
Abstract
Fermi-surface (FS) nesting originating from Peierls’ idea of electronic instabilities in one-dimensional materials is a key concept to stabilize charge-density wave (CDW), whereas its applicability to two-dimensional (2D) materials is under intensive debate. Here we report unusual CDW associated with the higher-order FS nesting in monolayer 2D VS2. Angle-resolved photoemission spectroscopy and scanning tunneling microscopy uncovered stripe CDW with \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) periodicity together with an energy-gap opening on the entire FS. We suggest that this CDW involves the higher-order FS-nesting vector 2q twice larger than that of the normal one (q), as supported by the anomalies in the calculated phonon dispersion and electronic susceptibility. The present results suggest that the cooperation of q and 2q nesting leads to the fully gapped CDW state unlike the case of conventional single q nesting which produces a partial gap, pointing to an intriguing mechanism of CDW transition.
Similar content being viewed by others
Introduction
The discovery of Dirac fermions in graphene has opened a new avenue to explore emergent quantum phenomena in two-dimensional (2D) materials1,2,3,4,5. Atomically thin transition-metal dichalcogenides (TMDs) offer a great opportunity to widen the functionality of 2D materials due to the tunability of band structure associated with the material variety, which has already manifested in the bulk counterparts as the emergence of rich electronic phases such as insulating, metallic, and superconducting phases. Besides the band tunability, two-dimensionalization of bulk TMDs further leads to unique quantum phenomena such as spin-valley Hall effect2, Ising superconductivity4, and robust Mott-insulator phase5, wherein the control of quantum confinement and interlayer interaction plays an essential role.
The most intensively studied quantum phenomenon in bulk and monolayer TMDs is the charge-density wave (CDW). The CDW in TMDs has attracted tremendous attention because it provides an ideal platform to access a long-standing issue on the applicability of the Fermi-surface (FS)-nesting scenario, where the shift of FS with respect to the original one by the CDW wave vector causes the energy gain by the gap opening at the Fermi level (EF). There is growing evidence that many of the CDWs observed in TMDs cannot be explained in terms of the simple FS nesting, and more exotic mechanisms (e.g., exciton formation, van-Hove band singularity, and k-dependent electron–phonon coupling) may be at work in TMDs6,7,8,9.
Among TMDs, vanadium dichalcogenides VXc2 (Xc = S, Se, and Te) are useful for investigating the interplay between FS nesting and CDW because this family of materials commonly undergoes CDW. Bulk 1T-VSe2 shows CDW with a 4 × 4 × ~3 periodicity triggered by the simple FS nesting10,11,12, whereas the monolayer counterpart exhibits CDW with various periodicities (e.g., 4 × 4, 4 × 1, \(\sqrt 7 \times \sqrt 3\), \(2 \times \sqrt 3\), \(4 \times \sqrt 3\))12,13,14,15,16,17,18,19,20. The situation is similar in 1T-VTe2 where the bulk exhibits a 3 × 1 × 3 CDW21 while the monolayer shows various CDWs with, e.g., 4 × 4, 4 × 1, 5 × 1, or \(2\sqrt 3 \times 2\sqrt 3\) periodicity22,23,24,25,26,27. This suggests that the simple FS-nesting scenario may be insufficient to account for a variety of CDW periodicities6.
To access this question, we have deliberately chosen monolayer VS2 because the comprehensive understanding of CDW of the VXc2 family may be essential to obtain insights into this problem. However, the nature of CDW in monolayer VS2 has not been well investigated, partially because of the technical difficulty in employing the sample-fabrication method so far adopted for VSe2 and VTe213,14,15,16,17,18,19,22,23,24,25,26,27 due to the high vapor pressure of sulfur atom. In fact, previous works utilized catalytic reaction28 or MBE with FeS source29,30 instead of MBE with V and S sources. While the CDW with local \(7 \times \sqrt 3 R30^\circ\) or \(9 \times \sqrt 3 R30^\circ\) periodicity30 was observed in such monolayer films, the electronic band structure relevant to the occurrence of CDW and the mechanism of CDW are still unclear. We have overcome the difficulty in obtaining high-quality single-crystalline films by employing a unique sample-fabrication method that combines MBE and topotactic-reaction technique, as detailed later. We investigated the electronic structure of grown monolayer film by micro-focused angle-resolved photoemission spectroscopy (μ-ARPES) and scanning tunneling microscopy (STM), together with first-principles band-structure calculations.
