
Abstract
Chemical upcycling of polyolefin plastic waste to lubricant, wax and fuel-range hydrocarbons over metal-based catalysts is a crucial technological solution to the enormous environmental threat posed by plastic waste. However, currently available methods are incompatible with chlorine-contaminated feedstocks. Here we report a two-stage strategy for upcycling chlorine-contaminated polypropylene. First, magnesia–alumina mixed oxide at 30 bar H2 and 250 °C serves as a chlorine trap by rapidly forming solid chloride, resulting in nearly complete chlorine extraction from the polyolefin melt. This enables the upcycling of plastic waste with up to 10% polyvinyl chloride content to lubricants over ruthenium-based catalysts, in the second stage. The strategy is also applicable to chlorinated aromatics and alkanes. The proposed strategy renders hydrocracking and hydrogenolysis catalysts less sensitive to the chlorine impurities in feedstocks while eliminating HCl emissions and chlorine contamination in products. It could incentivize further progress in plastics upcycling.
Similar content being viewed by others

Main
Since the 1950s, several billion tons of plastic have been generated, with ~60% discarded into landfills1. The recycling of plastic waste (PW) is relatively minuscule compared with the waste generation2, especially of single-use packaging films3,4,5. Most PW leaks into the environment, destroying ecosystems2,6. An important problem in PW management is its mixed nature, consisting of several polymers, including polyolefins (POs) (polypropylene (PP) and polyethylene (PE)), polyvinyl chloride (PVC) and polyethylene terephthalate (PET), blended with various additives such as plasticizers, dyes and antioxidants7. This makes mechanical recycling very challenging8. Pyrolysis of PO is less sensitive to the nature of the material but requires high reaction temperatures and high energy, producing low-value products (char and light gases)9. Hydrogenolysis of pure POs (PP and PE) over metal-based catalysts such as Pt/SrTiO3 (ref. 10), Ru/C (refs. 11,12) and Ru/TiO2 (ref. 13) or hydrocracking over metal–acid bifunctional catalysts, such as Pt/WO3/ZrO2 (ref. 14), makes valuable products, such as lubricant, wax and jet fuel. Unfortunately, contaminants in actual PO waste probably deactivate these catalysts. One of the harshest impurities, PVC, is present in PW at ~4% concentration (if easily recyclable polyethylene terephthalate is excluded from the calculation)15. The Cl atoms poison the Ru and Pt active sites. Decomposition of PVC during the reaction produces HCl, a highly corrosive and toxic compound. Lubricants and fuels contaminated with even several ppm of HCl or chlorinated organics are unsuitable for use.
Moving chemical recycling and upcycling real-world PW forward requires PVC-tolerant catalysts or an efficient Cl removal step. Such technology will also eliminate the necessity of labour-intensive sorting of small amounts of PVC from PW. Recent reports show several approaches to Cl removal based on solvothermal treatment16 and pyrolysis over a zeolite catalyst17.
In this Article, we propose a two-stage absorptive dechlorination/hydrogenolysis or hydrocracking (Fig. 1). Instead of extensive sorting, cleaning or extraction of impurities, the feedstock reacts with a dechlorinating agent, magnesium–aluminium mixed oxide (Mg3AlO4.5), to trap Cl as inert MgCl2. Mg3AlO4.5 provides a high density of basic sites18,19 to bind HCl formed from PVC. Importantly, we show that dechlorination protects hydrogenolysis and hydrocracking catalysts from poisoning, prevents Cl leakage into the liquid reaction products and eliminates HCl emissions. The broader scope of halogen-containing plastic additives, such as fire retardants20 and plasticizers21, can be treated similarly. Ultimately, the proposed two-step technology makes hydrogenolysis PVC tolerant, bringing the PO-to-products process closer to practice. Sustainable PO hydroconversion will rely on this pretreatment step to eliminate HCl poisoning of the catalyst and its emission or leakage into the product mixture. This will generally reduce the burden for waste feedstock purification, facilitating widespread adoption of waste catalytic hydroconversion. Recently discussed plastic product design strategies22 could create a stable pipeline of relatively clean PO feedstock, further incentivizing development of catalytic upcycling technologies (such as conversion of PP to lubricants13 or PE to aromatics23).

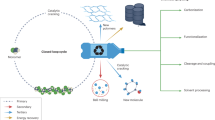
Current scheme of hydrotreatment of PVC-contaminated plastic waste and proposed absorptive dechlorination. LDPE, low-density PE; PS, polystyrene; HDPE, high-density PE.
Results
PP–PVC mixture hydrogenolysis
The Ru/TiO2 catalyst is highly active for pure PP hydrogenolysis (Fig. 2a), consistent with previous reports13. Liquid products formed over Ru/TiO2 showed lubricant properties, similar to group I base oil13. The liquid yield reaches 66%; the major side product is gas (a yield of 28%), consisting of 76% methane, due to secondary terminal C–C bond breaking (Supplementary Table 1). In this context, liquid in PP hydrogenolysis refers to the fluid fraction of the product that is easily separable from the solid residue. For PP mixed with 10 wt% of PVC, Ru/TiO2 shows almost no activity, with the residual solid yield reaching 99% (Fig. 2a). During PVC thermal decomposition, HCl and –[CH = CH]– polyene form, potentially poisoning the Ru active sites. HCl was also detected in the gas phase of the reactor headspace using Fourier transform infrared (FTIR) spectroscopy (Fig. 2b).

a, Product distribution for pure PP over Ru/TiO2 (i), PP–PVC mixture over Ru/TiO2 (ii), PP–PVC over Mg3AlO4.5 mixed with Ru/TiO2 (iii), PP–PVC over Ru/Mg3AlO4.5 (iv), PP–PVC via the two-stage strategy (v). b, FTIR spectra of the product gas for each case, with the HCl spectrum as a reference. c, 1H NMR spectra of liquid products formed with pure PP (i) and PP–PVC via the two-stage strategy (v). d, GPC chromatograms of liquid products. e,f, Two-dimensional 1H–13C heteronuclear single quantum coherence NMR of liquid products formed from pure PP (e) and PVC–PP via the two-step strategy (f). Dechlorination conditions: 250 °C, 30 bar H2, 6 h, 0.2 g PVC, 0.2 g Mg3AlO4.5, 1.8 g PP. Reaction conditions: 250 °C, 30 bar H2, 16 h, 0.1 g Ru/TiO2, dechlorinated solid.
