
Abstract
Progress in sequencing technologies and clinical experiments has revolutionized immunotherapy on solid and hematologic malignancies. However, the benefits of immunotherapy are limited to specific patient subsets, posing challenges for broader application. To improve its effectiveness, identifying biomarkers that can predict patient response is crucial. Machine learning (ML) play a pivotal role in harnessing multi-omic cancer datasets and unlocking new insights into immunotherapy. This review provides an overview of cutting-edge ML models applied in omics data for immunotherapy analysis, including immunotherapy response prediction and immunotherapy-relevant tumor microenvironment identification. We elucidate how ML leverages diverse data types to identify significant biomarkers, enhance our understanding of immunotherapy mechanisms, and optimize decision-making process. Additionally, we discuss current limitations and challenges of ML in this rapidly evolving field. Finally, we outline future directions aimed at overcoming these barriers and improving the efficiency of ML in immunotherapy research.
Similar content being viewed by others


Introduction
The immune system is crucial in monitoring cancer and identifying neoantigens produced by tumor cells that can trigger cellular immune responses1. But tumor cells have developed strategies to evade immune surveillance2. To address this, cancer immunotherapy was developed, aiming to stimulate the immune system or create lab-engineered substances that restore the ability to recognize and eliminate cancer cells. Immunotherapy options include immune checkpoint inhibitors (ICI), cancer vaccines, adoptive cellular therapies (ACT), cytokine, tumor-infecting viruses, targeted antibodies, and adjuvants. While immunotherapy has significantly improved patient outcomes, its effectiveness is confined to a small and unpredictable subset of patients within a given cancer diagnosis3, and immune-related adverse events (irAEs) may occur4. Therefore, precise identification of a patient’s tumor microenvironment (TME) and of the ability to predict its immunotherapy response are essential for enhancing overall immunotherapy effectiveness.
Current prediction of immunotherapy response relies on biomarkers such as immune-cell infiltration5, tumor mutational burden (TMB)6, PD-1/PD-L1 expression7, CTLA-4 expression8, mismatch repair (MMR) and microsatellite instability (MSI)9. However, existing clinical practices based on simplistic threshold-based methods lack accuracy. In this context, machine learning (ML) technologies have emerged as valuable tools, offering the potential to refine the precision of immunotherapy response prediction. By harnessing sophisticated algorithms and analyzing extensive datasets, ML models can discern intricate patterns and interactions among various molecular biomarkers, providing a more nuanced understanding of the complex immunotherapy tumor microenvironment. These state-of-the-art ML models not only capture subtle relationships between individual biomarkers but also adapt to the dynamic nature of immune responses, offering a more comprehensive and adaptable approach than traditional threshold-based methods. Indeed, ML-based approaches have shown capacity in various oncology applications, including early diagnosis10, cancer type classification11, the complexity and plasticity of TME and immune system deciphering12, response and prognosis prediction13, and potential neoantigen detection14.
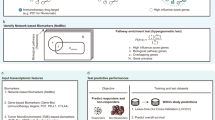
This review summarizes the application of ML in molecular analyses related to immunotherapy, including prediction of immunotherapy responses, identification of response-associated biomarkers, and analysis of the TME (Fig. 1). It also explores ML approaches developed to optimize the identification of crucial neoantigens in personalized immunotherapy. Additionally, this review discusses the challenges and opportunities encountered in current research endeavors, aiming to enhance understanding and recognition of the significant contribution of ML in advancing cancer immunotherapy.

We provide an overview of machine learning methodologies applicable to different aspects of tumor immunotherapy including prediction of immunotherapy responses, identification of response-associated biomarkers, and analysis of the TME. ML machine learning, DEG differentially expressed gene, RFE recursive feature elimination, UAF univariate association filtering, LASSO the least absolute shrinkage and selection operator, LR logistic regression, SVM support vector machine, RF random forest, FCNN fully-connected neural network, CNN convolutional neural network, RNN recurrent neural network, PFS progression-free survival, TME tumor microenvironment.
Employing machine learning for predicting immunotherapy response and identifying biomarkers associated with response
While immunotherapy has brought significant benefits to cancer treatments, its effectiveness remains confined to a small and unpredictable subset of patients with a given cancer diagnosis15. Moreover, the treatment process often imposes substantial financial, physical and mental care burden on patients. Acknowledging these challenges, researchers are increasingly directing their efforts toward identifying valuable molecular biomarkers capable of predicting immunotherapy outcomes and improving its overall utility16. Considering the complex omics space, conducting extensive sampling through experimental methods is impractical. Consequently, in silico approaches, including those employing ML algorithms, provide an opportunity to address this critical need.
Tumors are caused by the accumulation of various genetic variations that regulate the way cells growth and multiplication17,18,19. In light of this, recent studies have turned on ML models to predict a patient’s response to immunotherapy by leveraging his genomic biomarkers and clinical features (Table 1, Fig. 2). Somatic mutations, including single-nucleotide variants (SNVs), insertions, and deletions, provide direct evidence documenting the driving forces behind tumorigenesis and tumor cell proliferation20. These mutations have demonstrated their ability in predicting immunotherapy responses. For example, Peng et al.21 used SNV data and convolutional neural network (CNN) model to classify anti-PD-1/PD-L1 therapy response from metastatic non-small-cell lung cancer (NSCLC) patients. Nonsynonymous mutations can alter transcription, subsequently impacting pathway activations and gene functions. Leveraging the distinct changes in gene expression levels, particularly in oncogenes and tumor suppressor genes, ML models can subtly predict immunotherapy response. According to our survey, RNA-based features, including bulk RNA sequencing22,23,24,25,26,27,28,29,30,31, single cell RNA sequencing (scRNA-seq)32,33, flow cytometry34, and circulating cell-free microRNA sequencing35,36, have been widely implemented in immunotherapy response prediction. Furthermore, the availability of numerous accessible RNA-seq datasets, coupled with the outstanding performance of models utilizing RNA-seq data, has been instrumental in advancing research. From RNA-seq data, many advanced features can be extracted from bioinformatic or ML tools to better characterize tumor genomic profiles, such as tumor-infiltrating lymphocytes (TILs)32,33, pathway activity28 and cell-cell communication23. It is worth noting that some studies have leveraged these features to extract high-level features, thereby enhancing predictive performance. For instance, Wang et al.22 utilized TMB information based on SNV data, gene expression information, and support vector machines-recursive feature elimination (SVM-RFE) algorithm to select gene features. Subsequently, they used least absolute shrinkage and selection operator (LASSO) logistic regression classifier to predict responses of urothelial carcinoma patients treated with the PD-L1 inhibitor atzolizumab using selected gene features. Their approach achieved an AUC of 93% in the test dataset. Additionally, they utilized generalized linear models (GLMs) to derive a TMB-related LASSO score (TLS) from the LASSO regression results. The TLS can serve as an effective indicator for immunotherapy response prediction like TMB. Lapuente-Santana et al.23 employed regularized multi-task linear regression (RMTLR) to identify interpretable biomarkers in relation to immune cells markers, intracellular networks, and intercellular networks for predicting immunotherapy response. On the other hand, Zeng et al.28 implemented a joint nonnegative matrix factorization (NMF) to decompose gene expression matrix, molecular phenotype matrix, and immunotherapy response matrix. This approach aims to identify pivotal genes correlated with immunotherapy response. Similar to RNA-seq data, Shang et al.37 and Filipski et al.38 have successfully employed DNA methylation profiles (CpG sites) for ICI response prediction in NSCLC37 and metastatic melanoma38 patients. Apart from these conventional biomarkers, clinical information39 and Raman spectroscopy data40 have shown promise as reliable biomarkers of ICI response prediction. In a separate study, Sidhom et al.41 integrated human leukocyte antigen (HLA) and T cell receptor (TCR) sequencing to predict ICI response in melanoma. Their approach involved employing a multiple-instance learning model that incorporated HLA into the featurization of the TCR sequences to provide a representation of a joint TCR-HLA antigenic latent space. The contextualization of TCR-HLA was then trained on multihead attention networks to learn the attention weights, which were used to predict the final immunotherapy response.