Results
Fabrication and characterization of monolayer VS2
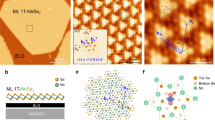
First, we explain the fabrication and characterization of monolayer VS2. We epitaxially grew monolayer VTe2 film on bilayer graphene/SiC(0001) by the MBE method (Fig. 1a) and then replaced Te atoms with S atoms by topotactic-reaction technique in which sulfurization was carried out by exposing the VTe2 film to a S atmosphere (for details, see “Methods”)31. The reflection high-energy electron diffraction (RHEED) pattern for VTe2 film (Fig. 1b) shows a sharp 1 × 1 streak pattern to confirm the monolayer nature of grown VTe2 film22 since it is known that multilayer films have a 2 × 1 periodicity32. After sulfurization, the RHEED image still maintains a sharp 1 × 1 feature (Fig. 1c). The intensity profile of the RHEED pattern in Fig. 1d reveals a slight outward shift of peak position relative to the central peak after sulfurization, corresponding to the shrinkage of lattice constant. The photoemission spectrum of the sulfurized film (Fig. 1e) signifies sharp peaks originating from the S 2p1/2 and 2p3/2 spin-orbit satellites at binding energy (EB) of 162–163 eV. Prominent Te 4d3/2 and 4d5/2 spin-orbit satellites at EB of 40–42 eV observed in monolayer VTe2 completely vanish after sulfurization (inset to Fig. 1e), indicative of the complete replacement of Te with S. STM measurements for this sulfurized film (Fig. 1f) signify monolayer islands on bilayer graphene with a step height of 0.9 nm (Fig. 1g), supporting its monolayer nature33,34 (note that the STM image shows a double-layer-like island in some areas due to the artifact associated with the shape of the tip, as reported in other monolayer TMDs35,36). These results indicate that a monolayer VS2 film with a negligible mixture of VTe2 was successfully grown on bilayer graphene/SiC(0001).

a Schematics of topotactic growth procedure for monolayer 1T-VS2 film. b, c RHEED patterns of VTe2 and VS2 films on bilayer (BL) graphene, respectively. d Direct comparison of RHEED intensity profiles between VTe2 and VS2. e Photoemission spectrum in wide EB range of monolayer VS2 measured with hν = 250 eV. Inset shows the comparison around the Te-4d core levels between VTe2 and VS2. f Constant current STM image of monolayer VS2 in the surface area of 25 × 50 nm2 measured at sample bias voltage VS = −2 V and set-point tunneling current It = 50 pA. g Height profile along a red dashed line of (f).
Electronic states of monolayer VS2
Figure 2a shows the valence-band ARPES intensity of monolayer VTe2 measured at T = 40 K along the KΓM cut of the hexagonal Brillouin zone (BZ). The band structure of monolayer VTe2 is characterized by highly dispersive holelike Te 5p bands and a weakly dispersive V 3d band near EF22. The topmost Te 5p band touches the V 3d band at the Γ point near EF. After sulfurization (Fig. 2b), the holelike bands shift downward, and a relatively flat feature appears at EB = 2.5–3.0 eV. This downward shift, which is attributed to the lower atomic energy level of the S 3p orbital relative to that of the Te 5p orbital, is better visualized in the ARPES-intensity plot near EF in Fig. 2c, d. The width of the V 3d band along the ΓM cut is enhanced after sulfurization, suggesting the increase in the transfer integral between V 3d orbitals due to the shrinkage of the lattice constant.

a, b ARPES-intensity plots of monolayer VTe2 and VS2 films, respectively, measured along the KΓΜ cut at T = 40 K. c, d Same as (a) and (b), but expanded around EF. Dashed curves in (a–d) are a guide for the eyes to trace the band dispersion. e Calculated band structure of monolayer 1T-VS2 obtained with first-principles band calculations. Note that the overall intensity profile of VS2 seems more diffuse than that of VTe2, likely due to the increase in chalcogen defects and disorders in the crystal after sulfurization and the resultant enhancement in the electron scattering.
Our first-principles band-structure calculations for free-standing monolayer 1T-VS2 capture the overall characteristics observed in the experiment. As seen from a comparison of Fig. 2b, e, the relatively flat feature at EB = 2.5–3.0 eV and the highly dispersive topmost S 3p band observed in the experiment are well reproduced in the calculation. This suggests that our monolayer VS2 film has the 1T structure. Also, the quantitative matching in the energy position of S 3p bands at the Γ point between experiments and calculations suggests a negligible charge transfer from the graphene substrate due to a weak van der Waals coupling at the interface. We also found small quantitative mismatches in the energy position and bandwidth between experiments and calculations. For example, the width of the V 3d band near EF is 0.5 eV in the experiment, whereas it is 0.75 eV in the calculation. This is likely due to the mass-renormalization effect associated with the correlation of V 3d electrons. We found no evidence for the energy splitting of V 3d bands at T = 40 K within our experimental uncertainty, consistent with the DFT calculation for the non-magnetic phase (Fig. 2e), suggesting the absence of ferromagnetism.
To clarify the electronic states near EF, we have performed ARPES measurements in 2D k-space for monolayer VS2. Figure 3a shows the contour plots at representative EB slices at T = 40 K, together with the calculated equi-energy plots (red curves) for monolayer 1T-VS2. At EB = EF, one can recognize a single bright intensity spot concentrated at the Γ point. This is in sharp contrast to the calculation that predicts a large ellipsoidal electron pocket centered at the M point. While such disagreement is seen up to EB ~ 0.2 eV, the ARPES intensity appears to follow the calculated contours at EB ≧ 0.3 eV. This can be recognized from the gradual movement of the intensity maxima from the Γ point toward the M point with increasing EB, which roughly follows the k location of the corner of ellipsoids. These results suggest that the spectral-weight suppression on the ellipsoidal pocket is associated with the energy-gap opening. To see more clearly the gap opening, we show in Fig. 3b the ARPES intensity at T = 40 K measured along representative k cuts (cuts 1–5 in Fig. 3a), which cross the ellipsoidal pocket. Along cut 5 (Fig. 3b5), which nearly passes the M points, one can see two U-shaped features corresponding to the electron bands bottomed at EB ~ 0.4 eV at each M point. These features are qualitatively reproduced by the calculation along the same k cut in Fig. 3c, although the bandwidth is narrower in the experiment due to the mass-renormalization effect. Intriguingly, although the calculation shows a clear EF-crossing of bands, the experimental dispersive feature loses its intensity around EF due to the gap opening. This suppression is also observed along the k cuts around Γ (e.g., cut 1), while the energy scale of the suppression is systematically reduced on approaching the Γ point (cut 5 to 1).

