
Abstract
We combine infrared absorption and Raman scattering spectroscopies to explore the properties of the heavy transition metal dichalcogenide 1T-HfS2. We employ the LO–TO splitting of the Eu vibrational mode along with a reevaluation of mode mass, unit cell volume, and dielectric constant to reveal the Born effective charge. We find \({Z}_{{\rm{B}}}^{* }\) = 5.3e, in excellent agreement with complementary first-principles calculations. In addition to resolving the controversy over the nature of chemical bonding in this system, we decompose Born charge into polarizability and local charge. We find α = 5.07 Å3 and Z* = 5.2e, respectively. Polar displacement-induced charge transfer from sulfur p to hafnium d is responsible for the enhanced Born charge compared to the nominal 4+ in hafnium. 1T-HfS2 is thus an ionic crystal with strong and dynamic covalent effects. Taken together, our work places the vibrational properties of 1T-HfS2 on a firm foundation and opens the door to understanding the properties of tubes and sheets.
Similar content being viewed by others

Introduction
While 3d transition metal oxides and chalcogenides display strong electronic correlations, narrow bandwidths, and robust magnetism, 4 and 5d systems are recognized for strong spin–orbit coupling, increased hybridization, and more diffuse orbitals. As a result, materials that contain 4- and 5d centers often have enhanced or emergent properties1,2,3,4,5,6,7,8,9. Transition metal dichalcogenides such as MoTe2, IrTe2, HfSe2, and PtSe2 are of great interest for their unconventional chemical bonding and hybridization, topology, multiferroicity, and tendency toward complex dimerization patterns10,11,12,13,14,15,16,17. Within this class of materials, 1T-HfS2 has attracted particular attention as an analog of HfO2—a highly polarizable gate dielectric18,19.
1T-HfS2 is a layered material with a \(P\bar{3}m1\) (#164) space group at 300 K 20. Each Hf4+ ion has D3d site symmetry and is located at the center of a S2− octahedron. The van der Waals gap is 3.69 Å, and the sheet thickness is 2.89 Å. Photoemission studies reveal an indirect bandgap of 2.85 eV between Γ and M/L, which varies slightly from the ≈ 2 eV optical gap21. 1T-HfS2 forms a high-performance transistor with excellent current saturation22. The carrier mobility is on the order of 1800 cm2 V−1 s−1—much higher than MoS2 and thickness-dependent as well23,24. Group theory predicts that at the Γ point, 1T-HfS2 has vibrational modes with symmetries of A1g + Eg + A2u + Eu. The A1g + Eg modes are Raman-active, and the A2u + Eu modes are infrared-active20,25. Despite many years of work, there are a surprising number of unresolved questions about 1T-HfS2—even in single-crystal form. In the field of vibrational spectroscopy, there is controversy about mode assignments, the role of resonance in creating hybrid modes, the presence or absence of surface phonons, and the use of these data to reveal the Born effective charge (\({Z}_{{\rm{B}}}^{* }\)). As an example, Born effective charges between 3.46e and 5.5e have been reported by various experimental26,27 and theoretical28 groups. Evidence for the degree of ionicity (or covalency) is both interesting and important because 5d orbitals tend to be more diffuse than those of their 3d counterparts. Within this picture, 1T-HfS2 has the potential to sport significant covalency. High-pressure Raman scattering spectroscopy reveals a first-order phase transition near 11 GPa and different ∂ω/∂P’s (and thus mode Gruneisen parameters) for the hybrid Eu and fundamental A1g modes29. At the same time, variable temperature Raman scattering spectroscopy shows a systematic blueshift of the spectral features down to 100 K, except for the large A1g mode ~330 cm−1, which redshifts24,29. In few- and single-layer form, 1T-HfS2 is suitable for high-performance transistors22,30,31, displays a direct gap (rather than indirect as in the bulk)32, exhibits photocatalytic behavior appropriate for water splitting33, reveals applications in photodetection34, is susceptible to strain effects35, and is useful in N, C, and P surface adsorption36. This system can be integrated into van der Waals heterostructures and grown vertically as well31,37,38.