Advancements in sequencing technologies have paved the way for exploring diverse approaches in immunotherapy response prediction. To improve efficiency and mitigate overfitting, dimensionality reduction and feature selection techniques are performed prior to model training. Multimodal models offer the flexibility to train on data with multiple modalities. These models first utilize sub-models to extract unimodal features from each data modality. Subsequently, a data fusion step transforms each extracted unimodal feature into a compact multimodal representation. Finally, a classification sub-model is implemented to infer response based on the integrated features.
Accompanied by the advancement of sequencing technologies, recent studies have focused on developing complex ML models incorporating multi-omics datasets for immunotherapy prediction42,43,44,45,46,47. Compared to single omics approaches, the integration of multiple omics data can provide a more comprehensive scope of tumor profile, from the original cause of tumors (genetic, environmental, or developmental) to the functional consequences48,49, and consequently leads to improved performance in immunotherapy response prediction47. In a recent approach50, researchers integrated RNA-seq data with somatic mutations, copy number alterations and protein expression alterations to comprehensively investigate various subcohorts within TME using a sparse hierarchical clustering model. By employing this method, they have successfully identified distinct subcohorts within the TME, each exhibiting unique responses to different cancer treatments, including immunotherapy. This model holds significant potential in guiding precise decision-making for combination therapy strategies. However, integrating and training multi-omics data, usually accrued from different platforms, is more challenging than training unimodal features. Addressing this challenge, a recent study by Vanguri et al.47 developed a dynamic deep attention-based multiple-instance learning model that integrates radiology, pathology, and multiomics data to predict the response of NSCLC patients treated with anti-PD-1/PD-L1 blockade. Comparisons demonstrated that the multimodal approach, integrating these features, enables higher accuracy than unimodal approach in the prediction of immunotherapy response. Notably, their multimodal model can also handle redundant information and missing values in combination with data from different modalities. In addition to ICI prediction, ML models have been utilized to predict the chimeric antigen receptor (CAR) T therapy response. Daniels et al.51 developed a DL model to utilize signaling motifs to evaluate the antitumor efficacy of a given CAR. Their DL framework takes the motif sequence of the CAR as the input and propagated the encoded sequence through two CNN layers, a long short-term memory (LSTM) network layer and seven fully connected neural network (FCNN) layers. This approach enables directly prediction of tumor stemness and cytotoxicity based on the motif combinations designed for CAR T cells, thereby guiding the design of the engineering of CAR signaling domains in CAR T therapies.
In our survey, to improve the computational efficiency and reduce noise and complexity of ML models, most studies utilized statistical or ML algorithms, or both to identify a subset of gene markers for model training. ML techniques for gene selection can handle high-dimensional data and identify patterns that may not be apparent through manual inspection or traditional analyses. These ML approaches, including LASSO29, random forest (RF)21,40, SVM-RFE22,26, NMF28, and logistic regression (LR)27, automatically assess the importance of each gene in relation to the immunotherapy response prediction. Using these extracted biomarkers, various algorithms, including LASSO, LR, RF, XGBoost, naive Bayes (NB), SVM, decision tree (DT) and NN, have demonstrated their ability to accurately predict immunotherapy responses. By focusing on this refined set of features, researchers can also enhance the interpretability and generalizability of the models, fostering a more effective integration of machine learning into the complex landscape of immunotherapy research.
Employing machine learning as a supplementary tool for the identification of biomarkers in the tumor microenvironment for immunotherapy
The TME refers to the intricate cellular landscape surrounding tumors, including immune cells, cancer cells, stroma cells, the inflammatory cytokines and chemokines, metabolites, acidity, cytokines and hypoxia52. It plays a critical role in supporting tumorigenesis and progression, and immunotherapy effectiveness53,54. Extensive studies have elucidated the complex interactions within TME, driving functions like angiogenesis55, metastasis56 and immunosuppression57. Although obtaining accurate datasets for TME factors such as hypoxia and low pH poses challenges, the integration of tumor omics data and the implementation of ML models have enabled the identification of TME characteristics directly or indirectly associated with cancer immunotherapy (Table 2, Fig. 3).

Various machine learning models have been developed to effectively identify biomarkers and comprehend the relationship between tumor microenvironment and immunotherapy, including risk, development and treatment. These models aim to improve the efficiency of immunotherapies by providing valuable insights and understanding. ML machine learning, TME tumor microenvironment, TIL tumor-infiltrating lymphocytes, CAF cancer-associated fibroblast, CSC cancer stem-like cell, MSI microsatellite instability, TMB tumor mutational burden.
Microsatellite instability and tumor mutational burden
MSI and TMB are FDA-approved biomarkers that predict immunotherapy response. While not directly related to the TME, MSI and TMB reflect genetic alterations occurring within tumor cells. MSI indicates microsatellite length polymorphism due to mismatch repair deficiency, while TMB represents the number of somatic mutations per million bases in the exome region58. Tumors with higher MSI or TMB tend to produce neoantigens recognized by the immune system, rendering them more responsive to immunotherapy. Given the high cost of large-scale genomic sequencing, using ML models to assess MSI or TMB based on a panel with limited genes can offer a more cost-effective approach. Zhou et al.59 successfully identified a 54-microsatellite-site biomarker using an RF classifier, allowing accurate classification of microsatellite instability-high (MSI-H) and microsatellite stable (MSS) tumors. In a similar vein, Lu et al.60 implemented LASSO regression to identify a gene-targeted panel capable of accurately assessing TMB levels. Recently, many deep learning (DL) models have been developed to use whole-slide images (WSIs) to predict MSI61,62 and TMB status63,64, which enables a more cost-effective means of predicting immunotherapy response without relying on genomic data.
Cancer stem-like cell
Cancer stem-like cells (CSCs) are a small population of cancer cells that can reconstitute and propagate tumors. They have been implicated in metastasis, relapse, and resistance to cancer therapies65,66. Numerous studies have focused on identifying and categorizing CSCs within tumor cell populations. Researchers such as Wei et al.67 and Wang et al.68 employed LASSO regression to identify stemness features in tumor samples using RNA-seq data. These identified stemness features have shown a strong correlation with the prognosis of immunotherapy and can serve as valuable biomarkers for predicting immunotherapy response.