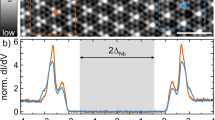
a Plots of ARPES intensity as a function of 2D wave vector at representative EB’s for monolayer VS2. Red solid curves indicate the calculated FS for monolayer 1T-VS2. kx and ky are defined according to the BZ of BL graphene. Black solid lines represent the BZ of monolayer VS2. b1–b5 ARPES-intensity plots as a function of the wave vector and EB, measured at T = 40 K along representative k cuts shown by red dashed lines in (a) (cuts 1–5). c Calculated band dispersions in the normal state corresponding to cut 5. d EDCs at T = 40 K at representative kF points on the elongated pocket (points 1–5 of inset). The kF points were determined by tracing the minimum gap locus of the EDCs. The peak position is indicated by dots. Inset shows a comparison of EDC around EF between gold (black curve) and monolayer VS2 (red curve) obtained at point 1. e Magnitude of CDW gap at different kF points (points 1–5) estimated from the peak position and leading-edge midpoint of the EDCs.
Characteristic k dependence of the gap is better visualized by the energy distribution curves (EDCs) at the Fermi wave vectors (kF’s) in Fig. 3d. At point 5 (which is almost on the MK line), one can find a broad hump at EB ~ 0.35 eV, reflecting a large energy gap. This hump gradually moves toward EF on approaching the Γ point and reaches EB ~ 0.1 eV at the corner of the ellipsoidal pocket (point 1), accompanied by a positive leading-edge shift as seen in the inset to Fig. 3d. This suggests that the energy gap opens on the entire pocket at T = 40 K and is strongly anisotropic, as also highlighted by the plot of gap size Δ and leading-edge midpoint against k (Fig. 3e), which always takes a positive value irrespective of k (note that the leading-edge shift serves as an effective energy gap under a strong spectral broadening; for details, see Supplementary Note 1). We have confirmed that the gap is robust against temperature variation, as seen from the fact that it survives even at room temperature at point 1 (see Supplementary Fig. 1). It is noted that the EDCs in Fig. 3d always show a residual spectral weight around EF, reminiscent of the pseudogap in underdoped high-Tc cuprate superconductors37. Such a pseudogap was also recognized in the CDW phase of other monolayer TMDs such as VSe2 and VTe210,14,15,16,17,22,23. While the exact mechanism to produce the pseudogap is unclear at the moment, we speculate that the strong damping of quasiparticle lifetime due to many-body interactions, such as coupling of electrons to CDW fluctuations and electron–electron scattering associated with electron correlation may play a role.
Stripe charge-density wave of monolayer VS2
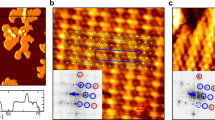
To clarify the origin of the energy gap, we have performed STM measurements. One can recognize a stripe feature associated with the CDW formation in the STM image at T = 4.8 K (Fig. 4a). The Fast Fourier transformation (FFT) image in Fig. 4b signifies the superstructure spots with a \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) periodicity. Intriguingly, this periodicity is apparently different from that of bulk crystal (\(3\sqrt 3 \times 3\sqrt 3 R30^\circ\))38. Further, it is also different from those of VSe2 and VTe2 monolayers in the CDW phase (4 × 4, 4 × 1, \(\sqrt 7 \times \sqrt 3\), \(2 \times \sqrt 3\), and \(4 \times \sqrt 3\) for VSe217,18; 4 × 4, 4 × 1, 5 × 1, and \(2\sqrt 3 \times 2\sqrt 3\) for VTe223,24,25,26,27). It is noted here that a recent STM study on monolayer VS230 has reported the CDW composed of \(7 \times \sqrt 3 R30^\circ\) and \(9 \times \sqrt 3 R30^\circ\) superstructures and its gapless behavior at EF. This is in contrast to the present study showing a full gap at EF associated with \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) CDW, which cannot be reproduced by the combination of \(7 \times \sqrt 3 R30^\circ\) and \(9 \times \sqrt 3 R30^\circ\) CDW (see Supplementary Fig. 2). Note that there exist intrinsic differences between our work and the work of ref. 30 regarding the experimental observation and interpretation of the CDW mechanism (for details, see Supplementary Note 3).