Basic oxides trap HCl during high-temperature thermolysis of PVC. Various compounds can be efficient Cl traps for pure PVC pyrolysis and dechlorination, including Fe3O4 (ref. 24), ZnO (ref. 25), mixed magnesia–alumina oxide and others26. Following ref. 26, we added Mg3AlO4.5 with an Al/Mg ratio of 0.33 and a surface area of 205 m2 g−1 to the hydrogenolysis reactor (Supplementary Fig. 1). On the basis of stoichiometry for complete chlorination of Mg3AlO4.5 to MgCl2, it should trap ~2.1 g PVC per gram of adsorbent. We used experimental loadings of 1 g gads–1 of PVC since full chlorination does not occur even at high reaction temperatures due to the formation of a stable chloride shell and the slow Cl diffusion in the bulk27. However, the activity did not increase, while the amount of HCl decreased 5–6 times (still detectable). The same happened when Ru was directly deposited on Mg3AlO4.5 (Fig. 2a). Thus, a single-step strategy seems inadequate. In the two-step strategy (Fig. 1), the mixture was first treated with Mg3AlO4.5 and then with Ru/TiO2. For dechlorination, the PVC–PP mixture was combined with Mg3AlO4.5 in a batch reactor at 30 bar H2 pressure, 250 °C for 6 h. The resulting solid was mixed with fresh Ru/TiO2 and reacted at the same conditions for 16 h. Mg3AlO4.5 was not separated from the polymer mixture after dechlorination (see Methods for details). The liquid yield reached ~70%, close to pure PP. Interestingly, the gas yield dropped to 2.2% with a gas consisting mainly of isobutane and isopentane (60% selectivity). The molecular weight distributions of the liquids are similar, with a weight-averaged Mw equal to 900 Da for pure PP and 610 Da for PVC + PP mixture (Fig. 2d). Nuclear magnetic resonance (NMR) of liquid products highlights differences in microstructure (Fig. 2c,e,f). For pure PP, the liquid consists mainly of polypropylene oligomers with distinct signals of CH3, CH2 and CH groups in the heteronuclear single quantum coherence spectra13. A small fraction of (CH2)n fragments and other defects in the PP sequence lead to the 41–19 ppm peaks13. For PP + PVC conversion, the signals of various CH3 groups have higher relative intensity due to shorter chain size, consistent with their smaller 13C chemical shift of 17.0–9.5 ppm range. The signals of CH2 groups in (CH2)n domains are much more intense than for pure PP. This finding points to a favourable detachment of methyl groups in PP via breaking the CH3–CH bond. Standard PP hydrogenolysis preferably breaks CH2–CH bonds in the first 3–16 h, while CH3–CH bonds are broken later, in 16–24 h. The stark difference in the relative content of CH2 groups was also quantitatively assessed by 1H NMR (Fig. 2c), which shows higher intensity in the 1.6–1.0 ppm region, especially the 1.3 ppm broad peak. Despite differences in the microstructure, both liquids correspond to lubricant-range hydrocarbons.
Direct conversion of a PVC-containing feed in a single step completely inhibits hydrogenolysis, consistent with previous reports showing Ru/C deactivation with only 0.1% PVC (ref. 28). The most stable catalyst tested so far is 0.125% Ru/CeO2, which raised the solid yield from 5% to 45% with 1% PVC contamination29. Our results show that the two-stage strategy effectively retains the Ru catalytic activity and gives 70% liquid yield with 10% PVC content in the mixture by effectively entrapping HCl in Mg3AlO4.5 and physically separating the dechlorination from the hydrogenolysis. Tests with high-density PE– (HDPE–) PVC mixture and amorphous PP show that the two-step procedure is applicable to other polymer compositions (Supplementary Fig. 2 and Table 2). Interestingly, the residual Cl content in the liquid depends on the type of polymer mixed with PVC. The amorphous PP–PVC mixture had Cl contamination, while the HDPE–PVC mixture was Cl free.
Dechlorination mechanism
During dechlorination, PVC is decomposed over Mg3AlO4.5 while PP remains intact (Supplementary Fig. 3). In the second stage, PP undergoes hydrogenolysis over Ru/TiO2, with some exposure to minor products of PVC decomposition from the first stage. This may affect the product distribution over Ru/TiO2. To assess this hypothesis, the dechlorination mechanism of pure PVC was studied by heating it over Mg3AlO4.5 at different temperatures at 30 bar H2.
Figure 3a shows that the initial Mg3AlO4.5 contained mostly a MgO phase with a broad (200) reflection and an impurity of κ-alumina. The initial hydrotalcite decomposition leads to the crystallization of MgO, with Al3+ being incorporated into the lattice19. Upon heating with PVC to 200 °C, new diffraction maxima due to MgCl2 emerge. As the temperature increases, the reflection becomes more refined owing to the growth of the MgCl2 crystals. The MgO peak decreases in intensity, reaching a residual value at ~250 °C due to saturation with chlorine. Unreacted MgO is left because we employed nearly double the stoichiometric Mg3AlO4.5 amount as complete chlorination may not occur due to forming a stable chloride shell and slow Cl diffusion in the bulk27. In practice, excess Mg3AlO4.5 increases the cost and may impede mixing. The 13C magic angle spinning (MAS) NMR (Fig. 3b) reveals that as MgCl2 forms, PVC undergoes decomposition, and the two PVC peaks at 57 and 47 ppm (–CHCl– and –CH2– groups) decrease. Simultaneously, new peaks at 128 and 32 ppm appear after heating at 250 °C (ref. 30). They correspond to –C = C– polyenic chain segments and aromatic hydrocarbons formed after HCl elimination from PVC. The 32 ppm peak corresponds to alkyl fragments closely attached to these unsaturated units. These species do not originate from PP (experiments with mixed PVC–Mg3AlO4.5 were conducted).

a, XRD patterns of PVC mixed with Mg3AlO4.5 heated at different temperatures. b, 13C MAS NMR of the same samples. c,d, Quantitative XPS results on Cl/Mg (c) and Al/Mg (d) ratios as a function of treatment temperature. e, Reaction scheme. Dechlorination conditions: 30 bar H2, 6 h, 0.2 g PVC, 0.2 g Mg3AlO4.5.
The initial PVC–Mg3AlO4.5 mixture does not have Cl atoms on the surface, as shown in X-ray photoemission spectroscopy (XPS) (Fig. 3c). At low treatment temperatures, PVC is covered mainly with Mg3AlO4.5 particles, leading to an apparent lack of Cl and C (Supplementary Fig. 4 shows Cl 2p and C 1s data). Cl appears on the surface at 200 °C and reaches a stable Cl/Mg ratio of ~1 at 250 °C. According to XPS, the surface Al content is constant, leading to an Al/Mg ratio of 0.35–0.40 at low treatment temperatures. At 250 and 275 °C, Al is absent from the XPS spectra (Supplementary Fig. 4), and the Al/Mg ratio drops to zero. Simultaneously, scanning electron microscope with energy-dispersive X-ray, more sensitive to the bulk, shows a constant Al content (Supplementary Table 3). Mg3AlO4.5, upon interaction with PVC and its decomposition products, will cover the surface with MgCl2 and carbonaceous deposits, while Al is in bulk, probably as unreacted Mg3AlO4.5 phase or a separate oxychloride phase, with particles too small to be detected by X-ray diffraction (XRD) (Fig. 3e). Formation of MgCl2 compared with AlCl3 is strongly preferred thermodynamically at these conditions, which drives surface segregation. Results of thermogravimetric analysis coupled with differential scanning calorimetry (TGA-DSC) (Supplementary Fig. 5) also show that Mg3AlO4.5 traps ~30% of HCl produced by PVC decomposition at 280–300 °C and atmospheric pressure; the remaining ~70% of HCl desorbs in the gas phase. Operation under 30 bar pressure in batch mode reinforces HCl–Mg3AlO4.5 interactions and shifts the HCl absorption temperature from ~300 °C (determined by TGA) to 250 °C (Fig. 3). It also increases the Cl trapping efficiency from 30% to ~100% since no HCl was detected in the reactor headspace using FTIR (Fig. 2b).
