from Thermo Fisher Scientific, New England Biolabs and Macherey–Nagel. Coenzyme A was bought from Roche Diagnostics. Materials and equipment for protein purification were obtained from GE Healthcare, Bio-Rad and Merck Millipore. Pyruvate kinase/lactic dehydrogenase, malic dehydrogenase, glucose-6-phosphate dehydrogenase, glucose dehydrogenase and PEP carboxylase were bought from Sigma–Aldrich.
Library generation of GCC variants
Plasmid libraries of randomly mutagenized GCC were created by megaprimer-based whole-plasmid PCR (MEGAWHOP)47. To generate randomized fragments of the β subunit of GCC M3 (pTE1412) for the first round of mutagenesis, error-prone PCR48 was performed using 2.5 U Taq-polymerase with Mg-free buffer (New England Biolabs; M0320S), 7 mM MgCl2, 0.4 mM dGTP and dATP each, 2 mM dCTP and dTTP each, 0.4 µM primer PccB_fw_P1, 0.4 µM primer PccB_rev_P2 (Supplementary Table 6), 10% (vol/vol) dimethyl sulfoxide, 50 ng template DNA of pTE1412 and 200–500 µM MnCl2 in a 50 µl reaction. The randomized fragments were digested with DpnI, purified by agarose gel electrophoresis and used as mega primers for a whole-plasmid PCR (MEGAWHOP), as described elsewhere47, or subjected to another error-prone PCR reaction to further increase the mutation rate. The MEGAWHOP reaction (50 µl) contained 1× KOD Hot Start reaction buffer (Novagen), 0.2 mM dNTPs, 1.5 mM MgSO4, 500 ng megaprimer, 50 ng template plasmid (GCC M3; pTE1412) and 2.5 U KOD Hot Start DNA polymerase (Novagen). The MEGAWHOP product was purified, digested with DpnI, again purified, and transformed into ElectroMAX DH5α (Thermo Fisher Scientific) to ensure a high number of transformants in the resulting libraries.
To estimate the mutation rate for the different concentrations of MnCl2 used in the error-prone PCR, the plasmids of six randomly picked clones after MEGAWHOP were purified, sequenced and analysed for nucleotide exchanges. Library 1_1 contained 3.6 mutations per kilobase pair (kbp) (two times 500 µM MnCl2 in subsequent error-prone PCR); library 1_2 contained 1.1 mutations per kbp (500 µM MnCl2); and library 1_3 contained 0.2 mutations per kbp (200 µM MnCl2).
For the second round of random mutagenesis, plasmid pTE3100 (GCC M4) was used as a template for error-prone PCR as well as for MEGAWHOP, following the procedure described above. Here, we aimed to create libraries with mutation rates of 0.2 and 1.1 per kbp and used 200 and 500 µM MnCl2, respectively.
Screen of GCC libraries in microtitre plates
The libraries 1_1, 1_2 and 1_3 were transformed into E. coli BL21_BirA (for the bacterial strains used, see the Supplementary Methods) and colonies were picked into 96-deep-well plates (PlateOne) with lysogeny broth (Miller) containing 100 µg ml−1 ampicillin and 50 µg ml−1 spectinomycin. The plates were incubated over night at 37 °C with subsequent transfer into fresh 96-deep-well plates with lysogeny broth (Miller), 100 µg ml−1 ampicillin, 50 µg ml−1 spectinomycin and 2 µg ml−1 biotin to an OD600 of 0.1. Protein expression was induced with 0.25 mM isopropyl β-d-1-thiogalactopyranoside at an OD600 of 0.4–0.6 and the cells were incubated over night at 25 °C. The cells were lysed using CelLytic B (Sigma–Aldrich) and stored in 20% glycerol at −80 °C. The enzyme activity was measured in a plate reader by the coupled enzyme assay with purified CaMCR (TCR). We used small-volume 384-well plates (Greiner Bio-One) with 2 µl cell extract, 100 mM 3-(N-morpholino)propanesulfonic acid (MOPS)/KOH (pH 7.8), 1 mM ATP, 50 mM KHCO3, 500 µg ml−1 CaMCR, 1 mM NADPH, 10 mM MgCl2 and 1 mM glycolyl-CoA in a reaction volume of 10 µl. The absorbance of NADPH was measured at 340 nm and 37 °C for 10 h with intervals of 47 s in a plate reader (Tecan Infinite M Plex).