Cancer-associated fibroblast
Cancer-associated fibroblasts (CAFs), also a critical component of TME, can modulate cancer metastasis through signaling interactions with cancer cells. They can also influence leukocyte infiltration, drug access and therapy responses69. To identify CAF related genes, Wang et al.70 applied ML models to classify tumor samples as either CAF-enriched (CAF+) or CAF-absent (CAF−). Their analysis revealed that the CAF− subtype was associated with longer overall survival and higher immune cell infiltration compared to the CAF+ subtype. These findings provide valuable insights for predicting the response to immunotherapy. Similarly, Tian et al.71 used LASSO regression to obtain six CAF-related genes that can be used to predict the response to anti-PD-1 therapy in melanoma patients. These studies demonstrate the utility of ML models in elucidating the role of CAFs and their associated genes in immunotherapy response prediction.
Tumor-infiltrating lymphocyte
TILs are highly specific immunological reactive lymphocytic cell populations that can recognize and kill tumor cells72. Their presence is crucial in mediating response to cancer therapy, and a higher abundance of TILs is often associated with better clinical outcomes after immunotherapy73,74,75. Currently, ML models have been broadly implemented to quantify various TIL-based biomarkers for immunotherapy response prediction. These biomarkers encompass RNA-seq data and somatic mutation features76, including protein-protein interaction (PPI) networks77, tumor-infiltrating immune cell-associated lncRNAs78,79, T cell signatures80,81, B cell signatures82 and immunophenotype-related DNA methylation signatures (iPMS)83. A general approach adopted in these models involves clustering tumor samples based on the tumor immune microenvironment, such as immunoactivity, disease stages, and survival outcomes. ML models are then employed to extract significant biomarkers for cluster classification. Subsequently, another ML model was then utilized for predicting immunotherapy response to validate the selected biomarkers for each cluster. Typically, TILs comprise both mononuclear and polymorphonuclear immune cells, including T cells, B cells, natural killer cells, macrophages, neutrophils, dendritic cells, mast cells, eosinophils, and basophils. Accurately assessing the abundance of each immune cell type within tumor tissues is essential for treatment decision-making and evaluating drug response. To this end, ML models have been developed to automatically estimate the abundance of these immune cells82,84,85,86,87, enabling precise deconvolution. In a recent study, Fernández et al.84 used their deconvolved proportions of 22 immune cells as the input feature, which could accurately predict the response of patients treated with ICI therapy.
Metabolism
Metabolism refers to the changes observed in cellular metabolic pathways in tumor cells. Typically, oncogenic transformation can induce cancer cells to adopt a well-characterized metabolic phenotype that can profoundly influence the TME88. Increasing evidence has highlighted the role of metabolism in tumor immunosuppressive responses and resistance to immunotherapy89,90. For instance, tumor cells can alter metabolism by increasing glucose uptake and fermentation of glucose to lactate, promoting tumor growth, survival, proliferation, and long-term maintenance91. To improve immunotherapy efficacy, researchers have proposed employing ML models to identify metabolic TME subtype that respond favorably to immunotherapy. Ge et al.92 conducted an analysis of lipid metabolism genes and immune-related genes of lung adenocarcinoma (LUAD) patients and identified two distinct subtypes, namely “metabolism phenotype” and “immunoactive phenotype”, using Cox regression. The “metabolism subtype” exhibited reduced sensitivity and poorer prognosis to immunotherapy. The identified metabolic features hold promise as potential biomarkers to predict immunotherapy response.
Neoantigen
Neoantigens are novel peptides that form in tumor cells due to certain somatic mutations. Neoantigens have the potential to be recognized by immune cells, triggering immune responses against tumor cells93,94. Immunogenic neoantigens have been identified as crucial for developing personalized neoantigen-targeted cancer immunotherapies95,96, including vaccines and adoptive T-cell therapies94. However, the process of neoantigen discovery and validation remains a daunting question that must be addressed before neoantigen-based immunotherapies can become prominent in cancer treatment97. For example, many tumor peptides lack immunogenicity, highlighting the importance and complexity of accurately identifying which neoantigens can effectively stimulate immune cell responses.
Recently, novel pipelines and state-of-the-art ML algorithms have been developed to identify T-cell neoantigens through major histocompatibility complex (MHC) class I and II presentations (Table 3, Fig. 4). Pipelines utilize genomics data, usually derived from whole-genome sequencing (WGS) or whole-exome sequencing (WES), obtained from tumor samples to infer the mutated peptides based on the somatic non-synonymous SNVs. To facilitate neoantigen prediction, researchers have conducted The Immune Epitope Database (IEDB), which provides experimentally characterized T cell epitopes and a comprehensive set of MHC-binding and MHC eluted ligand (EL) data for humans98. These resources significantly enhance the convenience and accuracy of neoantigen prediction. Based on our review, the majority of ML models focus on identifying class I MHC alleles, which have the ability to bind peptides derived from intracellular proteins and present them on the cell surface to CD8 + T cells. Some studies employ ML models to predict neoantigens by estimating the binding affinity between a given mutated peptide and a class I MHC molecule, known as peptide-MHC (pMHC) binding affinity99,100,101,102,103,104,105,106. These models can be categorized into two groups based on their output. The first group predicts a score representing the relative binding affinity between a peptide and MHC99,100,101,102,103. Among these models, NetMHC99 and NetMHCpan100 used the FCNN framework. While NetMHC was trained solely on binding affinity datasets and can predict peptide binding to specific MHC alleles, NetMHCpan integrated information from both binding affinity data and mass spectrometry (MS) EL data, allowing it to predict binding for a wider range of MHC molecules with high accuracy. Different from NetMHC and NetMHCpan, MHCflurry101 added two one-dimensional convolutional layers before fully connected layers, resulting in higher accuracy. EDGE102, on the other hand, used three peptide-extrinsic features (RNA abundance, flanking sequence, per-gene coefficients) captured from MS data as the input, propagating them into three locally connected layers respectively before merging them into fully connected layers for binding affinity prediction. This approach extracts more information than using a single input, resulting in higher positive predictive values (PPV) compared to benchmarked models. Another model, MHCRoBERTa103, built a transfer learning model by pre-training on the UniprotKB/Swiss-Prot dataset and fine-tuning on IEDB dataset. This strategy allows the model to maintain high accuracy and efficiency simultaneously. The second form of binding affinity prediction in these models involves providing a binary classification result to determine whether a given peptide is a binder104,105,106. In most studies, a threshold of <500 nM of the IC50 value is used to define candidate peptides that are likely to bind to MHC. Notably, to improve performance, ForestMHC105 considered six different sequence-related features and their combinations as input features to select the optimal feature subset. Similarly, Anthem106 collected five published sequence scoring functions that can calculate a binding probability based on sequence information. These scoring functions, along with their combinations, were used as input features to select the optimal subset of scoring functions for binder classification. Considering the distinct advantages and limitations of each binding affinity model, Gartner et al.107 built a random forest-based model that integrates known class I candidate human tumor neoantigens predicted by other models and next-generation sequencing (NGS) data from individuals with metastatic cancers. This model can rank the candidate neoantigens, providing a ranked list that can serve as therapeutic targets and facilitate studies aimed at developing more effective immunotherapies. Recently, increasing evidence indicates that CD4 + T cells can recognize cancer-specific antigens and control tumor growth. As a result, MHC class II neoantigen prediction has become important in immunotherapies like vaccine design and targeted therapy development. However, unlike MHC class I molecules that are highly specific and bind a limited set of peptides of a narrow length distribution108, MHC class II molecules are highly polymorphic and the size of the peptides presented are promiscuous109, making it more challenging for neoantigen prediction. In response to this challenge, several models have been established to predict the binding affinity of pMHC class II complexes110,111,112,113,114. Compared with class I binding affinity prediction models, the MHC class II prediction models were generally trained on more complicated datasets, such as the IEDB MHC class II cell surface receptor (IEDB MHC-DR) restricted peptide-binding dataset. In particular, MHC class II prediction models need to consider longer or even variable peptide lengths as their inputs.