In order to explore the vibrational properties of 1T-HfS2, we measured the infrared absorption and Raman scattering response and employed the results to evaluate Born effective charge. We find that \({Z}_{{\rm{B}}}^{* }\) = 5.3e, in excellent agreement with our complementary first-principles calculations. In order to understand how \({Z}_{{\rm{B}}}^{* }\) relates to the nominal 4+ charge of the Hf center, we employ a Wannier function analysis to project out the different orbital contributions. This analysis reveals that the sulfur p orbital transfers charge to the cation and that this contribution enhances the Born charge beyond the nominal value. Decomposing \({Z}_{{\rm{B}}}^{* }\) into polarizability and local charge, we find that there is strong ionicity as well as significant covalency in this system. Both are quite different than in 2H-MoS2—probably on account of spin–orbit coupling. We also identify two weak structural distortions near 210 and 60 K evidenced by subtle frequency shifts of the Eg and A1g vibrational modes as well as changes in the phonon lifetimes. Taken together, these findings resolve the controversy over the nature of chemical bonding in 1T-HfS2 and clarify the role of the 5d center in the process.
Results and discussion
First-principles predictions of charge and bonding
Figure 1a displays the projected density of states of 1T-HfS2 computed using density functional theory and atom-centered local projectors39. The bands can be assigned Hf and S characters easily, and the degree of hybridization between the atoms is not dominant (albeit nonzero). This reveals the strong ionic nature of this system: the valence band is composed mainly of S-p orbitals, whereas the conduction band is predominantly Hf-d in character. We find a bandgap of 2.05 eV using the HSE06 functional. This is consistent with the small electronegativity of the Hf (1.3 on the Pauling scale) compared to that of S (2.6 on the Pauling scale).

a The electronic density of states of 1T-HfS2. Different colors indicate different orbitals of the two types of atoms. b The phonon spectrum of 1T-HfS2 and its density of states. The Γ-point phonon mode frequencies are marked with dashed lines. c Displacement patterns of several Γ-point phonon modes. Blue atoms are S and the red atoms are Hf. The arrows are not scaled to real displacement amplitudes. Numerical details are available in Supplementary Information.
Despite the apparent ionicity of the density of states, the dynamical Born effective charges in 1T-HfS2 are anomalous. We find an in-plane value for the Hf ions as \({Z}_{\mathrm{B},xx}^{* }=+6.4e\). By contrast, the out-of-plane value for Hf \({Z}_{{\mathrm{B}},zz}^{* }\) is only 2.0e. This reveals that either (i) the Hf ions are strongly polarizable, or (ii) the small degree of covalency is strongly dependent on ionic displacements40.
Figure 1b displays the phonon dispersions of 1T-HfS2. While the spectrum is highly dispersive within the plane (for instance, in the Γ–M direction), it is much less so in the out-of-plane direction (for instance, along Γ–A). This difference is a natural consequence of the layered crystal structure and is the origin of the spikes in the phonon density of states (right panel, Fig. 1b). One aspect of these predictions that will be important for later discussion is the mode order around the A1g fundamental. Notice that the Eu feature is predicted to be below the A1g mode, whereas the A2u mode is predicted to be above the A1g fundamental. These features are labeled in Fig. 1b. Theoretical phonon frequencies are in excellent agreement with our measured results (Table 1).
Lattice dynamics can be used to gain information about the chemical bonding in crystals. In 1T-HfS2, the Eu optical mode is due to the in-plane vibrations of the Hf cations against the S anions (Fig. 1c). The frequency difference between the longitudinal and transverse optical modes (the LO–TO splitting) depends on the permittivity, as well as the dynamical charges of the ions. Formally, the LO–TO splitting stems from a nonanalytic term added to the dynamical matrices of ionic insulators at the zone center41:
Here the s, t are atomic indices, and α, β are cartesian directions. \({Z}_{B}^{* }\) is the Born effective charge and q is the wavevector. With this extra term present in the dynamical matrix, the twofold degeneracy of the Eu optical modes is lifted:
where ui is the displacement vector of Eu(TO) mode. By considering the fact that \({Z}_{{\mathrm{Hf}},xx}^{* }\) is always equal to \(2\cdot {Z}_{{\mathrm{S}},xx}^{* }\) because of charge neutrality, and the symmetry imposed form of the Eu displacement pattern, we can write this equation for \({\bf{q}}\parallel \hat{x}\) as
where m* is the effective mass, determined by
in this case, the effective mass is 47.02 u. A general expression for effective mass is provided in Supplementary Information. We use this result along with Eq. (3) to analyze the experimental Born effective charge below. Gaussian units are employed.