Sequential reconstruction of the TaCo pathway
For sequential reconstruction of the TaCo pathway, the enzymes were added one after the other to enable the detection of the intermediates of the reaction. The assay was run at 37 °C and contained 100 mM MOPS/KOH (pH 7.8), 5 mM ATP, 10 mM MgCl2, 2 mM coenzyme A, 4 mM NADPH, 50 mM NaH13CO3, 2 mM glycolate, 200 mM phosphocreatine, 10 U ml−1 creatine phosphokinase (CPK) and 50 µg ml−1 myokinase. Before starting the reaction, a t0 sample was taken and immediately quenched with 1% HCl. All subsequent samples were treated in the same way. To start the reaction, 0.5 mg ml−1 GCS was added. After 20 min, the next sample was taken, followed by the addition of 0.6 mg ml−1 GCC M4. After 20 min, the next sample was taken, followed by the addition of 0.7 mg ml−1 TCR. After 20 min, the final sample was taken. Samples were analysed for glycolyl-CoA and tartronyl-CoA by ultraperformance liquid chromatography–high-resolution mass spectrometry (UPLC–hrMS) and for glycerate using the derivatization method via UPLC with tandem mass spectrometry (UPLC-MS/MS) (for detailed UPLC-MS/MS methods, see the Supplementary Methods).
In vitro reconstruction from 2-PG/3-PGA (malate read-out)
The assays of the in vitro reconstruction of the TaCo pathway starting from 2-PG and 3-PGA were run at 37 °C and initially contained 100 mM MOPS/KOH (pH 7.8), 10 mM MgCl2, 2 mM NADPH, 2 mM NADH, 50 mM NaH13CO3, 2 mM coenzyme A, 200 mM phosphocreatine, 20 mM polyphosphate, 10 mM glucose-6-phosphate, 0.26 mg ml−1 enolase, 0.29 mg ml−1 Gpm, 0.03 mg ml−1 2-PG phosphatase, 4.8 U ml−1 creatine kinase, 4.8 U ml−1 malate dehydrogenase, 4.8 U ml−1 glucose-6-phosphate dehydrogenase and 0.19 mg ml−1 polyphosphate kinase II-2 (PPKII-2). The assay for the negative control additionally contained 0.58 mg ml−1 GlxK. The assay containing the TaCo enzymes additionally contained 0.58 mg ml−1 GlxK, 0.46 mg ml−1 GCS, 0.69 mg ml−1 GCC M4 and 1.07 mg ml−1 TCR. The assays for the no GlxK control additionally contained 0.46 mg ml−1 GCS, 0.69 mg ml−1 GCC M4 and 1.07 mg ml−1 TCR. All of the assays were started with the addition of 5 mM ATP, 1 mM 2-PG and 1 mM 3-PGA. Aliquot samples were withdrawn at different time points and immediately quenched with 1% HCl. The samples were centrifuged at 17,000g for 20 min at 4 °C, derivatized with 3-NPH and analysed via UPLC-MS/MS for malate and glycerate (for detailed UPLC-MS/MS methods, see the Supplementary Methods).
Ethylene glycol conversion by the TaCo pathway
The conversion of ethylene glycol by the TaCo pathway was determined by measuring glycerate formation. All of the assays were run at 37 °C for 2 h. Samples were withdrawn at different time points and immediately quenched with 1% HCl. The samples were centrifuged at 17,000g for 20 min at 4 °C, derivatized with 3-NPH and analysed via UPLC-MS/MS for glycerate (for detailed UPLC-MS/MS methods, see the Supplementary Methods).
Ethylene glycol experiment 1
The assays contained 100 mM MOPS/KOH (pH 7.8), 2 mM NAD+, 2 mM coenzyme A, 50 mM KHCO3, 10 mM ATP, 13.3 mM MgCl2, 5 mM NADPH, 100 mM ethylene glycol, 1.7 mg ml−1 FucO, 0.4 mg ml−1 PduP, 0.4 mg ml−1 GCC M4 and 0.7 mg ml−1 TCR.