a High-resolution STM image in surface area of 8 × 8 nm2 for monolayer VS2 at T = 4.8 K. White broken rhombus indicates the unit cell of \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure, whereas the red solid rhombus corresponds to the original 1 × 1 unit cell. b FFT image of (a). White arrows indicate primitive reciprocal lattice vectors g1 and g2 for the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure. c Calculated FS of monolayer 1T-VS2 obtained by the first-principles band calculation. White arrows indicate g1 and g2. The absolute value of Fermi velocity vF is depicted by gradual coloring. d The \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure obtained by the structural optimization. a1 and a2 indicated by black arrows are primitive lattice vectors of the original 1 × 1 1T structure. The black dashed rhombus corresponds to the unit of the cell of the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure. e Calculated band structure of monolayer 1T-VS2 for the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure, folded back to the original 1 × 1 BZ. Red broken curves represent the band structure for the 1 × 1 phase. f, g Comparison between the calculated band structure for the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure and the ARPES intensity at T = 40 K along the KΜΚ cut. The white dashed curve in (f) corresponds to the calculated band dispersion for the 1 × 1 phase. h Calculated spectral weight for the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure along the KΓM cut. i Corresponding ARPES intensity near EF at T = 40 K. White dashed curves in (h) correspond to calculated band dispersion for the 1 × 1 phase.
Reciprocal lattice vectors of the observed superlattice, g1 and g2 (white arrows in Fig. 4b), are written as g1 = (2/9)b1 − (1/9)b2 and g2 = −(1/9)b1 + (5/9)b2 with the original 1 × 1 reciprocal lattice vectors, b1 and b2. The corresponding crystal structure drawn after the structural optimization in our first-principles band-structure calculations is shown in Fig. 4d, in which both V and S atoms are slightly displaced to form a superlattice with a large parallelogram unit cell. To clarify the influence of CDW on the band structure, we have performed band calculations for this superstructure, and the obtained band structure projected onto the original 1 × 1 BZ is shown in Fig. 4e (note that we assumed the existence of three domains rotated by 120° from each other, by taking into account the crystal symmetry). One can recognize that although the overall spectral weight in the CDW phase traces the band structure of the original 1 × 1 lattice (dashed red curves), there exist characteristic deviations. For example, while the original V 3d band crosses EF midway between the M and K points, the spectral weight in the CDW phase is suppressed around the crossing point, accompanied by a marked deviation of the band position from the original one. Such a CDW signature in the calculation is better visualized in an expanded view near EF of the calculated spectral weight along the MK cut in Fig. 4f. Corresponding ARPES intensity shown in Fig. 4g also signifies the spectral-weight suppression near EF. To clarify the influence of CDW on the ARPES intensity in more detail, we compare in Fig. 4h, i the calculated spectral weight in the CDW phase with the ARPES intensity at T = 40 K along the KΓM cut. Although the bandwidth of the topmost V 3d band is smaller in the experiment due to the mass-renormalization effect associated with the electron correlation, the overall ARPES intensity shows a better agreement with the calculated spectral weight for the CDW phase in comparison to the calculated band structure for the undistorted 1T phase (white dashed curves). Specifically, the V 3d band located slightly below EF at the Γ point is well reproduced by the calculation for the CDW phase, whereas the calculated band for the undistorted phase is located above EF. Such a critical difference can also be confirmed by the band dispersion along the ΓK cut (left half of Fig. 4i), which signifies the existence of a dispersive V 3d band below EF associated with the band folding due to the superstructure-induced periodic potential, consistent with the calculation for the CDW phase, but inconsistent with that for the 1 × 1 phase showing fully unoccupied bands along the ΓK cut (Fig. 4h). These results suggest that the anisotropic gap observed by ARPES originates from CDW, and some other mechanisms such as electron correlation are unlikely responsible for the gap opening (see Supplementary note 4). We also found from the quantitative analysis that the calculated energy gap along cut 1 (the ΓM cut) in Fig. 3a is 110 meV, whereas that along cut 5 (the MM cut) is 260 meV, qualitatively consistent with the anisotropic gap observed by ARPES (Fig. 3d). The CDW origin of the energy gap is also supported by the STM image which exhibits clear charge reversal between positive and negative bias voltages, which is a typical characteristic of the CDW (see Supplementary Fig. 5).
While we cannot completely rule out the possibility of a non-nesting-originated CDW mechanism at the moment, a natural starting point to understand the CDW mechanism may be the FS nesting which connects parallel segments of the elongated pocket. The regular nesting vector is q = g1 or q = g2, as widely discussed in many 2D and quasi-2D materials. In both cases, the k space spanned by g1 or g2 reproduces the superlattice spots in the FFT image of STM data. We shifted the calculated FS of monolayer VS2 with respect to the original one by q = g2 (Fig. 5a) and found that the parallel segments of FSs are not well connected; it only produces several crossings of original and shifted FSs at discrete k points as shown by orange circles. This would be natural because the g2 vector is not parallel to the 1D array of superspots in the FFT image and, thereby, unsuited to represent the observed CDW periodicity. On the other hand, the parallel segments of the vertically elongated pockets centered at M1 appear to show a rather good nesting condition with q = g1, whereas a large area of side pockets at M2 and M3, as well as the curved region of pockets around the Γ point remains un-nested. Obviously, the normal FS nesting with q = g1 (as well as q = g2) is insufficient to explain the gap opening on the entire FS, unlike the case of some other TMDs where the simple FS nesting plays a dominant role10,11 (note that the consideration of 3q nature of CDW is still insufficient to explain the gap opening at the curved region of the pockets; for details, see Supplementary Fig. 6).