Infrared properties of 1T-HfS2
Figure 2a summarizes the infrared response of single-crystalline 1T-HfS2. We assign the vibrational modes, symmetries, and displacement patterns based on prior literature as well as our complementary lattice dynamics calculations (Table 1)24,25,29,42. There are two fundamental infrared-active phonons. The Eu symmetry mode is extremely broad and centered at 155 cm−1. It is ascribed to an in-plane, out-of-phase motion of the sulfur layers against the hafnium. As we will discuss below, the maximum corresponds to the transverse optical (TO) phonon frequency. In the bulk form, peaks in absorption always correspond to the TO component of the mode. The A2u mode at 336 cm−1 is much weaker and narrower. This feature is due to an out-of-plane + in-phase sulfur layer stretching, and although it is polarized in the c direction, it appears in the nascent form here probably due to surface flakiness or slight misalignment. In any case, it is very small. There is also a minute structure near 310 cm−1 which, as we shall see below, corresponds to the weakly activated longitudinal optical (LO) frequency of the Eu mode. Of course, each polar phonon has separate LO and TO components, and the LO frequency is always higher than that of the TO frequency due to the polarizability of the surrounding medium. Displacement patterns for each phonon are shown in Fig. 1c and summarized in Table 1.

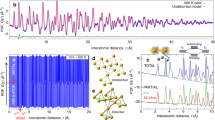
a Infrared absorption spectra of 1T-HfS2 as a function of temperature. The color scheme emphasizes the different phases, and the curves are offset for clarity. b Close-up view of the infrared response of 1T-HfS2 on an absolute scale. The inset displays the phonon lifetime of the Eu mode as a function of temperature. Error bars are on the order of the size of the data points.
Figure 2a also displays the infrared response of 1T-HfS2 as a function of temperature. What is striking about these results is the overall lack of temperature-induced change over the 300–8 K range—in both peak position and intensity. Traditionally, modeling of frequency vs. temperature effects provides important information on anharmonicity in a solid as well as the various force constants. The idea is to bring together frequency vs. temperature plots along with one of several different equations that depend upon the situation. The absence of mode hardening with decreasing temperature suggests, however, that anharmonic effects in 1T-HfS2 are modest and that energy scales are high. Oscillator strength sum rules are obeyed, as expected. Below 100 K, there is a small resonance in the form of a dip that develops on top of the 155 cm−1 Eu symmetry phonon. This structure is one of the spectroscopic signatures of weak local lattice distortion. Because the evidence for the effect is stronger in the Raman response, we will defer this discussion until later. Partial sum rules on the Eu vibrational mode are obeyed, as expected.
Another way to reveal temperature effects is to examine phonon lifetimes (Fig. 2b). These values, which are expressions of the Heisenberg uncertainty principle, can be calculated from the linewidth of each vibrational mode as \(\tau =\frac{\hslash }{{{\Gamma }}}\), where Γ is the full width at half-maximum and ℏ is the reduced Planck’s constant 43. We find that the phonon lifetime of the Eu mode is 0.03 ps—exceptionally short compared to that of the A2u mode. The lifetime of the Eu mode is nearly insensitive to temperature as well, meaning that this mode dissipates energy very well—even at low temperatures. In other words, τ is large because there are many scattering events. On the other hand, the lifetime of the 336 cm−1A2u feature is on the order of 0.4 ps. Overall, these phonon lifetimes are shorter than those of 2H-MoS2 (τ = 5.5 and 2.3 ps for the E1u and A2u modes, respectively) as well as those of polar semiconductors like GaAs, ZnSe, and GaN (which tend to be between 2 and 10 ps)44. Likewise, silicon has a phonon lifetime between 1.6 and 2 ps, depending on the carrier (hole) density45. This means that carrier–phonon scattering is of greater importance in 1T-HfS2 than in 2H-MoS2 or the traditional semiconductors. Employing a characteristic phonon velocity of 4700 ms−1(see ref. 32), we find mean free paths in 1T-HfS2 of 143 pm and 1740 pm for the Eu and A2u modes, respectively. The mean free path for the Eu mode is slightly smaller than all of the characteristic length scales in the system, including the 369 pm van der Waals gap, the 289 pm sheet thickness, and the 253 pm Hf–S bond length, whereas the mean free path for the A2u mode is four or five times larger than these characteristic length scales.