Ethylene glycol experiment 2
The assays contained 100 mM MOPS/KOH, 2 mM NAD+, 2 mM coenzyme A, 50 mM KHCO3, 10 mM ATP, 20 mM MgCl2, 4 mM NADPH, 100 mM ethylene glycol, 10 mM phosphocreatine, 0.5 mg ml−1 Gox0313, 0.3 mg ml−1 PduP, 0.7 mg ml−1 GCC M4, 1.5 mg ml−1 TCR, 3.3 U ml−1 CPK and 0.04 mg ml−1 Nox.
Ethylene glycol experiment 3
The assays contained 100 mM MOPS/KOH (pH 7.8), 2 mM NAD+, 2 mM coenzyme A, 50 mM KHCO3, 5 mM ATP, 20 mM MgCl2, 4 mM NADPH, 119 mM ethylene glycol, 200 mM phosphocreatine, 0.5 mg ml−1 Gox0313, 0.3 mg ml−1 PduP, 0.7 mg ml−1 GCC M4, 1.5 mg ml−1 TCR, 6.6 U ml−1 CPK and 0.08 mg ml−1 Nox.
TaCo coupled to CETCH
For the coupling of the TaCo pathway to the CETCH cycle, we performed three experiments in the course of optimization.
CT experiment 1
The assay for CT experiment 1 was run as described earlier for CETCH (version 5.4)2 with the following changes. The assay mix did not contain Mcl or GlcB and additionally contained 63 µg ml−1 PPK2-II, 100 mU Gox1801, 0.4 mg ml−1 GCS, 0.6 mg ml−1 GCC M3 and 0.2 mg ml−1 TCR. Aliquot samples were withdrawn at different time points, quenched in 4% formic acid and centrifuged at 17,000g for 20 min at 4 °C. The glycerate concentration of the samples was measured using derivatization with 3-NPH following analysis via UPLC-MS/MS (for detailed UPLC-MS/MS methods, see the Supplementary Methods).
CT experiment 2
The assay for CT experiment 2 was run at 30 °C and 450 r.p.m. and contained 200 mM MOPS/KOH (pH 7.5), 10 mM MgCl2, 1 mM coenzyme A, 20 mM ATP, 10 mM NADPH, 40 mM formate, 40 mM polyphosphate, 100 mM NaHCO3 and 1.6 µg ml−1 carbonic anhydrase. We used double the amounts of enzymes as described previously for CETCH (version 5.4)2. Instead of GlcB and Mcl, we added 200 mU Gox1801. The reaction was started with the addition of 2 mM propionyl-CoA. After 120 min, the TaCo assay mix (final concentrations: 10 mM ATP, 0.5 mM coenzyme A, 2 mg ml−1 GCS, 2 mg ml−1 GCC M4 and 1 mg ml−1 TCR) was added in a 1:2 ratio and the reactions were transferred to 37 °C. For the negative control, buffer was added, corresponding to the concentrations in the TaCo mix. Aliquot samples were withdrawn at different time points, quenched in 4% formic acid and centrifuged at 17,000g for 20 min at 4 °C. The glycerate concentration of the samples was measured using derivatization with 3-NPH following analysis via UPLC-MS/MS (for detailed UPLC-MS/MS methods, see the Supplementary Methods).
CT experiment 3
The assay for CT experiment 3 contained 100 mM HEPES (pH 7.5), 5 mM MgCl2, 0.5 mM CoA, 2 mM ATP, 5 mM NADPH, 20 mM glucose, 20 mM polyphosphate, 50 mM NaHCO3, 250 µM propionyl-CoA, 0.8 µg ml−1 carbonic anhydrase and the CETCH enzymes in the amounts previously described2 (CETCH version 5.4). Instead of GlcB, Mcl and Fdh, we added 100 mU Gox1801 and glucose dehydrogenase (Sigma–Aldrich). The assay was run for 3 h at 30 °C and then diluted in a 3:1 ratio with the TaCo master mix (final concentration: 100 mM HEPES-HCl (pH 7.5), 5 mM ATP, 10 mM MgCl2, 2 mM coenzyme A, 2 mM NADPH, 30 mM bicarbonate, 100 mM phosphocreatine, 6.6 U ml−1 CPK, 22 µg ml−1 myokinase, 0.9 mg ml−1 GCS, 1.4 mg ml−1 GCC M4 and 1.4 mg ml−1 TCR). Aliquot samples were withdrawn at different time points, quenched in 4% formic acid and centrifuged at 17,000g for 20 min at 4 °C. The glycerate concentration of the samples was measured using derivatization following analysis via UPLC-MS/MS (for detailed UPLC-MS/MS methods, see the Supplementary Methods).