The identification of tumor neoantigens involves the elution of MHC epitopes from tumor cells and the extraction of somatic mutations from sequencing data. Machine learning algorithms are then processed to model the binding affinity between mutant peptides and MHC proteins, allowing for the prediction of candidate neoantigens. To improve performance, some models incorporate TCR sequencing data to screen for candidate neoantigens with high proportions that interact with TCR and induce T cell responses. MHC major histocompatibility complex, TCR T-cell receptor, APC antigen-presenting cell, pMHC peptide-MHC, WES whole-exome sequencing, WGS whole-genome sequencing, LC/MS liquid chromatography/mass spectrometry.
Typically, identifying binding affinity between MHC and peptides alone is insufficient for accurate neoantigen predictions with high confidence. To overcome the limitation, some studies have focused on assessing the immunogenicity of the predicted binding molecules115,116,117,118. Immunogenicity refers to the ability of protein products to provoke an immune response, and it depends on several factors, including protein expression, peptide-MHC binding affinity and stability, peptide competition for MHC binding, and more94,119. Among these models, DeepHLApan117 designed a multi-task neural network model consisting of three layers of bidirectional Gated Recurrent Unit (BiGRU) with an attention layer. This model can simultaneously predict the binding affinity and the immunogenicity. Similar to DeepHLApan, DeepNetBim116 used a CNN model with an attention layer to predict binding affinity and binary immunogenic categories. In comparison to DeepHLApan, DeepNetBim incorporated an additional layer to merge the two independent outputs together, namely the binding affinity and the binary immunogenic category, in order to calibrate the final immunogenicity prediction. Seq2Neo118 took a different approach by developing an end-to-end software that directly utilize raw sequencing data (WES/WGS, RNA) in FASTQ, SAM and BAM formats to predict the immunogenicity directly through a CNN-based model. In contrast, Charoentong et al.120 did not focus on developing a state-of-the-art DL model like most approaches. Instead, they designed a comprehensive biomarker consisting of 127 features, including somatic mutation features, class-I and class-II MHCs, immune inhibitory and stimulatory genes, adaptive immunity cells and innate immunity cells from integrated WES, RNA-seq and clinical data. Their results demonstrated that proper feature extraction could achieve a high accuracy for tumor immunogenicity prediction using only an RF classifier. In addition to assessing the immunogenicity of the predicted binding molecules, some studies have explored the integration of TCR sequence to predict the likelihood of peptide-TCR interaction for neoantigen prediction. Besser et al.121 proposed using CD8 + T cell responses as the task of their models to detect neoantigens. To accomplish this, they employed an additional step in their ML models, training them on the Tantigen dataset122, a comprehensive database of tumor T cell antigens. Through this step, they were able to learn the changes in key parameters and features associated with T cell response, enabling them to predict whether a given MHC class I peptide was positive for inducing CD8 + T cell response. Likewise, iTTCA-Hybrid123 utilized the tumor T cell antigen dataset from Tantigen122 and non-tumor T cell antigen dataset from IEDB98 to train an ensemble model capable of classifying tumor and non-tumor T cell antigens. More recently, DLpTCR124 and pMTnet125 suggested that assessing the propensity of CD8 + TCR to recognize the pMHC complex is crucial for neoantigen prediction, as most in silico predicted antigen peptides fail to elicit immune responses in vivo. Both models take peptide and TCR sequences as input data, and their output is a binary classification of whether the TCR-pMHC has an interaction. To achieve better performance, DLpTCR124 designed an ensemble strategy based on three deep learning models: FCNN, LeNet-5 and ResNet. On the other hand, pMTnet125 utilized an autoencoder and an LSTM network to obtain the hidden encoding of the TCR sequence and peptide sequence, respectively. These encodings were then fed into an FCNN classifier for final prediction.
It is worth noting that peptide sequence encoding plays a crucial role in neoantigen prediction. Two commonly employed methods for encoding are one-hot encoding and BLOcks SUbstitution Matrix (BLOSUM) encoding (Table 3). Among them, BLOSUM is more prevalent as it offers insights into the homologies between protein sequences. In addition, personalized sequencing encoding techniques utilizing ML algorithms have also gained popularity. These include byte pair encoding103, skip-gram encoding104, principal component analysis (PCA) encoding124 and physicochemical properties (PCP) encoding124.
In conclusion, ML has emerged as a promising approach for evaluating TME, identifying TME related biomarkers and unraveling the intricate relationship between TME and immunotherapy. The biomarkers derived from ML approaches hold great potential for predicting clinical outcomes of immunotherapy and enhancing personalized immunotherapy strategies, thereby facilitating the advancement and wider application of immunotherapy in cancer treatment.
Challenges and opportunities
Despite the extensive application of ML in immunotherapy studies, several challenges remain to be addressed. These challenges pertain to gaining a mechanistic understanding of how immunotherapies target and eradicate tumor cells126 and the neoantigens that can be recognized by immune cells127. Whether and how ML models prompt the progression of immunotherapy will depend on how these challenges, as discussed below, are met in the future.
Insufficient amount of available data
Immunotherapy has emerged as a promising cancer treatment, driving numerous clinical trials worldwide128. Nevertheless, current clinical trials have primarily focused on PD-1/PD-L1 therapy, result in limited data for other treatment like CTLA-4 and CAR T therapy (Table 1). This data scarcity poses a significant barrier for developing ML models, particularly DL models that require substantial training data to avoid overfitting and enhance model performance129. To mitigate the limitations, the generation of pseudo databases has emerged as a potential solution. State-of-the-art generative models, such as generative adversarial network (GAN)130 and diffusion models131, have shown promise in computer vision and can generate synthetic data to supplement training datasets, mitigating overfitting issues. Likewise, Sové et al.132 developed a model using an ML approach to capture interpatient diversity in clinical trials, allowing the simulation of virtual patients. By leveraging these virtual patients, it becomes possible to mimic a virtual clinical trial scenario to quantitatively assess the efficacy of ICI treatments in a controlled environment.
Multi-omics data integration and analysis
The advent of multi-omics technologies has revolutionized our understanding of the biological mechanisms of driving immunotherapy. However, analyzing these large multi-omics data, particularly those from single-cell-based133 and spatial-based134 technologies, has brought new computational challenges. One challenge is the batch effects, resulting from diverse platforms used for data generation. To ensure accurate downstream analyses, removing platform-specific noise is crucial. Recently, ML models, particularly joint dimension reduction algorithms such as negative matrix factorization (NMF), PCA, singular value decomposition (SVD), canonical correlation analysis (CCA), have emerged as powerful tools for encoding data from diverse platforms into a shared latent space, thereby enabling effective batch effect removal135. Additionally, the training data often exhibit distinct statistical modalities. To tackle this challenge, multimodal learning with specialized modelling strategies has gained attention for integrating diverse data modalities, such as medical imaging and genomics41,47. By harnessing the strengths of multiple modalities, multimodal learning models offer the potential to address immunotherapy-related questions.