Revealing Born effective charge via infrared spectroscopy
The Born effective charge of transition metal dichalcogenides has been of sustained interest20,26,27,44,46. This is because Born charge can be calculated from first principles as summarized in the previous section and revealed directly from spectroscopic data by taking into account the relationship between the longitudinal and transverse optic phonon frequencies as indicated in Eq. (3). As a reminder, ωLO and ωTO are the longitudinal and transverse optic phonon frequencies, c is the speed of light, N is the number of formula units in the unit cell, V is the volume of the 1T-HfS2 formula unit, e is the electronic charge, ϵ0 is the permittivity of free space, ε(∞) is the dielectric constant after the phonons, and mk is the effective mass. Employing Eq. (4), we find mk = 47.02 u for 1T-HfS2. We also use ωLO = 310 cm−1, ωTO = 155 cm−1, ε(∞) = 6.20, N = 1, and V = 66.44 Å3. In the absence of a robust experimentally determined volume, we used a theoretically predicted value27. \({Z}_{{\rm{B}}}^{* }\) is extremely sensitive to the value of mk and the choice of ε(∞). Employing these numbers, we find \({Z}_{{\rm{B}}}^{* }\) = 5.3e.
Interestingly, prior studies have led to a variety of Born effective charges for 1T-HfS2 with values from 3.46 to 5.5e26,27,28. These findings are summarized in Table 2. The variations in \({Z}_{{\rm{B}}}^{* }\) are due to differences in mk, cell volume, and (to a lesser extent) ϵ(∞) as well as variability ωLO and ωTO. Our value of 5.3e is consistent with the large LO–TO splitting, which signals robust ionicity. By comparison, transition metal dichalcogenides like 2H-MoS2 have much smaller LO–TO splitting and \({Z}_{{\rm{B}}}^{* }\) = 1.11e in the ab plane46,47.
Born effective charge can be decomposed into polarizability (α) and local effective charge (Z*) as
and
Here, η is the depolarization constant. For two-dimensional systems, η = 1/348,49. We find α = 5.07 Å3 and Z* = 5.2e from our experimental value of the Born charge in the ab plane. These values compare well with those obtained from our theoretical value of \({Z}_{{\rm{B}}}^{* }\): α = 6.85 Å3 and Z* = 6.2e. These results along with literature values are summarized in Table 3.
Polarizability entails the sum of all cationic and anionic electron cloud volume contributions, whereas local effective charge is related to short-range interactions indicative of ionic displacement and chemical bonding. These values reveal the nature of chemical bonding. Typically, covalent systems such as silicon have Z* close to zero, whereas ionic materials have larger local effective charges. For example, Z* = 0.15e for 2H-MoS246, Z* = 1.14e for MnO50, and Z* = 0.8e for NaCl51. Higher effective and ionic charge in MnO is consistent with the distinction of a Reststrahlen band. 1T-HfS2 clearly has the largest local charge in this series.
Origin of anomalous Born effective charge
In a completely ionic crystal where electrons are attached to ions and displaced along with them by the same exact amount, the dynamical Born effective charge is equal to the formal ionic charge—which would have given \({Z}_{{\rm{B}}}^{* }\)=4 for Hf. 1T-HfS2 is closer to this limit due to the low electronegativity of the cation (1.3 in the Pauling scale). This is lower than any 3d transition metal, including Mo, which has an electronegativity of 2.2. The electronegativity of S is 2.6, so the Mo–S bonds in MoS2 are highly covalent. Nevertheless, we note that the Born effective charge of Hf in the in-plane direction is >50% larger than the nominal charge of +4. This anomalous Born charge signals either (i) covalency between the cations and anions or (ii) cation polarizability40. Uchida explored the issue in terms of static and dynamic charge26. We can address the question more robustly with contemporary tools.
Maximally localized Wannier functions can be utilized to explain the origin of anomalous Born effective charges52. The macroscopic electronic polarization can be expressed in terms of the center of localized Wannier functions as
where Wn(r) is the Wannier function and the sum is over the filled Wannier orbitals. By displacing the Hf atoms in the in-plane and out-of-plane directions, it is possible to calculate the shift of the center of each Wannier function, and hence get an orbital-by-orbital or band-by-band decomposition of the Born effective charges.