Cryo-EM sample preparation and data collection
For cryo-EM sample preparation, 4.0 µl of the purified complex (1 mg ml−1) containing 1 mM β,γ-imidoadenosine 5′-triphosphate, 2 mM MgCl2 and 4 mM glycolyl-CoA was applied to glow-discharged Quantifoil 2/1 grids, blotted for 3.5 s with force 4 in a Vitrobot Mark IV (Thermo Fisher Scientific) at 100% humidity and 4 °C and then plunge frozen in liquid ethane that had been cooled by liquid nitrogen.
For MePCC, cryo-EM data were collected on an FEI Glacios transmission electron microscope operated at 200 keV using the SerialEM software package49. A total of 2,637 video frames were recorded at a calibrated pixel size of 1.18 Å using a magnification of 36.000×. The total dose was 50.2 e− Å−2, distributed over 40 frames. Micrographs were recorded in a defocus range of −0.5 to −3.5 μm.
All subsequent imaging processing steps were performed in the cryoSPARC software package. The dose-fractionated videos were gain-normalized, aligned and dose-weighted using the patch motion algorithm. Defocus values were estimated using the CTFFIND4 implementation, and 168,488 particles were automatically chosen using a reference-free blob picker. Particle sorting and reference-free two-dimensional classification were performed to remove non-particle candidates and damaged particles. Ab initio model generation using the stochastic gradient descent algorithm was used to prevent any model bias using a completely data-driven starting model. The particles were further classified using heterogeneous refinement into three classes. The best-aligning class was subsequently subjected to three-dimensional refinement yielding 3.67 Å global resolution and a B factor of −99 Å2. After per-particle defocus and higher-order contrast transfer function (CTF) correction in the Refinement_New algorithm, the resolution improved to 3.48 Å with a B factor of −86.8 Å2.
For GCC M5, cryo-EM data were acquired with an FEI Titan Krios transmission electron microscope using SerialEM software49. Video frames were recorded at a nominal magnification of 105,000× (calibrated physical pixel size: 0.8512 Å px−1) using a K3 direct electron detector (Gatan) in correlated double sampling mode and a GIF quantum energy filter (Gatan) at a 20-eV slit width. The total electron dose of ~55 e− Å−2 was distributed over 35 frames. Micrographs were recorded in a defocus range of −0.5 to −2.7 μm.
Cryo-EM micrographs were processed on the fly using the Focus software package50 if they passed the selection criteria (iciness < 1.05; drift 0.4 Å < x < 70 Å; defocus 0.5 µm < x < 5.5 µm; estimated CTF resolution < 5 Å). Micrograph frames were aligned using MotionCor2 (ref. 51) and the CTF was determined using GCTF52. Using Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/), 5,398,283 particles were picked template free on 20,057 acquired micrographs. Candidate particles were extracted with a pixel box size of 340 using RELION 3.1 (ref. 53) and cleaned using reference-free two-dimensional classification. A total of 2,863,953 particles were imported into cryoSPARC54, used for ab initio construction of initial models and subjected to multiple rounds of heterogeneous refinement to obtain the best-aligning 2,181,317 particles. Non-uniform refinement resulted in a reconstruction with an estimated resolution of 2.25 Å and a temperature factor of −83.9 Å2. Using several rounds of per-particle defocus-estimated and higher-order CTF refinements, the final refinement yielded a global resolution of 1.96 Å and an improved temperature factor of 71.7 Å2 (gold-standard FSC analysis of two independent half-sets at the 0.143 cut-off). Local resolution estimation revealed a local resolution of 1.9 Å in the β subunit core of the molecule and a flexible amino-terminal α subunit. Visual inspection of the resulting electron density map revealed protein features that were consistent with the determined high resolution (for example, holes in the aromatic ring systems and the zigzag structure of extended sidechains such as Arg and Lys). Additionally, in the density maps, coordinated water molecules were visible (Supplementary Fig. 12).
Structural modelling and analysis
Homology modelling was performed using SWISS-MODEL13. As a template for homology modelling of EryACS1, the structure of an ACS mutant of S. enterica12 (PDB ID 2P2B; 62% amino acid identity) was used, which had an adenosine-5′-monophosphate-propyl ester (propyl-AMP) bound—an analogue of the reaction intermediate propionyl-AMP. Based on the position of the propyl-AMP in the S. enterica ACS, a glycolyl-AMP was modelled into the active site of the EryACS1 homology model.