The high H2 pressure of dechlorination enables low-temperature operation and efficient Cl removal, which are hard to achieve under atmospheric pressure attempted before31. Reports on PVC decomposition over ZnO (refs. 31,32) show that more-intimate contact between the oxide and polymer reduces the decomposition temperature from 250 °C to 200 °C, preventing direct HCl evolution in the gas phase.
In addition, we tested a set of Mg-containing oxides for Cl trapping (Supplementary Table 4 and Fig. 6). Characterization shows that MgO is the active component in Mg3AlO4.5 adsorbent. At the same time, Al2O3 does not participate in Cl trapping. This was further confirmed by experiments with MgAlO2.5 mixed oxide, which shows HCl leakage in the gas phase after dechlorination. Dispersion of MgO on silica also leads to efficient HCl capture. According to literature reports, other oxides, such as ZnO, should have comparable properties to MgO (ref. 33).
Impact of dechlorination temperature and pressure
Dechlorination with PP–PVC mixtures followed by hydrogenolysis showcased that the reaction outcome strongly depends on the dechlorination temperature (Supplementary Fig. 7). As the dechlorination temperature increased, the accompanying PP solid yield decreased. It requires >200 °C to effectively extract Cl from the PVC to the MgCl2 phase, consistent with the pure PVC–Mg3AlO4.5 results (Fig. 3). Given the high thermodynamic favourability of Mg3AlO4.5 chlorination with HCl, the initial thermal decomposition of PVC to polyenes and HCl is probably rate determining. TGA of pure PVC also shows that the peak decomposition happens at ~280 °C (Supplementary Fig. 5).
To elucidate the role of hydrogen in PVC decomposition and Cl trapping, we replaced H2 with He. The hydrogenolysis stage over Ru/TiO2 was unaltered. Figure 4a shows that the liquid yield in He is 1.4 times lower. Gel permeation chromatography (GPC) analysis (Fig. 4b) reveals that the liquid has approximately five times higher Mw when using He than when using H2. These results clearly indicate that the Ru/TiO2 catalyst is poisoned and produces less liquid with higher Mw when dechlorination occurs in He. Higher H2 pressure in dechlorination (Fig. 4c) decreases the Mw of the liquid product down to ~600 Da, typical for PVC-free hydrogenolysis13, and maximizes the liquid yield to 69% at 30 bar H2; beyond that, it slightly decreases the liquid due to excessive formation of gas (Supplementary Table 5).

a,b, Product distribution and Mw of liquid after the two stages with dechlorination conducted in H2 or He. c, Liquid yield and Mw at various H2 pressures. d, Reaction scheme for the formation of chlorinated alkanes. Dechlorination conditions: 250 °C, 30 bar He or H2, 6 h, 0.2 g PVC, 0.2 g Mg3AlO4.5, 1.8 g PP. Reaction conditions: 250 °C, 30 bar H2, 16 h, 0.1 g Ru/TiO2, dechlorinated solid.
The distribution of Cl in the solid, liquid and gas products of PP is vital for their use. Supplementary Table 6 shows that most Cl is segregated selectively on the solid residue, leading to a low Cl content in the liquid (<0.1 wt%). Overall, optimization of the dechlorination conditions (Supplementary Fig. 7 and Tables 5 and 6) shows that this step requires 30 bar H2, 250 °C and ~6 h for completion.
The promoting role of H2 on dechlorination is rationalized in Fig. 4d. PVC decomposition without hydrogen leads to HCl and unsaturated polyenes. HCl is either trapped in MgCl2 or undergoes a Markovnikov-type addition to C=C bonds of polyenes to chlorinated alkanes or chloroaromatic compounds, emblematic of PVC pyrolysis24. Chloroalkanes are poisonous for noble metals, including Ru/TiO2 (ref. 34). Hydrogenation of polyenes and other unsaturated compounds during dechlorination is key. This partial hydrogenation rationalizes why the liquid Mw is lower in H2 than in He. The Mg3AlO4.5 mixed oxides stabilize chemisorbed hydrogen atoms, leading to ethanol condensation to butanol, which involves intermediate hydrogenation of butenal19. A similar interaction between surface H atoms and strongly bonded unsaturated compounds may explain the minor hydrogenating ability of Mg3AlO4.5. This is also supported by the 13C MAS NMR spectra (Fig. 3b), where a substantial fraction of carbonaceous residues is saturated alkyls rather than the aromatic or polyenic compounds expected in direct PVC decomposition.
The negative role of chlorinated alkanes was further demonstrated with PP contaminated with 10% 1-chlorooctadecane (a model compound; Supplementary Fig. 8) without dechlorination. This decreased the yield of the liquid from 66% to 40%, the gas from 28% to ~1% and the methane selectivity from 76% in pure PP to just 4%; the main gas product was isobutane (52%). Overall, the gas product distribution (Supplementary Fig. 8) is similar to that in PP–PVC conversion.
The C–Cl bond hydrogenolysis in 1-chlorooctadecane over Ru produces HCl, similar to the chlorinated alkanes produced from PVC (Fig. 4d). Binding of HCl to the Ru surface may lead to Bronsted acid sites, catalysing cracking of hydrogenolysis intermediates, produced from PP. A similar process was detected for the HI-promoted Pd/Al2O3 catalyst35. HCl-catalysed isomerization of adsorbed hydrocarbon radicals followed by β-scission leads to isobutane, similar to hydrocracking, where isobutane is a major product36. Thus, these experiments with 1-chlorooctadecane show that chlorinated alkanes, formed from PVC, modify the Ru surface, reducing hydrogenolysis activity and gas yield. Dechlorination in H2 does not eliminate chlorinated alkanes altogether; the residual HCl binds to Ru/TiO2 and changes the gas formation mechanism.
Air-free XPS characterization of Ru/TiO2 treated with 1-chlorooctadecane and PP mixture reveals that Ru remains in the metallic state, but the surface contains chloride anion on the TiO2 support (Supplementary Fig. 9). FITR of adsorbed CO shows the formation of polycarbonyls from CO dissociation on the Ru particles. Interestingly, the peak position for the spent catalyst changes strongly compared with the fresh Ru/TiO2, probably due to changes in the Ru electronic state. A minor amount of newly formed Brønsted acid sites was detected with DRIFTS of adsorbed pyridine (Supplementary Fig. 10). These sites forming on the support or during the reaction on Ru could catalyse cracking to C4–C5 gas products, instead of methane (Supplementary Table 1).