Meta-analysis
In the field of immunotherapy response prediction, the definitions of “response” vary across studies. For example, Vanguri et al.47 and Chowell et al.42 employed Response Evaluation Criteria in Solid Tumors (RECIST)136 as their criterion for defining response, whereas Filipski et al.38 utilized survival (defined as the time from start of ICI treatment to date of decease) to characterize response. The disparate use of these distinct criteria underscores the considerable variability in how the concept of “response” is operationalized across studies, posing a challenge to the synthesis of studies and the establishment of a standardized framework for meta-analysis. Standardization the definition and harmonization data are necessary to achieve a consensus on common criteria or thresholds for defining immunotherapy response.
Neoantigen prediction
With ongoing developments of new algorithms, the field of cancer neoantigen identification holds promise for immunotherapies94. Given the uniqueness of the neoantigen landscape to each individual, the accurate targeting of neoantigens establishes a solid foundation for conducting systematic studies in precision medicine and providing clinical decision support for cancer immunotherapy. Computational models, especially ML algorithms, are commonly used for immunogenic neoantigen prediction. However, comparative studies have revealed that, thus far, none of the existing studies have achieved accurate identification of immunogenic neoantigens127. Factors such as tumor heterogeneity, diversity within the TCR repertoire, and the absence of true labeled data contribute to this inaccuracy. Future studies should focus on developing more comprehensive models integrating both pMHCs and TCR sequencing data to improve predictive performance of neoantigen identification. It is worth noting that certain studies have explored targeting tumor-specific gene fusion137 and MHC gene loss of heterozygosity (LOH)138 to improve immune recognition in neoantigen identification. Incorporating these factors could augment neoantigen predictions and contribute to higher accuracy in future studies.
Model generalizability and interpretability
While numerous ML models have been developed for immunotherapy response prediction, they often struggle to adapt well to unseen data. Their performance on new data is often moderate or deficient, indicating a lack of generalizability. Moreover, these models typically employ ML or statistical approaches to select marker genes. However, the selected marker genes vary between studies and may have limited effectiveness within specific datasets. To address these challenges, recent studies have employed transfer learning algorithms for immunotherapy response prediction. By leveraging pre-trained models and applying them to train on new, similar datasets39, this approach can enhance the efficiency and robustness139. In addition, the interpretability of ML models in immunotherapy remains a persistent concern, ML algorithms often function as black boxes, making it difficult to understand the decision-making process and the underlying biological rationale behind their predictions. To improve the generalizability, researchers are exploring feature insights and interactions through explainable AI (XAI) models140. XAI approaches can provide global and local explanations, enabling a deeper understanding of predictions and facilitating effective fine-tuning on new data.
Models in handling continual incremental datasets with real-time adaptation
In our studies we reviewed, almost all models applied for immunotherapy analyses are traditional batch learning approaches. These methods utilized entire datasets simultaneously for training, deploying the trained model for inference without frequent updates. However, they usually encounter high retraining cost when adapting to new training data141. With the growing of clinical and genomics data during the patient treatment, there is a need to develop models with the capacity to conduct incremental datasets and adapt in real-time to new information. Online learning emerges as a scalable and efficient approach that learn to continuously updates the model based on feedback on its decisions in the form of a sequence of examples141,142,143, demonstrating premium performance in clinical applications144. This approach holds the potential to significantly assist clinicians via providing diagnoses or making management decisions.
Clinical translation
While numerous ML models have been developed for predicting immunotherapy outcomes, our review reveals that almost none of these models have undergone clinical testing. Furthermore, contemporary ML-based clinical decision support systems, such as IBM Watson Health145 and Google DeepMind Health146, encounter obstacles hindering the smooth transition of models from research settings to standard clinical practice. This discrepancy underscores the critical necessity for rigorous clinical validation to evaluate the real-world efficacy and reliability of these predictive models. The complexity of the immune system, the dynamic nature of immunological responses, the lack of data quality and standardization, and the absence of highly reliable biomarkers all contribute to the challenges impacting the performance of these models. Conducting comprehensive clinical trials and validation studies is crucial to bridging the gap between theoretical concepts and practical applications in the field of immunotherapy.
Opportunities
Despite the limited number of databases, there are still a growing number of resources available for immunotherapy research. The Cancer Genome Atlas (TCGA)147 is a prevalent curated database containing genomic, epigenomic, transcriptomic, proteomic and whole slide imaging data across 33 cancer types. Among them, a significant number of patients were treated with immunotherapy, and these samples have been widely used in training ML models as reviewed in this study. In addition, the medical images (MRI, CT, digital histopathology, etc.) of some of these patients can be downloaded from The Cancer Imaging Archive (TCIA)148 database, enabling the multi-modality analysis of immunotherapy studies. Tumor Immunotherapy Gene Expression Resource (TIGER)149 and ICBatlas150 are comprehensive resources for integrative analysis of the transcriptome profiles related to tumor immunology. The Cancer Immunome Atlas120 is a web-accessible database that characterizes the intratumoral immune landscapes and the cancer antigenomes of 20 solid cancers. This database has also developed an immunophenoscore to quantify tumor immunogenicity from genomic features, which helps inform cancer immunotherapy and facilitate the development of precision immuno-oncology. To ensure safe cancer treatment, Wang et al.151 developed an irAE data resource consisting of a total of 893 irAEs. They also performed comparative analyses on these irAEs, making it more intuitive to identify and understand how off-targets of ICIs are involved in irAEs. In addition to clinical resources, there are datasets available for other immunotherapy-related collections. IEDB98 and Tantigen122 provide a comprehensive set of data related to antibody, B and T cell epitopes for humans, along with tools to assist in the prediction and analysis of neoantigens for immunotherapy. In summary, these resources and databases have facilitated the generation of new research tools, diagnostic techniques, vaccines and therapeutics that were previously used in immunotherapy studies.
Conclusions
Immunotherapy holds promise for cancer treatment, but the rapid accumulation of immunotherapy-related data has raised challenges. This review summarizes the use of ML approaches in addressing these challenges. Conventional ML algorithms (LR, RF, SVM, LASSO, XGBoost) have demonstrated their versatility in handling various omics datasets, including mutations, CNVs, methylation profiles, and expression profiles, to predict immunotherapy responses. ML models also analyze TME to identify biomarkers and subcohorts with distinct immunotherapy responses. Unsupervised clustering algorithms are typically utilized for subcohort identification, while LASSO regression is employed to identify subcohort biomarkers. Notably, DL approaches are extensively implemented for handling the sequencing data in neoantigen prediction. Natural language processing-related models, including word-to-vector models, are broadly used for sequence encoding, whereas recurrent neural networks-based models or transformers are commonly utilized for task training. Moreover, we highlight the prevailing challenges, emphasizing the need for ML models to handle multi-modal data to facilitate the rapid accumulation of imaging and omics data. Ultimately, this review aims to inspire cutting-edge ML research in maximizing the potential of immunotherapies.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors hereby declare that all pertinent data has already been displayed within the article. Additional data can be accessed upon request to the corresponding author.
References
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Rabinovich, G. A., Gabrilovich, D. & Sotomayor, E. M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 25, 267–296 (2007).
Drake, C. G., Lipson, E. J. & Brahmer, J. R. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 11, 24–37 (2014).
Schneider, B. J. et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J. Clin. Oncol. 39, 4073–4126 (2021).
Simoni, Y. et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Garon, E. B. et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med 372, 2018–2028 (2015).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996).