For initial homology modelling of the β subunit of MePCC, the structure of the β subunit of PCC from Ruegeria pomeroyi (PDB ID 3N6R; 72% amino acid identity)19 was used.
Initial cryo-EM map fitting was performed in UCSF Chimera version 1.14 (ref. 55) using homology models based on PDB ID 3N6R19. Automatic refinement of the structure was done using phenix.real_space_refine of the PHENIX 1.17.1-3660 suite56. Manual refinements and water picking were performed with COOT 0.8.9.2 (ref. 57).
All structural depictions were created using PyMOL (the PyMOL Molecular Graphics System; version 1.8; Schrödinger). Modelling of propionyl-CoA and glycolyl-CoA into the active sites of the MePCC and GCC, respectively, was based on the position of methylmalonyl-CoA in the structure of a methylmalonyl-CoA carboxytransferase from Propionibacterium freudenreichii58 (PDB ID 1ON3; 52% amino acid identity). Manual fitting and adjustments of the CoA thioesters reflecting differences in active-site architectures were done with COOT and PyMOL.
Flux balance analysis
To compare the natural and synthetic photorespiration pathways in terms of consumption of ATP, NAD(P)H and ferredoxin, as well as required turns of RuBisCO, a stoichiometric analysis was performed by applying flux balance analysis with COBRApy59. Simplified models were constructed including the reactions of CBB, the specific reactions of each photorespiration pathway and cofactor regeneration reactions (for example, ADP + inorganic phosphate → ATP, NAD+ → NADH and NADH + NADP+ → NAD+ + NADPH), as well as key interconversion reactions, such as adenylate kinase. The ratio between the carboxylation and oxygenation reactions catalysed by RuBisCO was set to 3:1 (25% oxygenation)60. The stoichiometric requirement of ATP, NADPH, ferredoxin and RuBisCO turns (that is, the sum of carboxylation and oxygenation reactions) was computed for the production of one 3-PGA molecule. The full code and the list of reactions of each model can be found at https://github.com/he-hai/PubSuppl within the 2020_TaCo directory.
Pathway thermodynamics analysis
MDF analysis22 was applied to evaluate the thermodynamics feasibility of the tartronyl-CoA module. The Python packages equilibrator_api, equilibrator_assets and equilibrator_pathway were used for the analysis. The change in Gibbs energy of the reactions was estimated using the component contribution method22. CO2 was considered as the substrate for the carboxylation reactions as its concentration is pH independent, unlike that of bicarbonate, thus simplifying the calculations. Metabolite concentrations were constrained to the range 1 µM to 10 mM, as descripted previously22. The pH was assumed to be 7.0, the ionic strength was assumed to be 0.25 M and –log[Mg2+] (pMg) was assumed to be 3. The scripts and details can be found at https://github.com/he-hai/PubSuppl within the 2020_TaCo directory.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The structural data that support the findings of this study have been deposited in the PDB with the accession codes 6YBP (MePCC wild type) and 6YBQ (GCC M5), and in the Electron Microscopy Data Bank with the accession codes EMD-10770 (MePCC wild type) and EMD-10771 (GCC M5). Other publicly available PDB structures used in this study are 2P2B, 3N6R and 1ON3. Other data supporting the findings of this study are available in the paper and Supplementary Information, or from the corresponding author upon reasonable request.
Code availability
For flux balance analysis of different photorespiratory pathways, the full code and list of reactions of each model, as well as the scripts and details of the pathway thermodynamics analysis, can be found at https://github.com/he-hai/PubSuppl within the 2020_TaCo directory.
References
Bar-Even, A., Noor, E., Lewis, N. E. & Milo, R. Design and analysis of synthetic carbon fixation pathways. Proc. Natl Acad. Sci. USA 107, 8889–8894 (2010).
Schwander, T., Schada von Borzyskowski, L., Burgener, S., Cortina, N. S. & Erb, T. J. A synthetic pathway for the fixation of carbon dioxide in vitro. Science 354, 900–904 (2016).
Kornberg, H. L. & Krebs, H. A.Synthesis of cell constituents from C2-units by a modified tricarboxylic acid cycle. Nature 179, 988–991 (1957).