a–c Calculated FS (red solid curves) and its shifted replicas (blue broken lines) that take into account the nesting vector q = g2, g1, and 2g1, respectively. g1 and g2 vectors are indicated by black solid arrows. Nesting vectors with positive and negative signs are indicated by blue solid and blue dashed arrows, respectively. FS segments where the original and shifted FSs overlap are indicated by orange circles and orange shades. d Schematics of nested FS region, which simultaneously takes into account q = g1 (same as (b)) and q = g2 (same as (c)) nesting vectors.
Discussion
Higher-order Fermi-surface nesting
We propose that a key to pinning down the mechanism of CDW in VS2 lies in consideration of a longer q vector, q’ = 2g1, termed here higher-order FS nesting vector, which also reproduces the superlattice points in the FFT image. The nesting vector twice (integer times in the more general form) larger than that of the original nesting vector q (= g1) is not usually taken into account as a CDW nesting vector. As shown in Fig. 5c, the original and shifted FSs show an overlap in a larger k area, not only in the straight segment part of the pockets at M2 and M3 but also in the curved region around the corner of these pockets. Although the nesting vector q’ (= 2g1) does not cover the whole pocket by the nesting with leaving the poorly nested region, e.g., along the pocket at M1, the cooperation of two nesting vectors q = g1 and q’ = 2g1 enables the full-gap opening as observed by ARPES, since the nested area almost fully covers the pocket as shown in Fig. 5d. Such a visual argument on the FS nesting is more quantitatively supported by our calculations. Calculated electronic susceptibility χ(q) shows a local enhancement (see Supplementary Fig. 7), and the calculated acoustic phonon dispersion shows a negative frequency (see Supplementary Figs. 8–10) at q’ = 2g1, supporting the CDW instability at this wave vector. These results suggest that the cooperation of q and 2q nesting is likely responsible for the occurrence of CDW in monolayer VS2. It is noted that the CDW gap monotonically increases on moving away from the BZ center, as shown in Fig. 3d, e. Since this seems to be not fully consistent with the fine structure of the FS-overlap condition discussed in Fig. 5d, one may need to take into account the influence of strong spectral broadening associated with the many-body interactions that smears out fine structures in the FS-overlap condition. Also, one may need to consider that the nesting vector of g1 and 2g1 plays primary and secondary roles, respectively, to the total magnitude of the CDW gap, while consideration of q-dependent electron–phonon coupling may be necessary to fully account for the observed k-dependence of the CDW gap. We will come back to this point later.
Difference between VS2 and VTe2
The 2q nesting proposed in this study captures the difference in the CDW behavior between monolayer VS2 and monolayer VTe2. The calculated χ(q) and phonon dispersion for monolayer VS2 show a local enhancement also at the commensurate wave vector of q ~ 1/2ΓM, which corresponds to the 4 × 4 periodicity. However, the 4 × 4 CDW is not realized in monolayer VS2. The calculated free energy for the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure per VS2 primitive unit cell is 18 meV lower than that of the original 1T structure. On the other hand, the free energy for the 4 × 4 superstructure is 4 meV lower than that of the original 1T structure30. This suggests the more stable nature of \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) CDW. As opposed to the case of monolayer VS2, the 4 × 4 CDW emerges in monolayer VTe223,24,25,26,27. A key to understanding this difference may lie in the difference in the FS topology. VS2 hosts an elongated electron pocket at the M point (Fig. 3a), whereas VTe2 hosts a triangular hole pocket at the K point, giving rise to a marked difference in the FS shape around the Γ point (see Supplementary Fig. 12). This causes the poorer nesting condition with the higher-order nesting vector so that \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) CDW is not stable in VTe2. In other words, the curved segments of the elongated pocket in VS2 around the Γ point are effectively nested by the 2q vector and contribute to the energy gain to stabilize CDW. As a consequence, a CDW gap opens on the entire FS in VS2 (Fig. 3d) by the cooperation of primary q and secondary 2q nesting vectors, in contrast to VTe2 where the corner of the triangular pocket remains gapless due to the insufficient nesting (see Supplementary Fig. 12). It is emphasized again that to account for the gap anisotropy of VS2, one may need to consider the primary and secondary roles of the q and 2q nesting vectors, respectively. In other words, the probability of electron scattering via 2q phonon is much lower than that via q phonon, so the latter plays a primary role in stabilizing CDW. The primary q vector, which connects parallel segments of the elongated pocket around the M1 point, effectively reduces the total energy to stabilize CDW, leading to the larger CDW gap along the MK cut. On the other hand, the 2q vector, which essentially connects curved segments of FS around the Γ point, plays a secondary role in stabilizing CDW so that the CDW-gap size around the Γ point is relatively small, leading to the anisotropic gap nature in monolayer VS2. It is noted, though, that this argument is based on our speculation and needs to be further corroborated by experiments that modulate the FS-nesting condition (by carrier doping, electrical gating, or epitaxial strain) to see the disappearance/weakening of CDW and also by the sophisticated theoretical modeling.