The difference in product yields and liquid microstructure (Fig. 2) observed for pure PP and PP–PVC mixture may be attributed to the Cl-induced modification of the Ru/TiO2 catalyst, evident from experiments with 1-chlorooctadecane. Previous work also showed that the Ru catalytic activity in the hydrogenolysis of PP is sensitive to the properties of the support37. Overall, variation in dechlorination conditions changes the outcome of hydrogenolysis by introducing polyene and Cl-containing species. Optimized dechlorination leads to high liquid yields, close to pure PP hydrogenolysis13.
Previously, two-step pyrolysis was proposed for a low-density PE–PVC mixture to decompose PVC at 300–400 °C (ref. 38). In our case, dechlorination at 250 °C over Mg3AlO4.5 traps HCl. Hence, the exhaust gas is free of Cl (Fig. 2b). A separate filtration to purify the pyrolysis oil from Cl impurities in conventional PVC pre-decomposition is unnecessary. A trace amount of HCl or chlorohydrocarbons in the oil will prevent its use as fuel or lubricant.
To draw comparison with previous reports38, mixed PP–PVC was thermally decomposed in He at atmospheric pressure and then converted over Ru/TiO2 at 30 bar H2. The liquid yield decreased to 26% (Supplementary Table 5). The 13C MAS NMR analysis (Supplementary Fig. 11) of the solid product showed a substantial contribution of polyenes and aromatics after treatment at ambient pressure. Compared with the data obtained after treatment at high pressure of H2, the relative content of polyenes increased by an order of magnitude. The liquid Cl content obtained after ambient pressure dechlorination (X-ray fluorescence (XRF) data; Supplementary Table 6) was 2.5 wt%, much higher than observed for high-pressure experiments. Cl content decreased to 0.77 wt% when dechlorination was conducted at 20 bar He with Mg3AlO4.5 in batch (Fig. 4), indicating the beneficial trapping effect of Mg3AlO4.5 and its inability to capture Cl fully without the high pressure of H2. In stark contrast, dechlorination at 30–50 bar H2 led to a Cl fraction in the liquid of 0.05–0.00%. High H2 pressure facilitates HCl trapping in MgCl2 and prevents trace amounts of Cl from entering the liquid through C=C bond chlorination and hydrogenation (Fig. 4d).
Hydrogenolysis of PP mixed with different chlorinated compounds
PW contains PVC and other Cl-containing species from fire retardants, antioxidants or other additives39. For example, brominated fire retardants during plastic pyrolysis at 450 °C contaminate the pyrolysis oil with bromophenols40. We assessed the two-stage Cl removal for PP mixed with polyvinylidene chloride (PVDC), 1,2,4-trichlorobenzene (TCB) and 1,1,2,2-tetrachloroethane (TCE). For TCB and TCE, the catalyst was active, producing 40–60% liquid with a small amount of gas (Supplementary Table 7), giving a wholly dechlorinated liquid. PVDC contamination, however, led to no catalyst activity. Bulk PVDC decomposition produces HCl slowly due to forming stable aromatic structures with several fused rings containing C–Cl groups41. Its dechlorination may require longer treatment times or higher temperatures to eliminate HCl, but we did not pursue this optimization.
Thus, Mg3AlO4.5 is capable of dechlorinating PP mixtures with small, chlorinated molecules, such as TCE and TCB, while failing with PVDC. This result shows that for sustainable catalytic PP conversion to lubricants, PVDC should first be removed. PVDC–PP mixtures can be separated using solvents42, or ideally, PVDC should be eliminated from the plastics products22.
In addition, dechlorination was tested with higher loadings of PVC (20 and 30 wt%). Results show (Supplementary Table 8) that the liquid yield drops to 37% with 20% PVC loading while forming Cl-free liquid. Higher amounts of PVC lead to negligible liquid production.
Hydrocracking of PVC-contaminated feedstock with dechlorination
An alternative method for polyolefin upcycling is hydrocracking over bifunctional metal–acid catalysts such as Pt/WO3/ZrO2 (ref. 43). Figure 5 shows that pure PP gives 87% liquid after 2 h reaction at 250 °C, with a product that is centred at C7–C8 alkanes. The PP was mixed with PVC and dechlorinated over Mg3AlO4.5, similar to the hydrogenolysis procedure. Then, the dechlorinated product was mixed with Pt/WO3/ZrO2 catalyst and produced 68% liquid with 24% solid residue after 2 h. The liquid product distribution is slightly shifted to C6–C7.

a, Product distribution from PVC–PP mixture in the two-stage and pure PP hydrocracking. b, Liquid and gas product distribution by carbon number. Dechlorination conditions: 250 °C, 30 bar H2, 6 h, 0.2 g PVC, 0.2 g Mg3AlO4.5, 1.8 g PP. Reaction conditions: 250 °C, 30 bar H2, 16 h, 0.1 g Pt/WO3/ZrO2, dechlorinated solid.
The position in the maximum of the product distribution (Fig. 5b) is sensitive to the ratio of metal (Pt) and acid (WO3/ZrO2) sites of the catalyst43. The decrease in the C8–C10 yield corresponds to a reduction in the relative fraction of metal sites. Probably, Pt clusters are more vulnerable than acid sites to poisoning by the PVC decomposition products. Nonetheless, the two-stage procedure retains ~80% of liquid alkanes produced over pure feedstock.
Adsorbent and catalyst regeneration
A sustainable two-stage strategy requires the Ru/TiO2 and Cl trap to be regenerable. A simple reduction in H2 flow restores the activity of Ru/TiO2, similar to standard PP hydrogenolysis (Supplementary Table 9)13. Here, Ru/TiO2 was regenerated in a mixture with MgCl2 and carbonaceous residues formed from the first cycle. This result demonstrates that MgCl2 from the first dechlorination does not impact the hydrogenolysis activity.
Chloride can be removed from the spent Mg3AlO4.5 to enable the reuse of the Cl trap. Calcination in air removes 90% of Cl, and steaming at 550 °C leads to negligible residual Cl content (Supplementary Table 10). Cl removal corresponds to the high-temperature MgCl2 hydrolysis reaction:
The HCl removed with steam can be separated from the water vapour in the downstream scrubber, following standard industrial practices. The Cl can be recuperated from the scrubber and recycled in PVC production or elsewhere. Despite changes in crystal size and morphology after steaming, the regenerated adsorbent showed complete HCl elimination from the gas phase, similar to fresh Mg3AlO4.5 (Supplementary Fig. 12).