Table 4 shows the band-by-band contributions to the Born effective charges of Hf. It turns out that despite the large electronegativity difference between Hf and S, the covalency of Hf–S bonds is the dominant reason behind the anomalous Born effective charge in this compound: the Wannier centers of the electrons in the S-3p bands displace significantly when the Hf ion is displaced. On the other hand, the Hf-5p electrons are displaced almost exactly as much as the Hf ion core itself. Thus, S-3p orbitals contribute most to the difference between Born effective and nominal charge.
Taking a closer look at the electrons in the S-3p orbitals, Fig. 3 shows one of the Wannier functions with and without the Hf ion displacement. In line with the strongly ionic density of states, the S-p electrons are mostly localized on S, with small lobes on Hf indicating hybridization. When Hf atoms are displaced in an in-plane direction, one of the three Hf–S bonds is shortened, while the other two are elongated. The shortened bond causes the Wannier function to be tilted, and its center shifted toward the Hf atom, shown in Fig. 3b. This explains why S-3p orbitals will contribute a positive polarization value when Hf atoms are moving in-plane. On the other hand, when Hf atoms move in the out-of-plane direction, the shape of the hybridized electron density on the Hf atom changes, showing a significant qualitative change in the hybridization, as well as a shift in the S-3p Wannier centers parallel to the Hf displacement. This leads to a negative dynamic contribution of S-3p to the out-of-plane Hf effective charge. Though only one S-3p orbital is shown here, the others are similar.

The blue atom is a sulfur center, whereas red atoms are bonded hafnium centers. The red and blue lobes of the orbital indicate the opposite signs of the wavefunction. No structural distortion is present in (a). Hf atoms are displaced in-plane and out-of-plane in (b) and (c), respectively.
As a comparison, the Born effective charge of Mo in 2H-MoS2 is 1.1–1.2e in the in-plane direction46, and theoretical results suggest a sign reversal of the cation Born charge53. This is likely because of a much stronger covalency in MoS2 than in HfS2, which results in more electrons transferred from S anions onto Mo cations when Mo ions are displaced. Note that there is a \(4{d}_{{z}^{2}}-{p}_{z}\) antibonding orbital in Mo–S bonds near the Fermi level because Mo’s 4d band is partially filled, and this orbital can result in large electron transfer in a way similar to π-backbonding in organic chemistry53. Our DFT calculation confirms the partially filled band structure as shown in the Supplementary Information.
Raman scattering reveals structural crossovers in 1T-HfS2
Figure 4a summarizes the Raman scattering response of 1T-HfS2 as a function of temperature. We assigned the spectral features based on our lattice dynamics calculations and prior literature (Table 1)25,29,42. The two Raman-active fundamentals at 259 and 336 cm−1 are even symmetry vibrational modes. We attribute these features to Eg symmetry (in-plane, out-of-phase sulfur-layer shearing) and the A1g symmetry (out-of-plane opposing sulfur motions leading to layer breathing), respectively. There is also a weak overtone mode near 650 cm−1 that is slightly less than twice the frequency of the A1g fundamental. This feature has also been referred to as a second-order mode25. There are several hybrid features as well. For instance, the Eu (TO) mode appears weakly in the Raman spectrum near 132 cm−1 due to an in-plane motion of the sulfur against the hafnium. Finally, the shoulder near 325 cm−1 has been the subject of some controversy in the literature. It was previously described as an in-plane shearing (Eu), an out-of-plane sulfur translation (A2u), or even a surface phonon24,25,29,42. Based on prior pressure studies, the position of this phonon diverges from that of the A1g mode near 325 cm−1 as pressure is increased 29. If this feature was a surface phonon, it would likely track parallel to the much larger mode. Because it does not, it is unlikely to be a surface phonon. With the help of prior literature as well as the common position with the Eu mode in the infrared studies, we assign this structure as an Eu symmetry phonon24,25,29,42. Additional justification for this assignment comes from our phonon density of states calculations and the predicted order of the hybrid modes around the A1g fundamental (Fig. 1b). We do not see the hybrid A2u mode in our spectra—even at low temperature, although different laser wavelengths should reveal it25.