Historically, the importance of higher-order nesting in CDW formation was theoretically proposed in the 1970s for a purely 1D system with a half-filled condition as the 4kF CDW39,40, which involves Umklapp scattering. In quasi-2D TMDs and quasi-1D conductors, the CDW, which involves a longer q vector extending the first BZ, was proposed to account for the lock-in transition between incommensurate and commensurate CDW41,42,43,44 and the unidirectional incommensurate CDW45,46. On the other hand, the cooperation of ordinary nesting and higher-order nesting has not been spectroscopically clarified, presumably because of the lack of a suitable material platform. In this regard, monolayer VS2, which shows a peculiar CDW with the incommensurate \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) periodicity, is a suitable platform to demonstrate such nesting because the FS curvature is well-optimized to promote the 2q nesting. The concept of higher-order-nesting-driven CDW proposed in this study may be applied to some other low-dimensional materials where a simple nesting scenario is hard to account for the peculiar CDW behaviors.
Methods
Sample preparation
Monolayer VS2 film was fabricated by two steps; MBE of VTe2 film and topotactic reaction to replace Te with S atoms. First, monolayer VTe2 film was prepared on bilayer graphene by MBE22,32. Bilayer graphene was fabricated by resistive heating of an n-type SiC(0001) single-crystal wafer at 1100 °C for 30 min. Subsequently, monolayer VTe2 film was grown by evaporating V atoms on the bilayer graphene substrate in a Te-rich atmosphere. The substrate was kept at 250 °C during epitaxy, and then the as-grown monolayer VTe2 film was annealed for 30 min. Second, we fabricated monolayer VS2 film by replacing Te with S atoms by using the topotactic reaction31,47. The monolayer VTe2 film was annealed in a S atmosphere under high vacuum (~1 × 10−7 Torr); the substrate was heated up to 300 °C during the sulfurization, and then the as-grown film was annealed for 30 min at 300 °C. In the sulfurization process, the sulfur source (99.999%) in a glass-tube cell equipped with a homemade evaporator was melted by heating by a ribbon heater. The sulfur flux was precisely controlled by tuning the leak rate of the variable leak valve. Although we were unable to estimate the exact sulfur flux, we monitored the relative flux by reading the vacuum inside the growth chamber during evaporation. To effectively proceed with the sulfurization, we tried several experimental conditions with different (1) sulfur flux, (2) annealing temperature, and (3) annealing time of the sample during and after the evaporation. As a result, we found that the optimized conditions are (1) 1 × 10−7 Torr in the growth chamber during the evaporation for the sulfur flux (note that the base pressure of the growth chamber is 1 × 10−10 Torr), (2) 300 °C for the annealing temperature during/after evaporation, and (3) at least 30 min for the annealing time. The growth process was monitored by RHEED and low-energy electron diffraction (LEED). After the growth, the film was transferred to the ARPES measurement chamber without exposing it to air.
ARPES and STM measurements
ARPES measurements were performed by using a DA-30 electron energy analyzer with a micro-focused synchrotron-radiation at the beamline BL-28A of Photon Factory (KEK). The beam spot was 10 × 12 μm248. Circularly polarized light of hν = 100 eV was used to excite photoelectrons. The energy and angular resolutions were set to be 25–50 meV and 0.2–0.3°, respectively. STM measurements were performed using a custom-made ultrahigh vacuum (UHV) STM system49 at T = 4.8 K under UHV below 2.0 × 10−10 Torr. Te capping with ~20 nm thickness was deposited on VS2 film to protect the surface. This capping was removed in the STM chamber by Ar-ion sputtering for 30 min and subsequent annealing at 250 °C for 120 min. We used PtIr tips. All STM images were obtained in constant current mode.
First-principles calculations
First-principles band-structure calculations were carried out by using Quantum ESPRESSO code50,51 with generalized gradient approximation52 with the Perdew–Burke–Ernzerhof (PBE) realization using PSLibrary53. A pseudopotential of the Augmented-Plane-Wave (APW) type was used. The plane-wave cutoff energy and uniform k-point mesh were set to be 60 Ry and 24 × 24 × 1, respectively. Also, the charge cutoff energy was set to be 700 Ry. The k grid in the DFT calculation for the supercell was set to be 6 × 3 × 1. All structures were fully relaxed for monolayer VS2 with lattice constant a = 3.18 Å. The thickness of inserted vacuum layer was set to be more than 15 Å to prevent interlayer interaction. We have carried out the unfolding of bands for the \(\sqrt {21} R10.9^\circ \times \sqrt 3 R30^\circ\) superstructure by using a method proposed in the previous literature54. All the atoms are fully relaxed until the Hellmann–Feynman force becomes lower than 1 × 10−4 eV/Å. Phonon calculations were carried out using the density functional perturbation theory (DFPT) implemented in Quantum ESPRESSO. The phonon dispersions were obtained by setting at 24 × 24 × 1 k-mesh and 12 × 12 × 1 q-grid. The electronic temperature (Fermi–Dirac smearing) was set at 0.01 Ry for entire calculations. The DFT calculation based on the CDW-induced superstructure unit cell was carried out by using ISODISTORT tools55. Specifically, we have taken into account 12 types of phonon modes from the consideration of crystal symmetry. By changing the initial atomic position of V sites, we have structurally optimized the atomic coordinates of all the S atoms in the unit cell.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Novoselov, K. et al. Two-dimensional gas of massless Dirac fermions in graphene. Nature 438, 197–200 (2005).