Discussion
Precious metal-based catalysts in the chemical recycling of polyolefins are severely poisoned by HCl and Cl-containing compounds, preventing treating POs that contain up to 1–4% PVC. A two-stage technology was introduced to avoid catalyst poisoning. Mixtures of PO with PVC are treated at high H2 pressure with Mg3AlO4.5 as a Cl trap. During this dechlorination treatment, PVC decomposes to polyenes and HCl, both adsorbed and absorbed on Mg3AlO4.5. HCl is absorbed as MgCl2. Polyenes undergo partial C=C bond hydrogenation in the first stage and hydrochlorination to chloroalkanes. This mixture then reacts over Ru/TiO2, leading to ≥65% yields of lubricant-range hydrocarbons, similar to the PVC-free case. A remarkable benefit is that methane is strongly suppressed due to the hydrogenolysis of the C–Cl bond and the release of HCl, which modifies the properties of Ru particles and TiO2 support. Due to Cl-induced modification of the catalyst, liquid products possess different structure than pure PP hydrogenolysis at the same molecular weight.
The catalysts in this study contain expensive noble metals Ru and Pt, an obvious economic obstacle. Indeed, techno-economic analysis44 for PE conversion to lubricants showed that precious metal affects the final product cost. Recent studies using Earth-abundant, cheap catalysts for hydroconversion, including nickel45, cobalt46 and even zirconia-based catalysts47, are very promising. While these materials are much cheaper, catalyst active sites would still be vulnerable to Cl poisoning, necessitating dechlorination. Application of the proposed dechlorination strategy to these catalysts is an important future direction.
The proposed dechlorination operates at 50–150 °C lower temperature than previous analogues, saving energy, producing liquids free of Cl and eliminating downstream energy-intensive purification. Unlike dechlorination traps operating at atmospheric pressure in an inert atmosphere, our Mg3AlO4.5 trap operates at high H2 pressure, facilitating HCl absorption and crystallization of MgCl2 at milder conditions and hydrogenation of polyenes. Importantly, the approach extends to various Cl-containing contaminants, including chloroalkanes and chloroarenes. Absence of HCl emissions in the gas phase helps reducing equipment corrosion, since HCl emission and absorption are conducted in the same vessel. Energy savings and elimination of downstream separations stemming from the liquid purity are important for sustainable manufacturing. Despite having lower dechlorination temperature, more traditional Cl removal procedures, such as pyrolysis33 or thermal extrusion48, could be preferred in some cases. For example, if dechlorination of PP–PVC mixture is followed by pyrolysis at 400–450 °C, some of the Cl from MgCl2 would leak at this temperature, leading to product contamination. Thus, it is better to dechlorinate at mild conditions before low-temperature catalytic hydroconversion. Importantly, when PP is contaminated with PVDC, low-temperature dechlorination does not work, and pyrolytic Cl removal should be considered25,49.
Notably, the catalyst and the trap are regeneratable, despite some morphological changes in the crystal size of Mg3AlO4.5 after steaming. The high regeneration temperature to steam crack MgCl2 imposes limitations that can counterbalance the benefits of the lower dechlorination temperature.
The importance of sorting out halogenated compounds in plastic recycling has been highlighted50. The ability to operate on mixed contaminated feedstock will make waste sorting substantially less challenging, improving the logistics of waste collection before catalytic upcycling. This in turn would greatly reduce the cost and enable a broader application of waste hydroconversion. Combined with the availability of green hydrogen, this opens the possibility of reducing CO2 emissions, currently associated with using hydrogen gas in hydroconversion.
Methods
Catalyst and adsorbents preparation
The Ru/TiO2 catalyst was prepared according to a previously published procedure13. Before the reaction test, the catalyst was pre-reduced in a tubular furnace at 300 °C for 3 h in a 50% H2/He gas mixture (heating rate 10 °C min−1). The Mg3AlO4.5 was prepared as follows. Commercial hydrotalcite Mg6Al2(CO3)(OH)16·4H2O (Sigma-Aldrich) was calcined in a muffle furnace in stagnant air at 550 °C for 6 h (heating rate 2 °C min–1).
The MgO/SiO2 was prepared by incipient wetness impregnation of aqueous Mg(NO3)2·9H2O solution (Sigma-Aldrich) on silica granules (Davisil 646, Brunauer–Emmett–Teller surface area (SBET 275 m2 g−1)). After impregnation, the sample was dried at room temperature for 3 h and 70 °C overnight. Then it was calcined at 550 °C in stagnant air for 6 h. The Mg–Al mixed oxides were prepared by nitrate co-precipitation according to ref. 19. Samples with Mg/Al ratios of 0.5 and 7.1 were calcined at 550 °C for 6 h and tested in dechlorination.
Adsorbents characterization
After calcination, the surface area and pore volume were estimated through Brunauer–Emmett–Teller and Barrett–Joyner–Halenda analysis of N2 sorption isotherms at −196 °C using an ASAP 2020 instrument (Micromeritics). Before measurements, samples were degassed at 300 °C for 3 h in vacuum. Elemental composition was measured using X-ray fluorescence on a Rigaku Supermini 200 WD XRF machine with a Pd anode.
Polymer feedstock
Several polymers were mixed with PVC to test process feasibility with different feedstocks: isotactic PP (weight-averaged molecular mass (Mw) 250 kDa, number-averaged molecular mass (Mn) 67 kDa; Sigma-Aldrich 427888), amorphous PP (Sigma-Aldrich 428175) and HDPE (Mw 108 kDa; Sigma-Aldrich 427985).
Dechlorination reaction
Granules of PP (Mw 250 kDa, Mn 67 kDa; Sigma-Aldrich 427888) were mixed with PVC (Mw 43 kDa, Mn 22 kDa; Sigma-Aldrich 389293) and loaded in a 50 ml high-pressure stainless-steel Parr reactor equipped with a glass liner. Then the required amount of Mg3AlO4.5 was added with a Teflon-covered stir bar; the reactor was sealed, purged with pure hydrogen (or helium) three times and then charged to attain the required pressure. The reactor was placed on a hot plate for magnetic stirring and heated using a 2 inch band heater for 20–25 min to the required dechlorination temperature. After fixed time intervals, the reactor was quenched in an ice bath, and gas products were removed into a 1 l Tedlar gas bag. The solid products were removed from the liner, separated from the stir bar and used later for hydrogenolysis experiments. Gravimetric estimation showed that solid yield in the dechlorination step was close to 98–100% in all cases.
To expand the scope of dechlorination, PVDC (GoodFellow, LS554616), 1,2,4-trichlorobenzene (99%; Sigma-Aldrich) and 1,1,2,2-tetrachloroethane (Sigma-Aldrich) were mixed with PP and Mg3AlO4.5.
Hydrogenolysis reaction
Solid products after dechlorination were placed in a separate 50 ml Parr reactor with a Teflon-covered magnetic stir bar and 100 mg of freshly reduced Ru/TiO2 catalyst. Then the reactor was sealed, purged with H2 five times and charged with 30 bar H2. It was heated with a band heater on the hot plate for 20 min to 250 °C and kept at this temperature for 16 h. After that, the reactor was quickly cooled in an ice bath.