Overman, M. J. et al. Nivolumab in patients with DNA mismatch repair deficient/microsatellite instability high metastatic colorectal cancer: Update from CheckMate 142. J. Clin. Oncol. 35 (2017).
Kourou, K., Exarchos, T. P., Exarchos, K. P., Karamouzis, M. V. & Fotiadis, D. I. Machine learning applications in cancer prognosis and prediction. Comput Struct. Biotechnol. J. 13, 8–17 (2015).
Li, Y. & Luo, Y. Performance-weighted-voting model: An ensemble machine learning method for cancer type classification using whole-exome sequencing mutation. Quant. Biol. 8, 347–358 (2020).
Ye, Z., Zeng, D., Zhou, R., Shi, M. & Liao, W. Tumor microenvironment evaluation for gastrointestinal cancer in the era of immunotherapy and machine learning. Front Immunol. 13, 819807 (2022).
Li, Y., Wu, X., Yang, P., Jiang, G. & Luo, Y. Machine learning for lung cancer diagnosis, treatment, and prognosis. Genom. Proteom. Bioinforma. 20, 850–866 (2022).
Lang, F., Schrors, B., Lower, M., Tureci, O. & Sahin, U. Identification of neoantigens for individualized therapeutic cancer vaccines. Nat. Rev. Drug Discov. 21, 261–282 (2022).
Yang, Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest 125, 3335–3337 (2015).
Spencer, K. R. et al. Biomarkers for immunotherapy: current developments and challenges. Am. Soc. Clin. Oncol. Educ. Book 35, e493–e503 (2016).
Ling, S. et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl Acad. Sci. USA 112, E6496–E6505 (2015).
Zhang, Y. et al. Genetic load and potential mutational meltdown in cancer cell populations. Mol. Biol. Evol. 36, 541–552 (2019).
Li, G. et al. Evolution under spatially heterogeneous selection in solid tumors. Mol. Biol. Evol. 39, msab335 (2022).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007).
Peng, J. et al. Deep learning to estimate durable clinical benefit and prognosis from patients with non-small cell lung cancer treated with PD-1/PD-L1 blockade. Front Immunol. 13, 960459 (2022).
Wang, Y. J. P., Chen, L., Ju, L. G., Xiao, Y. & Wang, X. H. Tumor mutational burden related classifier is predictive of response to PD-L1 blockade in locally advanced and metastatic urothelial carcinoma. Int. Immunopharmacol. 87, 106818 (2020).
Lapuente-Santana, O., van Genderen, M., Hilbers, P. A. J., Finotello, F. & Eduati, F. Interpretable systems biomarkers predict response to immune-checkpoint inhibitors. Patterns (N. Y) 2, 100293 (2021).
Lu, Z. et al. Prediction of immune checkpoint inhibition with immune oncology-related gene expression in gastrointestinal cancer using a machine learning classifier. J. Immunother. Cancer 8, e000631 (2020).
Polano, M. et al. A pan-cancer approach to predict responsiveness to immune checkpoint inhibitors by machine learning. Cancers (Basel) 11, 1562 (2019).
Ahmed, Y. B., Al-Bzour, A. N., Ababneh, O. E., Abushukair, H. M. & Saeed, A. Genomic and transcriptomic predictors of response to immune checkpoint inhibitors in melanoma patients: a machine learning approach. Cancers (Basel) 14, 5605 (2022).
Jin, W. et al. Ensemble deep learning enhanced with self-attention for predicting immunotherapeutic responses to cancers. Front. Immunol. 13, 1025330 (2022).
Zeng, Z. et al. Machine learning on syngeneic mouse tumor profiles to model clinical immunotherapy response. Sci. Adv. 8, eabm8564 (2022).
Wiesweg, M. et al. Machine learning reveals a PD-L1-independent prediction of response to immunotherapy of non-small cell lung cancer by gene expression context. Eur. J. Cancer 140, 76–85 (2020).
Banchereau, R. et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat. Commun. 12, 3969 (2021).
Luo, Z. et al. Development of a metastasis-related immune prognostic model of metastatic colorectal cancer and its usefulness to immunotherapy. Front Cell Dev. Biol. 8, 577125 (2020).
Liu, R., Dollinger, E. & Nie, Q. Machine learning of single cell transcriptomic data from anti-PD-1 responders and non-responders reveals distinct resistance mechanisms in skin cancers and PDAC. Front Genet 12, 806457 (2021).
Kang, Y., Vijay, S. & Gujral, T. S. Deep neural network modeling identifies biomarkers of response to immune-checkpoint therapy. iScience 25, 104228 (2022).
Rodin, A. S. et al. Dissecting response to cancer immunotherapy by applying bayesian network analysis to flow cytometry data. Int J. Mol. Sci. 22, 2316 (2021).
Zhang, Y. et al. Machine learning-based exceptional response prediction of nivolumab monotherapy with circulating microRNAs in non-small cell lung cancer. Lung Cancer 173, 107–115 (2022).
Bustos, M. A. et al. A pilot study comparing the efficacy of lactate dehydrogenase levels versus circulating cell-free microRNAs in monitoring responses to checkpoint inhibitor immunotherapy in metastatic melanoma patients. Cancers (Basel) 12, 3361 (2020).
Shang, S. et al. MeImmS: predict clinical benefit of Anti-PD-1/PD-L1 treatments based on DNA methylation in non-small cell lung cancer. Front Genet 12, 676449 (2021).
Filipski, K. et al. DNA methylation-based prediction of response to immune checkpoint inhibition in metastatic melanoma. J. Immunother. Cancer 9, e002226 (2021).
Przedborski, M., Smalley, M., Thiyagarajan, S., Goldman, A. & Kohandel, M. Systems biology informed neural networks (SBINN) predict response and novel combinations for PD-1 checkpoint blockade. Commun. Biol. 4, 877 (2021).
Paidi, S. K. et al. Raman spectroscopy and machine learning reveals early tumor microenvironmental changes induced by immunotherapy. Cancer Res. 81, 5745–5755 (2021).
Sidhom, J. W. et al. Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy. Sci. Adv. 8, eabq5089 (2022).
Chowell, D. et al. Improved prediction of immune checkpoint blockade efficacy across multiple cancer types. Nat. Biotechnol. 40, 499–506 (2022).
Zhang, Z. et al. Integrated analysis of single-cell and bulk RNA sequencing data reveals a pan-cancer stemness signature predicting immunotherapy response. Genome Med. 14, 45 (2022).
Hwang, M. et al. Peripheral blood immune cell dynamics reflect antitumor immune responses and predict clinical response to immunotherapy. J. Immunother. Cancer 10, e004688 (2022).
Zheng, K., Gao, L., Hao, J., Zou, X. & Hu, X. An immunotherapy response prediction model derived from proliferative CD4(+) T cells and antigen-presenting monocytes in ccRCC. Front. Immunol. 13, 972227 (2022).
Kong, J. et al. Network-based machine learning approach to predict immunotherapy response in cancer patients. Nat. Commun. 13, 3703 (2022).
Vanguri, R. S. et al. Multimodal integration of radiology, pathology and genomics for prediction of response to PD-(L)1 blockade in patients with non-small cell lung cancer. Nat. Cancer 3, 1151–1164 (2022).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83 (2017).
Civelek, M. & Lusis, A. J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet 15, 34–48 (2014).