Mak, K. et al. Control of valley polarization in monolayer MoS2 by optical helicity. Nat. Nanotechnol. 7, 494–498 (2012).
Qian, X., Liu, J., Fu, L. & Li, J. Quantum spin Hall effect in two-dimensional transition metal dichalcogenides. Science 346, 1344–1347 (2014).
Xi, X. et al. Ising pairing in superconducting NbSe2 atomic layers. Nat. Phys. 12, 139–143 (2016).
Nakata, Y. et al. Robust charge-density wave strengthened by electron correlations in monolayer 1T-TaSe2 and 1T-NbSe2. Nat. Commun. 12, 5873 (2021).
Rossnagel, K. On the origin of charge-density waves in select layered transition-metal dichalcogenides. J. Phys. Condens. Matter 23, 213001 (2011).
Rice, T. M. & Scott, G. K. New mechanism for a charge-density-wave instability. Phys. Rev. Lett. 35, 120–123 (1975).
Zhu, X. et al. Classification of charge density waves based on their nature. Proc. Natl Acad. Sci. USA 112, 2367–2371 (2015).
Johannes, M. D., Mazin, I. I. & Howells, C. A. Fermi-surface nesting and the origin of the charge-density wave in NbSe2. Phys. Rev. B 73, 205102 (2006).
Terashima, K. et al. Charge-density wave transition of 1T-VSe2 studied by angle-resolved photoemission spectroscopy. Phys. Rev. B 68, 155108 (2003).
Sato, T. et al. Three-dimensional Fermi-surface nesting in 1T-VSe2 studied by angle-resolved photoemission spectroscopy. J. Phys. Soc. Jpn. 73, 3331 (2004).
Strocov, V. N. et al. Three-dimensional electron realm in VSe2 by soft-X-ray photoelectron spectroscopy: origin of charge-density waves. Phys. Rev. Lett. 109, 086401 (2012).
Zhang, D. et al. Strain engineering a 4a×√3a charge-density-wave phase in transition-metal dichalcogenide 1T-VSe2. Phys. Rev. Mater. 1, 024005 (2017).
Umemoto, Y. et al. Pseudogap, Fermi arc, and Peierls-insulating phase induced by 3D–2D crossover in monolayer VSe2. Nano Res. 12, 165–169 (2019).
Chen, P. et al. Unique gap structure and symmetry of the charge density wave in single-layer VSe2. Phys. Rev. Lett. 121, 196402 (2018).
Feng, J. et al. Electronic structure and enhanced charge-density wave order of monolayer VSe2. Nano Lett. 18, 4493–4499 (2018).
Duvjir, G. et al. Emergence of a metal–insulator transition and high-temperature charge-density waves in VSe2 at the monolayer limit. Nano Lett. 18, 5432–5438 (2018).
Wong, P. K. J. et al. Evidence of spin fluctuation in a vanadium diselenide monolayer magnet. Adv. Mater. 31, 1901185 (2019).
Chen, G. et al. Correlating structural, electronic, and magnetic properties of epitaxial VSe2 thin films. Phys. Rev. B 102, 115149 (2020).
Si, J. G. et al. Origin of the multiple charge density wave order in 1T-VSe2. Phys. Rev. B 101, 235405 (2020).
Bronsema, D. K. et al. The crystal structure of vanadium ditelluride, V1+xTe2. J. Solid State Chem. 53, 415 (1984).
Sugawara, K. et al. Monolayer VTe2: incommensurate Fermi surface nesting and suppression of charge density waves. Phys. Rev. B 99, 241404(R) (2019).
Wang, Y. et al. Evidence of charge density wave with anisotropic gap in a monolayer VTe2 film. Phys. Rev. B 100, 241404(R) (2019).
Wong, P. K. J. et al. Metallic 1T phase, 3d1 electronic configuration and charge density wave order in molecular beam epitaxy grown monolayer vanadium ditelluride. ACS Nano 13, 12894–12900 (2019).
Liu, M. et al. Multimorphism and gap opening of charge-density-wave phases in monolayer VTe2. Nano Res. 13, 1733–1738 (2020).
Miao, G. et al. Real-space investigation of the charge density wave in VTe2 monolayer with broken rotational and mirror symmetries. Phys. Rev. B 101, 035407 (2020).
Wu, Q. et al. Orbital-collaborative charge density waves in monolayer VTe2. Phys. Rev. B 101, 205105 (2020).
Arnold, F. et al. Novel single-layer vanadium sulphide phases. 2D Mater. 5, 045009 (2018).
Kim, H. J. et al. Electronic structure and charge-density wave transition in monolayer VS2. Curr. Appl. Phys. 30, 8 (2021).
van Efferen, C. et al. A full gap above the Fermi level: the charge density wave of monolayer VS2. Nat. Commun. 12, 6837 (2021).