After the reaction and product extraction, the reactor was thoroughly washed with hexane, acetone and deionized water. Then it was filled with 0.05 M HCl water solution and stirred for 3 h to remove contamination. Afterwards, the reactor was washed with water and acetone, dried and scrubbed with an abrasive pad.
Experiments with a model compound, 1-chlorooctadecane (98%; Sigma-Aldrich), were conducted. We mixed 1.8 g of PP with 0.1 g of freshly reduced Ru/TiO2 catalyst in a Parr reactor. Then 0.3 g of 1-chlorodecane were added, and the reactor was closed, purged with pure H2 and charged with 30 bar H2. After 16 h of reaction at 250 °C, product extraction and analysis were conducted as usual.
Dechlorination in a flow reactor
For comparison, dechlorination was conducted in a flow reactor at ambient pressure. We mixed PP with PVC and Mg3AlO4.5 in 1.8:0.2:0.2 proportions by weight. The mixture was loaded in a quartz boat in a horizontal furnace. It was then heated in He flow (100 ml min–1) to 250 °C for 6 h. Then the sample was cooled to room temperature and transferred to the Parr reactor, mixed with 0.1 g of Ru/TiO2 hydrogenolysis catalyst.
Product analysis
Product analysis was conducted according to a previously published procedure37. Briefly, gas from the reactor headspace was taken to a Tedlar gas bag. Liquid and solid products were separated using filtration with CH2Cl2, used as a solvent. The solid residue was then dried at room temperature. The liquid was cleaned from the solvent on a rotary evaporator. Gas products were quantified using a gas chromatography with flame ionization detector (HP-Plot Q 30 m column), calibrated with C1–C4 gas standards (Supelco 303100-U, 307300-U). The solid and liquid fraction yields were quantified gravimetrically. The yield of the ith group of products was calculated according to equation (2):
where mi is the mass of the gas, liquid or solid, minitial corresponds to the initial mass of PP and PVC, and the mass of solid was corrected for the mass of catalyst and adsorbent. Repetitive runs show yield deviation within ~10%.
The weight of the solid residue was calculated gravimetrically by subtracting the catalyst and Mg3AlO4.5 masses. The total weight of all products was used to calculate the mass balance:
where MB is the mass balance, mi is the mass of the ith product group (gas, liquid or solid residue) and minitial is the mass of PP and PVC. In all two-step dechlorination experiments, MB was above 95%.
The total Cl content in the product was calculated as the sum of Cl in the solid residue and in the liquid. Tests with Mg3AlO4.5 did not produce any detectable amounts of HCl or chlorohydrocarbons in the gas phase. The Cl balance (ClB) was calculated as follows:
where \({m}_{{{\mathrm{Cl}}}}^{{{\mathrm{initial}}}}\) is the initial mass of Cl in PVC (equal to 56.8 wt% of PVC loading); \({m}_{{{\mathrm{liq}}}}^{{{\mathrm{Cl}}}}\) is the mass of liquid products, \({\omega }_{{{\mathrm{liq}}}}^{{{\mathrm{Cl}}}}\) is the Cl weight fraction in liquid products estimated by XRF, \({m}_{{{\mathrm{solid}}}}^{{{\mathrm{Cl}}}}\) is the mass of solid residue,\(\,{m}_{{{{\mathrm{Mg}}}}_{3}{{\mathrm{Al}}}{{\mathrm{O}}}_{4.5}}\) is the initial mass of Mg3AlO4.5 and \({\omega }_{{{\mathrm{solid}}}}^{{{\mathrm{Cl}}}}\) is the Cl weight fraction in solid residue estimated by XRF. In all experiments with Mg3AlO4.5 as Cl trap, ClB reached ~90%.
Liquid products were analysed using GPC with high-resolution (HR) 4, HR 3 and HR 0.5 columns, tetrahydrofuran as a mobile phase and a refractive index detector. The retention time was calibrated using a polystyrene standards kit (Waters, WAT058931).
Adsorbent–PVC interactions
The PVC was mixed and dechlorinated directly with Mg3AlO4.5 without adding PP. The powder formed after dechlorination was dried overnight at room temperature. The XRD patterns were collected on Bruker D2 Phaser instrument with Cu tube, 0.05° 2θ step size and 3 s dwell. The 13C MAS NMR spectra were acquired on an 11.7 T Bruker Avance III NMR spectrometer with a 4 mm HX probe with a magic angle spinning at 14 kHz. Direct 13C excitation was achieved with a 3.5 μs 90° pulse. The XPS spectra were measured on a Thermo K-alpha instrument with Al Kα radiation. Due to charging, the peak position was not corrected, and only the intensity was used to monitor the change in elemental composition on the surface. Scanning electron microscope images and energy-dispersive X-ray analysis were acquired on an Auriga 60 CrossBeam high-resolution microscope with accelerating voltage at 10 kV with the sample deposited on carbon tape.
Characterization of spent Ru/TiO2 catalysts
Changes in Ru/TiO2 catalyst induced by chlorine were analysed after the reaction. Initially, 1.8 g of PP mixed with 0.3 g of 1-chlorooctadecane reacted over Ru/TiO2 at 250 °C for 16 h at 30 bar H2. After the reaction, the solid was separated with filtration and dried. Then the solid was transferred to a glass vial, mixed with 20 ml toluene and heated to 90 °C for 30 min, until all PP was dissolved. The liquid toluene was decanted with a glass pipette. The remaining powder was primarily spent Ru/TiO2, contaminated with organic residue and Cl. This powder was dried in a vacuum oven overnight at 90 °C to remove residual toluene.
The sample was transferred to a tubular quartz reactor and heated in 10% H2/He flow at 300 °C for 2 h (ramp rate 10 °C min–1). Then the powder was moved to a glovebox (<100 ppm oxygen) and loaded on copper tape in a vacuum transfer vessel. Then XPS spectra were measured using Thermo Fisher K-Alpha Instrument. The binding energy standard was O 1s at 529.9 eV, typical for TiO2. Transmission FTIR spectra of adsorbed CO were recorded on Nicolet 8700 spectrometer with a mercury cadmium telluride detector and a homemade tubular flow Pyrex cell with KBr windows. Samples were pressed in self-supported wafers (1.26 cm2, 80 bar cm−2 pressure). The sample was pre-reduced in 10% H2/He flow at 300 °C for 1 h, purged with pure He for 1 h and then cooled to 35 °C. Then the sample was contacted with 1% CO/He mixture for 10 min and flushed with pure He for 5 min followed by spectra recording. DRIFT spectra were recorded using a Harrick Praying Mantis cell, following a similar sample pre-reduction. After purging with He for 1 h at 300 °C, the sample was cooled to 150 C, and 2 μl liquid pyridine was injected in a He stream. Spectra are reported as differences between the sample before and after pyridine chemisorption. Sequential injections were used to ensure complete saturation of the surface with pyridine.