Li, X. et al. Precision combination therapies based on recurrent oncogenic coalterations. Cancer Discov. 12, 1542–1559 (2022).
Daniels, K. G. et al. Decoding CAR T cell phenotype using combinatorial signaling motif libraries and machine learning. Science 378, 1194–1200 (2022).
Anderson, N. M. & Simon, M. C. The tumor microenvironment. Curr. Biol. 30, R921–R925 (2020).
Whiteside, T. L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912 (2008).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018).
Crawford, Y. et al. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 15, 21–34 (2009).
Murgai, M. et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med 23, 1176–1190 (2017).
Paiva, A. E. et al. Pericytes in the premetastatic niche. Cancer Res. 78, 2779–2786 (2018).
Li, Y. & Luo, Y. Optimizing the evaluation of gene-targeted panels for tumor mutational burden estimation. Sci. Rep. 11, 21072 (2021).
Zhou, T. et al. MSIFinder: a python package for detecting MSI status using random forest classifier. BMC Bioinforma. 22, 185 (2021).
Lu, M. et al. A genomic signature for accurate classification and prediction of clinical outcomes in cancer patients treated with immune checkpoint blockade immunotherapy. Sci. Rep. 10, 20575 (2020).
Kather, J. N. et al. Deep learning can predict microsatellite instability directly from histology in gastrointestinal cancer. Nat. Med. 25, 1054 (2019).
Yamashita, R. et al. Deep learning model for the prediction of microsatellite instability in colorectal cancer: a diagnostic study. Lancet Oncol. 22, 132–141 (2021).
Niu, Y. et al. Predicting tumor mutational burden from lung adenocarcinoma histopathological images using deep learning. Front Oncol. 12, 927426 (2022).
Zhang, H. et al. in 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 920–925, https://ieeexplore.ieee.org/document/8983139 (2019).
Chang, J. C. Cancer stem cells role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 95, S20–S25 (2016).
Chen, K., Huang, Y. H. & Chen, J. L. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharm. Sin. 34, 732–740 (2013).
Wei, C. et al. Characterization of gastric cancer stem-like molecular features, immune and pharmacogenomic landscapes. Brief. Bioinform. 23, bbab386 (2022).
Wang, Z. et al. Machine learning revealed stemness features and a novel stemness-based classification with appealing implications in discriminating the prognosis, immunotherapy and temozolomide responses of 906 glioblastoma patients. Brief. Bioinform. 22, bbab032 (2021).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Wang, M. et al. Identification of cancer-associated fibroblast subtype of triple-negative breast cancer. J. Oncol. 2022, 6452636 (2022).
Tian, L. et al. A cancer associated fibroblasts-related six-gene panel for Anti-PD-1 therapy in melanoma driven by weighted correlation network analysis and supervised machine learning. Front. Med. (Lausanne) 9, 880326 (2022).
Paijens, S. T., Vledder, A., de Bruyn, M. & Nijman, H. W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 18, 842–859 (2021).
Stanton, S. E. & Disis, M. L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 4, 59 (2016).
Gentles, A. J. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med 21, 938–945 (2015).
Syn, N. L., Teng, M. W. L., Mok, T. S. K. & Soo, R. A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 18, e731–e741 (2017).
Bao, X., Shi, R., Zhao, T. & Wang, Y. Immune landscape and a novel immunotherapy-related gene signature associated with clinical outcome in early-stage lung adenocarcinoma. J. Mol. Med (Berl.) 98, 805–818 (2020).
Ma, J., Jin, Y., Tang, Y. & Li, L. DeepTI: A deep learning-based framework decoding tumor-immune interactions for precision immunotherapy in oncology. SLAS Discov. 27, 121–127 (2022).
Zhang, H. et al. Machine learning-based tumor-infiltrating immune cell-associated lncRNAs for predicting prognosis and immunotherapy response in patients with glioblastoma. Brief. Bioinform. 23, bbac386 (2022).
Zhang, N. et al. Machine learning-based identification of tumor-infiltrating immune cell-associated lncRNAs for improving outcomes and immunotherapy responses in patients with low-grade glioma. Theranostics 12, 5931–5948 (2022).
Arra, A. et al. Immune-checkpoint blockade of CTLA-4 (CD152) in antigen-specific human T-cell responses differs profoundly between neonates, children, and adults. Oncoimmunology 10, 1938475 (2021).
Failmezger, H., Zwing, N., Tresch, A., Korski, K. & Schmich, F. Computational tumor infiltration phenotypes enable the spatial and genomic analysis of immune infiltration in colorectal cancer. Front Oncol. 11, 552331 (2021).
Reiman, D. et al. Integrating RNA expression and visual features for immune infiltrate prediction. Pac. Symp. Biocomput 24, 284–295 (2019).
Pan, X. et al. Epigenome signature as an immunophenotype indicator prompts durable clinical immunotherapy benefits in lung adenocarcinoma. Brief. Bioinforma. 23, bbab481 (2022).
Fernandez, E. A. et al. Unveiling the immune infiltrate modulation in cancer and response to immunotherapy by MIXTURE-an enhanced deconvolution method. Brief. Bioinform. 22, bbaa317 (2021).
Park, C. et al. Tumor immune profiles noninvasively estimated by FDG PET with deep learning correlate with immunotherapy response in lung adenocarcinoma. Theranostics 10, 10838–10848 (2020).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Vander Heiden, M. G. & DeBerardinis, R. J. Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 (2017).
Li, X. et al. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 16, 425–441 (2019).
Bader, J. E., Voss, K. & Rathmell, J. C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 78, 1019–1033 (2020).
Liberti, M. V. & Locasale, J. W. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 41, 211–218 (2016).
Gu, X., Wei, S., Li, Z. & Xu, H. Machine learning reveals two heterogeneous subtypes to assist immune therapy based on lipid metabolism in lung adenocarcinoma. Front Immunol. 13, 1022149 (2022).
Yarchoan, M., Johnson, B. A. 3rd, Lutz, E. R., Laheru, D. A. & Jaffee, E. M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 569 (2017).
De Mattos-Arruda, L. et al. Neoantigen prediction and computational perspectives towards clinical benefit: recommendations from the ESMO precision medicine working group. Ann. Oncol. 31, 978–990 (2020).
Carreno, B. M. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
The problem with neoantigen prediction. Nat. Biotechnol. 35, 97 (2017).
Vita, R. et al. The immune epitope database 2.0. Nucleic Acids Res 38, D854–D862 (2010).
Andreatta, M. & Nielsen, M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32, 511–517 (2016).
Jurtz, V. et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 199, 3360–3368 (2017).
O’Donnell, T. J. et al. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. 7, 129–132.e124 (2018).
Bulik-Sullivan, B. et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 37, 55–63 (2019).
Wang, F. X. et al. MHCRoBERTa: pan-specific peptide-MHC class I binding prediction through transfer learning with label-agnostic protein sequences. Brief. Bioinforma. 23, bbab595 (2022).
Vang, Y. S. & Xie, X. HLA class I binding prediction via convolutional neural networks. Bioinformatics 33, 2658–2665 (2017).
Boehm, K. M., Bhinder, B., Raja, V. J., Dephoure, N. & Elemento, O. Predicting peptide presentation by major histocompatibility complex class I: an improved machine learning approach to the immunopeptidome. BMC Bioinforma. 20, 7 (2019).