Schada von Borzyskowski, L. et al. Marine Proteobacteria metabolize glycolate via the β-hydroxyaspartate cycle. Nature 575, 500–504 (2019).
Krakow, G., Barkulis, S. S. & Hayashi, J. A.Glyoxylic acid carboligase: an enzyme present in glycolate-grown Escherichia coli. J. Bacteriol. 81, 509–518 (1961).
Bauwe, H., Hagemann, M. & Fernie, A. R. Photorespiration: players, partners and origin. Trends Plant Sci. 15, 330–336 (2010).
Walker, B. J., VanLoocke, A., Bernacchi, C. J. & Ort, D. R. The costs of photorespiration to food production now and in the future. Annu. Rev. Plant Biol. 67, 107–129 (2016).
Trudeau, D. L. et al. Design and in vitro realization of carbon-conserving photorespiration. Proc. Natl Acad. Sci. USA 115, E11455–E11464 (2018).
Starai, V., Celic, I., Cole, R., Boeke, J. & Escalante-Semerena, J. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 (2002).
Starai, V. J. & Escalante-Semerena, J. C. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J. Mol. Biol. 340, 1005–1012 (2004).
Starai, V. J., Gardner, J. G. & Escalante-Semerena, J. C. Residue Leu-641 of acetyl-CoA synthetase is critical for the acetylation of residue Lys-609 by the protein acetyltransferase enzyme of Salmonella enterica. J. Biol. Chem. 280, 26200–26205 (2005).
Reger, A. S., Carney, J. M. & Gulick, A. M. Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46, 6536–6546 (2007).
Arnold, K., Bordoli, L., Kopp, J. & Schwede, T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 (2006).
Hügler, M., Menendez, C., Schägger, H. & Fuchs, G. Malonyl-coenzyme A reductase from Chloroflexus aurantiacus, a key enzyme of the 3-hydroxypropionate cycle for autotrophic CO2 fixation. J. Bacteriol. 184, 2404–2410 (2002).
Diacovich, L. et al. Crystal structure of the β-subunit of acyl-CoA carboxylase: structure-based engineering of substrate specificity. Biochemistry 43, 14027–14036 (2004).
Arabolaza, A. et al. Crystal structures and mutational analyses of acyl-CoA carboxylase β subunit of Streptomyces coelicolor. Biochemistry 49, 7367–7376 (2010).
Liu, X. et al. Characterization and directed evolution of propionyl-CoA carboxylase and its application in succinate biosynthetic pathway with two CO2 fixation reactions. Metab. Eng. 62, 42–50 (2020).
Beneyton, T. et al. Out-of-equilibrium microcompartments for the bottom-up integration of metabolic functions. Nat. Commun. 9, 2391 (2018).
Huang, C. S. et al. Crystal structure of the α6β6 holoenzyme of propionyl-coenzyme A carboxylase. Nature 466, 1001–1005 (2010).
Tong, L. Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci. 70, 863–891 (2013).
Noor, E., Haraldsdóttir, H. S., Milo, R. & Fleming, R. M. Consistent estimation of Gibbs energy using component contributions. PLoS Comput. Biol. 9, e1003098 (2013).
Noor, E. et al. Pathway thermodynamics highlights kinetic obstacles in central metabolism. PLoS Comput. Biol. 10, e1003483 (2014).
Noor, E., Flamholz, A., Liebermeister, W., Bar-Even, A. & Milo, R. A note on the kinetics of enzyme action: a decomposition that highlights thermodynamic effects. FEBS Lett. 587, 2772–2777 (2013).
Mordhorst, S., Siegrist, J., Müller, M., Richter, M. & Andexer, J. N. Catalytic alkylation using a cyclic S‐adenosylmethionine regeneration system. Angew. Chem. Int. Ed. 56, 4037–4041 (2017).
Nocek, B. et al. Polyphosphate-dependent synthesis of ATP and ADP by the family-2 polyphosphate kinases in bacteria. Proc. Natl Acad. Sci. USA 105, 17730–17735 (2008).
Kebeish, R. et al. Chloroplastic photorespiratory bypass increases photosynthesis and biomass production in Arabidopsis thaliana. Nat. Biotechnol. 25, 593–599 (2007).
Yu, H., Li, X., Duchoud, F., Chuang, D. S. & Liao, J. C. Augmenting the Calvin–Benson–Bassham cycle by a synthetic malyl-CoA-glycerate carbon fixation pathway. Nat. Commun. 9, 2008 (2018).