Cl distribution analysis
Chlorine has been measured using XRF for liquid and solid samples. For liquids, decalin and 1,2,4-trichlorobenzene (Sigma-Aldrich) were used to calibrate Cl Kα signal intensity in a broad concentration range. For solids, PVC mixed with parent Mg3AlO4.5 was used to calibrate Cl, Al and Mg content in the solid residue after the reaction. In the gas phase, the HCl content was estimated using FTIR spectroscopy on a Nicolet iS50 spectrometer with a 100 mm gas cell equipped with 38 mm CaF2 windows (Pike Technologies). The cell was evacuated at ~1 Torr and connected to a 1 l Tedlar gas sampling bag. Then the cell was filled with a sample gas and closed. Spectra were acquired at 4 cm−1 resolution, 32 scans with a deuterated triglycine sulfate detector.
Catalyst and adsorbent regeneration
To simulate severe Cl contamination, Mg3AlO4.5 was treated with pure PVC, and 0.4 g Mg3AlO4.5 and 0.4 g PVC were loaded in a Parr reactor, charged with 30 bar H2, heated at 250 °C for 6 h. Then the reactor was cooled in an ice bath, the gas was vented and the powder was removed and later calcined in a muffle furnace at 550 °C for 4 h in stagnant air. To remove traces of Cl, the sample was steamed after calcination. The sample was packed in a 12.7 mm quartz tubular reactor with quartz wool and purged with pure He (5 ml min–1) for 30 min. Then it was heated to 550 °C in He with a ramp rate of 5 °C min−1. Liquid water was administered into the reactor using a syringe pump operating at 0.5 ml min−1 at 550 °C for 2 h. After that, the sample was purged with pure He for 1 h and cooled to room temperature.
After the standard hydrogenolysis reaction, spent Ru/TiO2 was mixed with spent Mg3AlO4.5, and the solid residue was washed with 50 ml CH2Cl2 and then dried overnight at 100 °C. This solid mixture was transferred to a tubular furnace, heated to 300 °C with a ramp rate of 10 °C min−1 and dwelled for 4 h in a 50% H2/He mixture. Then the mixture was mixed with fresh 2 g PP and loaded in a Parr reactor. The reactor was charged with 30 bar H2 and then heated to 250 °C for 16 h. Products were analysed according to the standard procedure.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data that support the findings in the current study are available within the Article and its Supplementary Information. Any additional data are available from the corresponding authors upon reasonable request.
References
Tsakona, M. & Rucevska, I. Baseline Report on Plastic Waste (UNEP, 2020); https://gridarendal-website-live.s3.amazonaws.com/production/documents/:s_document/554/original/UNEP-CHW-PWPWG.1-INF-4.English.pdf?1594295332
Jambeck, J. et al. Plastic waste inputs from land into the ocean. Science 347, 768–771 (2015).
Hinton, Z. R. et al. Innovations toward the valorization of plastics waste. Annu. Rev. Mater. Res. 52, 249–280 (2022).
Yousef, S. et al. Pyrolysis kinetic behavior and TG-FTIR-GC–MS analysis of metallised food packaging plastics. Fuel 282, 118737 (2020).
Karayılan, S., Yılmaz, Ö., Uysal, Ç. & Naneci, S. Prospective evaluation of circular economy practices within plastic packaging value chain through optimization of life cycle impacts and circularity. Resour. Conserv. Recycl. 173, 105691 (2021).
Borrelle, S. B. et al. Predicted growth in plastic waste exceeds efforts to mitigate plastic pollution. Science 369, 1515–1518 (2020).
Jang, Y.-C., Lee, G., Kwon, Y., Lim, J.-H. & Jeong, J.-H. Recycling and management practices of plastic packaging waste towards a circular economy in South Korea. Resour. Conserv. Recycl. 158, 104798 (2020).
Vogt, B. D., Stokes, K. K. & Kumar, S. K. Why is recycling of postconsumer plastics so challenging? ACS Appl. Polym. Mater. 3, 4325–4346 (2021).
Kots, P. A., Vance, B. C. & Vlachos, D. G. Polyolefin plastic waste hydroconversion to fuels, lubricants, and waxes: a comparative study. React. Chem. Eng. 7, 41–54 (2022).
Celik, G. et al. Upcycling single-use polyethylene into high-quality liquid products. ACS Cent. Sci. 5, 1795–1803 (2019).
Rorrer, J. E., Troyano-Valls, C., Beckham, G. T. & Román-Leshkov, Y. Hydrogenolysis of polypropylene and mixed polyolefin plastic waste over Ru/C to produce liquid alkanes. ACS Sustain. Chem. Eng. 9, 11661–11666 (2021).
Jia, C. et al. Deconstruction of high-density polyethylene into liquid hydrocarbon fuels and lubricants by hydrogenolysis over Ru catalyst. Chem Catal. 1, 437–455 (2021).
Kots, P. A. et al. Polypropylene plastic waste conversion to lubricants over Ru/TiO2 catalysts. ACS Catal. 11, 8104–8115 (2021).
Liu, S., Kots, P. A., Vance, B. C., Danielson, A. & Vlachos, D. G. Plastic waste to fuels by hydrocracking at mild conditions. Sci. Adv. 7, eabf8283 (2021).
Patel, P. The Future of Plastic (ACS C&EN, 2020).
Kusakabe, K. et al. Dechlorination of polyvinyl chloride via solvothermal treatment with glycerol. Processes 10, 2047 (2022).
Marino, A. et al. ZSM-5 zeolites performance assessment in catalytic pyrolysis of PVC-containing real WEEE plastic wastes. Catal. Today 390, 210–220 (2022).
Xu, M. & Wei, M. Layered double hydroxide‐based catalysts: recent advances in preparation, structure, and applications. Adv. Funct. Mater. 28, 1802943 (2018).
Di Cosimo, J., Dıez, V., Xu, M., Iglesia, E. & Apesteguıa, C. Structure and surface and catalytic properties of Mg–Al basic oxides. J. Catal. 178, 499–510 (1998).
Wilkie, C. A. & Morgan, A. B. Fire Retardancy of Polymeric Materials (CRC Press, 2009).
Beach, E. S., Weeks, B. R., Stern, R. & Anastas, P. T. Plastics additives and green chemistry. Pure Appl. Chem. 85, 1611–1624 (2013).
Lange, J.-P. Managing plastic waste—Sorting, Recycling, Disposal, and Product Redesign. ACS Sustain. Chem. Eng. 9, 15722–15738 (2021).
Zhang, F. et al. Polyethylene upcycling to long-chain alkylaromatics by tandem hydrogenolysis/aromatization. Science 370, 437–441 (2020).
Bhaskar, T., Negoro, R., Muto, A. & Sakata, Y. Prevention of chlorinated hydrocarbons formation during pyrolysis of PVC or PVDC mixed plastics. Green. Chem. 8, 697–700 (2006).