Mei, S. T. et al. Anthem: a user customised tool for fast and accurate prediction of binding between peptides and HLA class I molecules. Brief. Bioinforma. 22, bbaa415 (2021).
Gartner, J. J. et al. A machine learning model for ranking candidate HLA class I neoantigens based on known neoepitopes from multiple human tumor types. Nat. Cancer 2, 563–574 (2021).
Yewdell, J. W. & Bennink, J. R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 17, 51–88 (1999).
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A. & Stevanovic, S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219 (1999).
Nielsen, M. & Lund, O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinforma. 10, 296 (2009).
Racle, J. et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 37, 1283–1286 (2019).
Abelin, J. G. et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity 51, 766–779.e717 (2019).
Jensen, K. K. et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406 (2018).
Chen, B. B. et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 37, 1332 (2019).
Kim, S. et al. Neopepsee: accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immunogenicity information. Ann. Oncol. 29, 1030–1036 (2018).
Yang, X., Zhao, L., Wei, F. & Li, J. DeepNetBim: deep learning model for predicting HLA-epitope interactions based on network analysis by harnessing binding and immunogenicity information. BMC Bioinforma. 22, 231 (2021).
Wu, J. C. et al. DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-Peptide binding and immunogenicity. Front. Immunol. 10, 2559 (2019).
Diao, K. et al. Seq2Neo: a comprehensive pipeline for cancer neoantigen immunogenicity prediction. Int J. Mol. Sci. 23, 11624 (2022).
Schumacher, T. N., Scheper, W. & Kvistborg, P. Cancer neoantigens. Annu Rev. Immunol. 37, 173–200 (2019).
Charoentong, P. et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017).
Besser, H., Yunger, S., Merhavi-Shoham, E., Cohen, C. J. & Louzoun, Y. Level of neo-epitope predecessor and mutation type determine T cell activation of MHC binding peptides. J. Immunother. Cancer 7, 135 (2019).
Olsen, L. R. et al. TANTIGEN: a comprehensive database of tumor T cell antigens. Cancer Immunol. Immunother. 66, 731–735 (2017).
Charoenkwan, P., Nantasenamat, C., Hasan, M. M. & Shoombuatong, W. iTTCA-Hybrid: improved and robust identification of tumor T cell antigens by utilizing hybrid feature representation. Anal. Biochem. 599, 113747 (2020).
Xu, Z. et al. DLpTCR: an ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief. Bioinform. 22, bbab335 (2021).
Lu, T. et al. Deep learning-based prediction of the T cell receptor-antigen binding specificity. Nat. Mach. Intell. 3, 864–875 (2021).
Chen, I., Chen, M. Y., Goedegebuure, S. P. & Gillanders, W. E. Challenges targeting cancer neoantigens in 2021: a systematic literature review. Expert Rev. Vaccines 20, 827–837 (2021).
Buckley, P. R. et al. Evaluating performance of existing computational models in predicting CD8+ T cell pathogenic epitopes and cancer neoantigens. Brief. Bioinform 23, bbac141 (2022).
Egen, J. G., Ouyang, W. & Wu, L. C. Human anti-tumor immunity: insights from immunotherapy clinical trials. Immunity 52, 36–54 (2020).
Rajkomar, A., Dean, J. & Kohane, I. Machine learning in medicine. N. Engl. J. Med 380, 1347–1358 (2019).
Goodfellow, I. et al. Generative adversarial networks. Commun. Acm 63, 139–144 (2020).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. Adv. Neural Inf. Process. Syst. 33, 6840–6851 (2020).
Sove, R. J. et al. Virtual clinical trials of anti-PD-1 and anti-CTLA-4 immunotherapy in advanced hepatocellular carcinoma using a quantitative systems pharmacology model. J. Immunother. Cancer 10, e005414 (2022).
Lee, J., Hyeon, D. Y. & Hwang, D. Single-cell multiomics: technologies and data analysis methods. Exp. Mol. Med 52, 1428–1442 (2020).
Rao, A., Barkley, D., Franca, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 596, 211–220 (2021).
Zeng, Z., Li, Y., Li, Y. & Luo, Y. Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 23, 83 (2022).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Weber, D. et al. Accurate detection of tumor-specific gene fusions reveals strongly immunogenic personal neo-antigens. Nat. Biotechnol. 40, 1276–1284 (2022).
Pyke, R. M. et al. A machine learning algorithm with subclonal sensitivity reveals widespread pan-cancer human leukocyte antigen loss of heterozygosity. Nat. Commun. 13, 1925 (2022).
Weiss, K., Khoshgoftaar, T. M. & Wang, D. A survey of transfer learning. J. Big Data 3, 1–40 (2016).
Arrieta, A. B. et al. Explainable artificial intelligence (XAI): concepts, taxonomies, opportunities and challenges toward responsible AI. Inf. Fusion 58, 82–115 (2020).
Hoi, S. C. H., Sahoo, D., Lu, J. & Zhao, P. L. Online learning: a comprehensive survey. Neurocomputing 459, 249–289 (2021).
Crammer, K., Dekel, O., Keshet, J., Shalev-Shwartz, S. & Singer, Y. Online passive-aggressive algorithms. J. Mach. Learn Res. 7, 551–585 (2006).
Parisi, G. I., Kemker, R., Part, J. L., Kanan, C. & Wermter, S. Continual lifelong learning with neural networks: a review. Neural Netw. 113, 54–71 (2019).
Lee, C. S. & Lee, A. Y. Clinical applications of continual learning machine learning. Lancet Digit Health 2, e279–e281 (2020).
Chen, Y., Argentinis, E. & Weber, G. IBM Watson: how cognitive computing can be applied to big data challenges in life sciences research. Clin. Ther. 38, 688–701 (2016).
Mesko, B. The role of artificial intelligence in precision medicine. Expert Rev. Precis Me 2, 239–241 (2017).
Cancer Genome Atlas Research, N. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet 45, 1113–1120 (2013).
Clark, K. et al. The Cancer Imaging Archive (TCIA): maintaining and operating a public information repository. J. Digit Imaging 26, 1045–1057 (2013).
Chen, Z. et al. TIGER: A Web Portal of Tumor Immunotherapy Gene Expression Resource. Genomics Proteomics Bioinformatics (2022).
Yang, M. et al. ICBatlas: a comprehensive resource for depicting immune checkpoint blockade therapy characteristics from transcriptome profiles. Cancer Immunol. Res. 10, 1398–1406 (2022).
Wang, Q. & Xu, R. Immunotherapy-related adverse events (irAEs): extraction from FDA drug labels and comparative analysis. JAMIA Open 2, 173–178 (2019).
Acknowledgements
This study is supported in part by National Institutes of Health of USA (Grant No. U01TR003528 and 1R01LM013337) awarded to Y.Luo and (Grant No. R01CA257520 and R01CA232347) to D.F.
Author information
Authors and Affiliations
Contributions
Y.Li conceived and designed the research, collected the data, drafted the manuscript and contributed to the figures. X.W. collected the data, drafted the manuscript and contributed to the figures. D.F. conceived and designed the research and provided critical revision. Y.Luo conceived and designed the research, and drafted the manuscript. All authors have read, edited and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Wu, X., Fang, D. et al. Informing immunotherapy with multi-omics driven machine learning. npj Digit. Med. 7, 67 (2024). https://doi.org/10.1038/s41746-024-01043-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41746-024-01043-6