Maier, A. et al. Transgenic introduction of a glycolate oxidative cycle into A. thaliana chloroplasts leads to growth improvement. Front. Plant Sci. 3, 38 (2012).
Shih, P. M., Zarzycki, J., Niyogi, K. K. & Kerfeld, C. A. Introduction of a synthetic CO2-fixing photorespiratory bypass into a cyanobacterium. J. Biol. Chem. 289, 9493–9500 (2014).
South, P. F., Cavanagh, A. P., Liu, H. W. & Ort, D. R. Synthetic glycolate metabolism pathways stimulate crop growth and productivity in the field. Science 363, eaat9077 (2019).
Hansen, R. W. & Hayashi, J. A. Glycolate metabolism in Escherichia coli. J. Bacteriol. 83, 679–687 (1962).
Doughty, C., Hayashi, J. A. & Guenther, H. Purification and properties of d-glycerate 3-kinase from Escherichia coli. J. Biol. Chem. 241, 568–572 (1966).
Boronat, A., Caballero, E. & Aguilar, J. Experimental evolution of a metabolic pathway for ethylene glycol utilization by Escherichia coli. J. Bacteriol. 153, 134–139 (1983).
Li, W. J. et al. Laboratory evolution reveals the metabolic and regulatory basis of ethylene glycol metabolism by Pseudomonas putida KT2440. Environ. Microbiol. 21, 3669–3682 (2019).
Sridhara, S., Wu, T. T., Chused, T. M. & Lin, E. C. C. Ferrous-activated nicotinamide adenine dinucleotide-linked dehydrogenase from a mutant of Escherichia coli capable of growth on 1,2-propanediol. J. Bacteriol. 98, 87–95 (1969).
Zarzycki, J., Sutter, M., Cortina, N. S., Erb, T. J. & Kerfeld, C. A. In vitro characterization and concerted function of three core enzymes of a glycyl radical enzyme-associated bacterial microcompartment. Sci. Rep. 7, 42757 (2017).
Zhang, X., Zhang, B., Lin, J. & Wei, D. Oxidation of ethylene glycol to glycolaldehyde using a highly selective alcohol dehydrogenase from Gluconobacter oxydans. J. Mol. Catal. B Enzym. 112, 69–75 (2015).
Nowak, C. et al. A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front. Microbiol. 6, 957 (2015).
Bar-Even, A. et al. The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 (2011).
Flamholz, A. I. et al. Revisiting trade-offs between Rubisco kinetic parameters. Biochemistry 58, 3365–3376 (2019).
Erb, T. J. Back to the future: why we need enzymology to build a synthetic metabolism of the future. Beilstein J. Org. Chem. 15, 551–557 (2019).
Obexer, R. et al. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 9, 50–56 (2017).
Chou, A., Clomburg, J. M., Qian, S. & Gonzalez, R. 2-Hydroxyacyl-CoA lyase catalyzes acyloin condensation for one-carbon bioconversion. Nat. Chem. Biol. 15, 900–906 (2019).
Siegel, J. B. et al. Computational protein design enables a novel one-carbon assimilation pathway. Proc. Natl Acad. Sci. USA 112, 3704–3709 (2015).
Chen, K. & Arnold, F. H. Engineering new catalytic activities in enzymes. Nat. Catal. 3, 203–213 (2020).
Erb, T. J., Jones, P. R. & Bar-Even, A. Synthetic metabolism: metabolic engineering meets enzyme design. Curr. Opin. Chem. Biol. 37, 56–62 (2017).
Miyazaki, K. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol. 498, 399–406 (2011).
Cirino, P. C., Mayer, K. M. & Umeno, D. Generating mutant libraries using error-prone PCR. Methods Mol. Biol. 231, 3–9 (2003).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Biyani, N. et al. Focus: the interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Li, X., Grigorieff, N. & Cheng, Y. GPU-enabled FREALIGN: accelerating single particle 3D reconstruction and refinement in Fourier space on graphics processors. J. Struct. Biol. 172, 407–412 (2010).
Scheres, S. H. A Bayesian view on cryo-EM structure determination. J. Mol. Biol. 415, 406–418 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A.cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Hall, P. R. et al. Transcarboxylase 12S crystal structure: hexamer assembly and substrate binding to a multienzyme core. EMBO J. 22, 2334–2347 (2003).