Okada, T., Sutoh, S., Sejima, K., Tomohara, H. & Mishima, S. A useful method for thorough dehydrochlorination of poly (vinylidene chloride–co–vinyl chloride) using zinc (II) oxide. Polym. Degrad. Stab. 171, 109040 (2020).
Zhou, Q., Tang, C., Wang, Y.-Z. & Zheng, L. Catalytic degradation and dechlorination of PVC-containing mixed plastics via Al–Mg composite oxide catalysts. Fuel 83, 1727–1732 (2004).
Lingaiah, N. et al. Catalytic dechlorination of chloroorganic compounds from PVC-containing mixed plastic-derived oil. Appl. Catal. A 207, 79–84 (2001).
Chen, L. et al. Effect of reaction conditions on the hydrogenolysis of polypropylene and polyethylene into gas and liquid alkanes. React. Chem. Eng. 24, 7332–7339 (2022).
Chen, L. et al. Disordered, sub-nanometer Ru structures on CeO2 are highly efficient and selective catalysts in polymer upcycling by hydrogenolysis. ACS Catal. 12, 4618–4627 (2022).
Shimokawa, S. & Yamada, E. A 13C NMR probe for high-temperature and high-pressure experiments. J. Magn. Reson. (1969) 51, 103–109 (1983).
Kosuda, T. et al. Characteristics and mechanism of low temperature dehydrochlorination of poly (vinyl chloride) in the presence of zinc (II) oxide. Polym. Degrad. Stab. 97, 584–591 (2012).
Zhang, B. et al. Thermogravimetric–mass spectrometric analysis of the reactions between oxide (ZnO, Fe2O3 or ZnFe2O4) and polyvinyl chloride under inert atmosphere. Mater. Trans. 41, 1342–1350 (2000).
Meng, T.-T., Zhang, H., Lü, F., Shao, L.-M. & He, P.-J. Comparing the effects of different metal oxides on low temperature decomposition of PVC. J. Anal. Appl. Pyrolysis 159, 105312 (2021).
Coq, B., Ferrat, G. & Figueras, F. Conversion of chlorobenzene over palladium and rhodium catalysts of widely varying dispersion. J. Catal. 101, 434–445 (1986).
Wu, D. et al. In situ generation of Brønsted acidity in the Pd–I bifunctional catalysts for selective reductive etherification of carbonyl compounds under mild conditions. ACS Catal. 9, 2940–2948 (2019).
Weitkamp, J. Catalytic hydrocracking—mechanisms and versatility of the process. ChemCatChem 4, 292–306 (2012).
Kots, P. A. et al. Electronic modulation of metal-support interactions improves polypropylene hydrogenolysis over ruthenium catalysts. Nat. Commun. 13, 5186 (2022).
Park, K.-B., Oh, S.-J., Begum, G. & Kim, J.-S. Production of clean oil with low levels of chlorine and olefins in a continuous two-stage pyrolysis of a mixture of waste low-density polyethylene and polyvinyl chloride. Energy 157, 402–411 (2018).
Hinton, Z. R. et al. Antioxidant-induced transformations of a metal–acid hydrocracking catalyst in the deconstruction of polyethylene waste. Green Chem. (2022).
Brebu, M. et al. The individual and cumulative effect of brominated flame retardant and polyvinylchloride (PVC) on thermal degradation of acrylonitrile–butadiene–styrene (ABS) copolymer. Chemosphere 56, 433–440 (2004).
Dacey, J. & Barradas, R. Structure of pyrolyzed polyvinylidene chloride. Can. J. Chem. 41, 180–190 (1963).
Zhao, Y.-B., Lv, X.-D. & Ni, H.-G. Solvent-based separation and recycling of waste plastics: a review. Chemosphere 209, 707–720 (2018).
Wang, C. et al. Polyethylene hydrogenolysis at mild conditions over ruthenium on tungstated zirconia. JACS Au 1, 1422–1434 (2021).
Cappello, V. et al. Conversion of plastic waste into high-value lubricants: techno-economic analysis and life cycle assessment. Green. Chem. 24, 6306–6318 (2022).
Vance, B. C., Kots, P. A., Wang, C., Granite, J. E. & Vlachos, D. G. Ni/SiO2 catalysts for polyolefin deconstruction via the divergent hydrogenolysis mechanism. Appl. Catal. B 322, 122138 (2023).
Zichittella, G. et al. Hydrogenolysis of polyethylene and polypropylene into propane over cobalt-based catalysts. JACS Au 2, 2259–2268 (2022).
Chen, S. et al. Ultrasmall amorphous zirconia nanoparticles catalyse polyolefin hydrogenolysis. Nat. Catal. 6, 161–173 (2023).
Fukushima, M. et al. Study on dechlorination technology for municipal waste plastics containing polyvinyl chloride and polyethylene terephthalate. J. Mater. Cycles Waste Manage. 12, 108–122 (2010).
Hubáček, J. et al. Dechlorination during pyrolysis of plastics: the potential of stepwise pyrolysis in combination with metal sorbents. Fuel Process. Technol. 231, 107226 (2022).
Delva, L., Hubo, S., Cardon, L. & Ragaert, K. On the role of flame retardants in mechanical recycling of solid plastic waste. Waste Manage. 82, 198–206 (2018).
Acknowledgements
We thank K. J. Korman and E. D. Bloch of the University of Delaware for providing access to the XRD instrument. This work was financially supported by the Center for Plastics Innovation (CPI), an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, award number DE-SC0021166. This research used instruments in the Advanced Materials Characterization Lab (AMCL) at the University of Delaware. MAS NMR measurements were made possible by the Delaware COBRE programme, supported by a grant from the National Institute of General Medical Sciences, NIGMS (5 P30 GM110758-02), from the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
P.A.K. conceived the idea, designed the project and conducted the experiments. B.C.V. and C.W. carried out some experiments and contributed to the discussions. C.M.Q. measured and analysed the MAS NMR spectra. D.G.V. supervised the project and acquired funding. P.A.K. and D.G.V. wrote the manuscript, and all authors discussed the results and assisted with the manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
P.A.K., B.C.V., C.W. and D.G.V. are authors of a patent application on polyolefin plastic waste upcycling to lubricants. C.M.Q. has no competing interests.
Peer review
Peer review information
Nature Sustainability thanks Ding Ma, Taylor Uekert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–12 and Tables 1–10.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kots, P.A., Vance, B.C., Quinn, C.M. et al. A two-stage strategy for upcycling chlorine-contaminated plastic waste. Nat Sustain 6, 1258–1267 (2023). https://doi.org/10.1038/s41893-023-01147-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41893-023-01147-z
This article is cited by
-
Tandem catalysis enables chlorine-containing waste as chlorination reagents
Nature Chemistry (2024)
-
Complete hydrogenolysis of mixed plastic wastes
Nature Chemical Engineering (2024)
-
Upcycling contaminated plastics
Nature Sustainability (2023)