Shigekawa, K. et al. Dichotomy of superconductivity between monolayer FeS and FeSe. Proc. Natl Acad. Sci. USA 116, 24470–24474 (2019).
Kawakami, T. et al. Electronic states of multilayer VTe2: quasi-one-dimensional Fermi surface and implications for charge density waves. Phys. Rev. B 104, 045136 (2021).
Sugawara, K. et al. Unconventional charge-density-wave transition in monolayer 1T-TiSe2. ACS Nano 10, 1341–1345 (2016).
Nakata, Y. et al. Monolayer 1T-NbSe2 as a Mott insulator. NPG Asia Mater. 8, e321 (2016).
Lo, W. K. & Spence, J. C. H. Investigation of STM image artifacts by in-situ reflection electron microscopy. Ultramicroscopy 48, 433 (1993).
Karn, A. et al. Modification of monolayer 1T-VSe2 by selective deposition of vanadium and tellurium. AIP Adv. 12, 035240 (2022).
Damascelli, A., Shen, Z.-X. & Hussain, Z. Angle-resolved photoemission spectroscopy of the cuprate superconductors. Rev. Mod. Phys. 75, 473 (2003).
Mulazzi, M. et al. Absence of nesting in the charge-density-wave system 1T-VS2 as seen by photoelectron spectroscopy. Phys. Rev. B 82, 075130 (2010).
Sólyom, J. The Fermi gas model of one-dimensional conductors. Adv. Phys. 28, 201–303 (1979).
Landau, L. D. & Lifshitz, M. Quantum Mechanics (Non relativistic Theory) 3rd edn (Pergamon, 1977).
McMillan, W. L. Landau theory of charge-density waves in transition-metal dichalcogenides. Phys. Rev. B 12, 1187–1196 (1975).
Nakanishi, K. & Shiba, H. Domain-like incommensurate charge-density-wave states and the first-order incommensurate-commensurate transitions in layered tantalum dichalcogenides. I. 1T-Polytype. J. Phys. Soc. Jpn. 43, 1839–1847 (1977).
Schmitteckert, P. & Werner, R. Charge-density-wave instabilities driven by multiple umklapp scattering. Phys. Rev. B 69, 195115 (2004).
Inagaki, K. & Tanda, S. Lock-in transition of charge density waves in quasi-one-dimensional conductors: reinterpretation of McMillan’s theory. Phys. Rev. B 97, 115432 (2018).
Fang, A., Ru, N., Fisher, I. R. & Kapitulnik, A. STM studies of TbTe3: evidence for a fully incommensurate charge density wave. Phys. Rev. Lett. 99, 046401 (2007).
Tomic, A. et al. Scanning tunneling microscopy study of the CeTe3 charge density wave. Phys. Rev. B 79, 085422 (2009).
Kanetani, K. et al. Ca intercalated bilayer graphene as a thinnest limit of superconducting C6Ca. Proc. Natl Acad. Sci. USA 109, 19610–19613 (2012).
Kitamura, M. et al. Development of a versatile micro-focused angle-resolved photoemission spectroscopy system with Kirkpatrick–Baez mirror optics. Rev. Sci. Instrum. 93, 1–8 (2022).
Iwaya, K. et al. Atomic-scale visualization of oxide thin-film surfaces. Sci. Technol. Adv. Mater. 19, 282 (2018).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Dal Corso, A. Pseudopotentials periodic table: from H to Pu. Comput. Mater. Sci. 95, 337 (2014).
Popescu, V. & Zunger, A. Extracting E versus k effective band structure from supercell calculations on alloys and impurities. Phys. Rev. B 85, 085201 (2012).
Campbell, B. J., Stokes, H. T., Tanner, D. E. & Hatch, D. M. ISODISPLACE: a web-based tool for exploring structural distortions. J. Appl. Cryst. 39, 607–614 (2006).
Acknowledgements
We thank C.-W. Chuang, M. Kitamura, K. Horiba, and H. Kumigashira for their assistance in the ARPES measurements. This work was supported by JST-CREST (no. JPMJCR18T1), JST-PRESTO (no. JPMJPR20A8), Grant-in-Aid for Scientific Research (JSPS KAKENHI Grant Numbers JP18H01821, JP20H01847, JP20H04624, JP21H01757, JP21K18888, JP21H04435, and JP22J13724), KEK-PF (Proposal No. 2020G669, 2021S2-001, and 2022G007), Foundation for Promotion of Material Science and Technology of Japan, and World Premier International Research Center, Advanced Institute for Materials Research. T.K. acknowledges support from GP-Spin at Tohoku University and JSPS.
Author information
Authors and Affiliations
Contributions
The work was planned and proceeded by discussion among T.K., K.S., K.N., and T.S. T.K. and K.S. fabricated ultrathin films. T.K. carried out the band-structure calculations. T.K., K.S., K.Y., S.S., and K.N. performed the ARPES measurements. H.O. and T.F. performed the STM measurements. T.K., K.S., T.T., and T.S. finalized the manuscript with inputs from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kawakami, T., Sugawara, K., Oka, H. et al. Charge-density wave associated with higher-order Fermi-surface nesting in monolayer VS2. npj 2D Mater Appl 7, 35 (2023). https://doi.org/10.1038/s41699-023-00395-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-023-00395-z