Ebrahim, A., Lerman, J. A., Palsson, B. O. & Hyduke, D. R. COBRApy: constraints-based reconstruction and analysis for Python. BMC Syst. Biol. 7, 74 (2013).
Sharkey, T. D. Estimating the rate of photorespiration in leaves. Physiol. Plant. 73, 147–152 (1988).
Acknowledgements
We dedicate this publication to our colleague and friend Arren Bar-Even, who was at the very heart of this project. We thank I. A. Berg and D. S. Tawfik for fruitful discussions throughout the project. J.M.S. and S.K.S. are grateful to E. Conti for scientific independence and great mentorship. This work was funded by the Max Planck Society (MaxSynBio) and FET-Open Grant 686330 (Future Agriculture). The work conducted by the US Department of Energy Joint Genome Institute (a DOE Office of Science User Facility) was supported under contract number DE-AC02-05CH11231. J.-C.B. acknowledges financial support from the Région Aquitaine and from the French Government ‘Investments for the Future’ programme, part of the University of Bordeaux Initiative of Excellence (IDEX Bordeaux; ANR-10-IDEX-03-02). J.M.S. is grateful to the DFG for an Emmy Noether grant (SCHU 3364/1-1). We thank E. Noor (ETH Zurich) for help with the thermodynamic calculations. We are thankful to the Core Facility for Protein Spectroscopy at the Institute of Cytobiology (Philipps University of Marburg), and especially to S.-A. Freibert and V. Schulz for the circular dichroism measurements of GCC variants.
Funding
Open access funding provided by Max Planck Society
Author information
Authors and Affiliations
Contributions
M.S., J.Z., S.A., A.B.-E. and T.J.E. conceived of the project. M.S., D.G.M. and J.Z. identified and tested the GCS, GCC and TCR candidates for the TaCo pathway, and Gox0313 and PduP for ethylene glycol conversion. P.P. tested the PCC homologues for GCC activity and the influence of biotin ligase on carboxylation activity. S.K.S., D.B. and J.M.S. performed the cryo-EM sample preparation and analysis. J.Z. refined the cryo-EM structures. J.L. constructed the GCS expression strain E. coli BL21 (DE3) AI ΔpatZ and performed the initial characterization of GCS. M.C. identified and tested CdSucD and Gox1801 for the CETCH cycle. J.Z., M.S. and T.J.E. designed active-site variants for GCS and GCC. M.S. and M.K. designed error-prone libraries for GCC. M.S., T.B. and J.-C.B. developed the microfluidics platform to screen the GCC libraries. M.S. and D.G.M. developed and performed microtitre plate assays to screen the GCC libraries, and isolated and characterized the GCC variants. M.S. performed all of the in vitro reconstruction experiments and optimizations. C.D. and M.S. performed the CETCH in vitro reconstruction experiments. N.C.S., C.D. and P.C. performed the mass spectrometric analyses. H.H. performed the flux balance analysis and thermodynamic calculations. The manuscript was written by M.S., J.Z. and T.J.E. with contributions from all other authors.
Corresponding author
Ethics declarations
Competing interests
T.J.E. and A.B.-E. have filed a patent covering the TaCo pathway. The other authors declare no competing interests.
Additional information
Peer review information Nature Catalysis thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Figs. 1–25 and Tables 1–8.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scheffen, M., Marchal, D.G., Beneyton, T. et al. A new-to-nature carboxylation module to improve natural and synthetic CO2 fixation. Nat Catal 4, 105–115 (2021). https://doi.org/10.1038/s41929-020-00557-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41929-020-00557-y
This article is cited by
-
Single-site iron-anchored amyloid hydrogels as catalytic platforms for alcohol detoxification
Nature Nanotechnology (2024)
-
Revealing reaction intermediates in one-carbon elongation by thiamine diphosphate/CoA-dependent enzyme family
Communications Chemistry (2024)
-
Design of microbial catalysts for two-stage processes
Nature Reviews Bioengineering (2024)
-
Structural insight into synergistic activation of human 3-methylcrotonyl-CoA carboxylase
Nature Structural & Molecular Biology (2024)
-
Metabolic and enzymatic engineering strategies for polyethylene terephthalate degradation and valorization
Reviews in Environmental Science and Bio/Technology (2024)