a Raman scattering response of 1T-HfS2 as a function of temperature. The mode symmetries are labeled, and the spectra are offset for clarity. b Contour plot showing subtle changes in the A1g mode as a function of temperature. Weak structural distortions at T1 and T2 are indicated. The color scale gives relative intensity. c Contour plot showing the development of the Eg mode with temperature. d Phonon lifetime of two Raman-active modes as a function of temperature. Data are available in Supplementary Information. T1 and T2 are denoted by the color scheme in (a) and the vertical blue lines in (d).
Figure 4b,c displays close-up views of the A1g and Eg symmetry Raman-active fundamentals as a function of temperature in contour form. While there is little frequency sensitivity in either feature (due to the high-energy scales in this system), there are noticeable linewidth effects. Focusing on the behavior of the A1g layer expansion mode in Fig. 4b, we see that the linewidth narrows considerably with decreasing temperature, with slight broadening across T1 ≈ 60 K and T2 ≈ 210 K. There is also a slight redshift across T1. Analysis of the Eg sulfur-layer stretching mode in Fig. 4c, again shows linewidth narrowing as the base temperature is approached, also with noticeable broadening across the two crossover regimes. A slight redshift below T1 is again present. We attribute the 60 and 210 K transitions in 1T-HfS2 to local lattice distortions involving a slight motion of the S centers with respect to the Hf ions so as to change the bond lengths and angles a little while maintaining the same overall space group.
Using these linewidth trends and the technique described previously, we calculated phonon lifetimes for the Raman-active vibrational modes of 1T-HfS2 (Fig. 4d). As a reminder, the A1g and Eg modes are the fundamentals. The lifetime of the A1g phonon rises steadily with decreasing temperature. The behavior of the Eg symmetry mode is different. It rises gradually below T2 and dramatically across T1. This suggests that carrier–phonon scattering is reduced with decreasing temperature. Overall, the lifetimes of the Raman-active even symmetry modes are similar to those of the infrared-active phonon modes in 1T-HfS2 (Fig. 2b)—with the exception of the Eu symmetry vibrational mode that is very lossy and therefore sports an extremely short lifetime. This is one surprising feature in 1T-HfS2 that is not replicated in more traditional systems like 2H-MoS2.
Toward chemical bonding in nanoscale analogs of 1T-HfS2
Overall, there has been surprisingly little discussion of chemical bonding and structural phase transitions in transition metal dichalcogenides in the literature. Much of the focus has instead been on oxides—particularly ferroelectrics—where questions of charge, polarizability, hybridization, and dynamic covalency are considered to be “solved problems”. The mechanism of polar displacement-induced charge transfer is, however, underexplored and may be responsible for enhanced Born charge compared to the nominal charge in oxides as well. This is not the case in transition metal dichalcogenides or complex chalcogenides, and there are many discrepancies in the literature—with estimates of simple quantities like Born charge varying by as much as 40% and almost no attention paid to the role of spin–orbit coupling in heavy chalcogenides. This work places the properties of 1T-HfS2 on a firm foundation for understanding (and even actuating) functionality under external stimuli. It also illustrates a broader approach to exploring chemical bonding in the chalcogenides and opens the door to examination of nanoscale analogs such as tubes, particles, and sheets, as well as effects of n- and p-type doping and intercalation. It will be interesting to learn whether Born effective charge is anomalously enhanced beyond the Madelung limit in these settings as well.
Methods
Crystal growth and spectroscopic measurements
1T-HfS2 single crystals were grown via chemical vapor transport by 2D Semiconductors, Inc. Prior to traditional infrared and Raman scattering measurements, the sample was surface-exfoliated to remove surface impurities and then mounted on a round pin-hole aperture exposing the ab plane. Far-infrared studies were performed using a Bruker IFS 113V Fourier-infrared spectrometer equipped with a bolometer detector covering the 20–700 cm−1 frequency range with 2 cm−1 resolution. The measured transmittance was converted to absorption as α(ω) = −\(\frac{1}{{d}}\)ln(\({\mathcal{T}}\)(ω)), where d is the thickness (in this case, ~8.4 × 10−4 cm). Raman scattering spectroscopy was carried out using a LabRAM HR Evolution Raman spectrometer (50–750 cm−1) using an excitation wavelength of 532 nm at a power of 0.1 mW with an 1800 line/mm grating. An open-flow cryostat was used to control the temperature between 4 and 300 K. Traditional peak fitting techniques were employed as appropriate.
First-principles calculations
First-principles calculations were performed using the projector-augmented wave approach as implemented in the Vienna Ab-initio Simulation Package (vasp)39,54,55. Both the Born effective charges and the high-frequency dielectric constant are determined from the response to finite electric fields. The high-frequency dielectric constant ϵ(∞), which only contains the electronic contribution, is obtained by differentiating the polarization with respect to the external electric field with clamped ions
where the polarization is calculated using the implementation of the Perturbation Expression After Discretization (PEAD) approach in the VASP package56,57. Electric fields of 2, 2, and 10 meV/Å are applied separately along a, b, and c axes to calculate the derivatives. Similarly, the Born effective charge is calculated through the derivatives of the polarization with respect to the ionic displacements. At this step, using the Hellman–Feynman forces enables the use of the more computationally efficient formula
Here, \({\mathcal{F}}\) is the electric enthalpy that is the sum of the Kohn–Sham energy and the energy gain due to the interaction between the polarization and the external electric field: \({\mathcal{F}}={E}_{KS}-{{\Omega }}{\bf{P}}\cdot {\bf{{\mathcal{E}}}}\). The Hellman–Feynman force F is given by \({F}_{i}=\frac{\partial {\mathcal{F}}}{\partial {u}_{i}}\). F solely depends on the ground-state wavefunction, and hence is easier to calculate than the polarization. As a reliability check, density functional perturbation theory (DFPT)58 combined with multiple functionals (including PBEsol59 and revised-TPSS meta-GGA60) with spin coupling is also performed to get the Born effective charge, which can be found in the Supplementary Information. Both approaches provide similar results for the dielectric constant and Born effective charge. We chose the latter method for its compatibility with Hartree–Fock method, and as a result, the hybrid functionals.
All the first-principles calculations are performed in the primitive three-atom unit cell with a 12 × 12 × 6 k-point grid and cut-off energy of 500 eV. The lattice constants and vectors are taken from the experimental literature, but the internal coordinates of the S ions are obtained through structural optimization of forces. The energy tolerance for self-consistency is set to 10−8 to get a well-converged wavefunction. To reproduce the experimental bandgap more closely, HSE hybrid functional is employed61. In the case of 1T-HfS2, an energy band gap of 2.05 eV can be achieved by using the screening parameter of 0.2, which is the so-called HSE06 approximation. Reports of the bandgap of HfS2 span values from 1.96 eV (from optical absorption)62 to 2.85 eV (from combined angle-resolved and inverse photoemission)21. Note that band structures calculated using PBEsol or meta-GGA functionals both underestimate the bandgap by at least a factor of two, which influences the prediction accuracy of electric field response. A comparison between different functionals is presented in the Supplementary Information. Since Hf is a heavy element with strong spin-orbital coupling (SOC)63, DFT calculations that take SOC into account were also performed, but no significant change of Born effective charges and phonon frequencies is observed. A detailed comparison of all theoretical results is provided in the Supplementary Information.
To further explain the origin of Born effective charge, we employed the maximum localized Wannier function (MLWF)64,65 to project the band-by-band contribution. The Wannier90 software package is used for this analysis66.
Data availability
The datasets generated and/or analyzed during this study are available from the corresponding author on reasonable request. Files related to the first principles calculation are available at this link: https://doi.org/10.13020/jyp2-qd29.
Code availability
The main components for our simulation are available at this GitHub address: https://github.com/lyglst/HfS2_results. Other computation details will be available from the corresponding authors upon request.
References
Kim, B. J. et al. Novel Jeff = 1/2 mott state induced by relativistic spin-orbit coupling in Sr2IrO4. Phys. Rev. Lett. 101, 076402 (2008).
Birol, T. & Haule, K. Jeff = 1 /2 Mott-insulating state in Rh and Ir fluorides. Phys. Rev. Lett. 114, 096403 (2015).
Zwartsenberg, B. et al. Spin-orbit-controlled metal-insulator transition in Sr2IrO4. Nat. Phys. 16, 290–294 (2020).
Cao, G. et al. Electrical control of structural and physical properties via strong spin-orbit interactions in Sr2IrO4. Phys. Rev. Lett. 120, 017201 (2018).
Cao, G. et al. Charge density wave formation accompanying ferromagnetic ordering in quasi-one-dimensional BaIrO3. J. Phys. C 113, 657–662 (2000).
Chen, Q. et al. Realization of the orbital-selective Mott state at the molecular level in Ba3LaRu2O9. Phys. Rev. Mater. 4, 064409 (2020).
Singleton, J. et al. Magnetic properties of Sr3NiIrO6 and Sr3CoIrO6: magnetic hysteresis with coercive fields of up to 55 T. Phys. Rev. B 94, 224408 (2016).
O’ Neal, K. R. et al. Spin-lattice and electron-phonon coupling in 3d/5d hybrid Sr3NiIrO6. npj Quant. Mater. 48, 1–6 (2019).
Ma, Y., Kou, L., Huang, B., Dai, Y. & Heine, T. Two-dimensional ferroelastic topological insulators in single-layer Janus transition metal dichalcogenides MSSe (M=Mo, W). Phys. Rev. B 98, 085420 (2018).
Yao, Q., Zhang, L., Bampoulis, P. & Zandvliet, H. J. Nanoscale investigation of defects and oxidation of HfSe2. J. Phys. Chem. C 122, 25498–25505 (2018).
Zhu, H. et al. Defects and surface structural stability of MoTe2 under vacuum annealing. ACS Nano 11, 11005–11014 (2017).
Chen, C. et al. Surface phases of the transition-metal dichalcogenide IrTe2. Phys. Rev. B 95, 094118 (2017).
Ideta, S. et al. Ultrafast dissolution and creation of bonds in IrTe2 induced by photodoping. Sci. Adv. 4, aar3867 (2019).
Pascut, G. L. et al. Dimerization-induced cross-layer quasi-two-dimensionality in metallic IrTe2. Phys. Rev. Lett. 112, 086402 (2014).
Jiang, K. et al. New pressure stabilization structure in two-dimensional PtSe2. J. Phys. Chem. Lett. 11, 73427349 (2020).
Kempt, R., Kuc, A. & Heine, T. Two-dimensional noble-metal chalcogenides and phosphochalcogenides. Angew. Chem. Int. Ed. 59, 9242–9254 (2020).
Kozhakhmetov, A. et al. Scalable BEOL compatible 2D tungsten diselenide. 2D Mater. 7, 015029 (2020).
Cheema, S. S. et al. Enhanced ferroelectricity in ultrathin films grown directly on silicon. Nature 580, 478–482 (2020).
Lee, H.-J. et al. Scale-free ferroelectricity induced by flat phonon bands in HfO2. Science 67, 1–10 (2020).
Iwasaki, T., Kuroda, N. & Nishina, Y. Anisotropy of lattice dynamical properties of ZrS2 and HfS2. J. Phys. Soc. Japan 51, 2233–22440 (1982).
Traving, M. et al. Combined photoemission and inverse photoemission study of HfS2. Phys. Rev. B 63, 035107 (2001).
Kanazawa, T. et al. Few-layer HfS2 transistors. Sci. Rep. 6, 22277 (2016).
Xu, K. et al. Ultrasensitive phototransistors based on few-layered HfS2. Adv. Mater. 27, 7881–7887 (2015).
Najmaei, S. et al. Cross-plane carrier transport in van der Waals layered materials. Small 14, 1–11 (2018).
Roubi, L. & Garlone, G. Resonance Raman spectrum of HfS2 and ZrS2. Phys. Rev. B 37, 6808–6812 (1988).
Uchida, S.-i & Tanaka, S. Optical phonon modes and localized effective charges of transition-metal dichalcogenides. J. Phys. Soc. Japan 45, 153–161 (1978).
Lucovsky, G., White, R. M., Benda, J. A. & Revelli, J. F. Infrared-reflectance spectra of layered group-IV and group-VI transition-metal dichalcogenides. Phys. Rev. B 7, 3859–3870 (1973).
Chen, J. Phonons in bulk and monolayer HfS2 and possibility of phonon-mediated superconductivity: a first-principles study. Solid State Commun. 237-238, 14–18 (2016).
Ibáñez, J. et al. High-pressure Raman scattering in bulk HfS2: comparison of density functional theory methods in layered MS2 compounds (M = Hf, Mo) under compression. Sci. Rep. 8, 1–10 (2018).
Fu, L. et al. van der Waals epitaxial growth of atomic layered HfS2 crystals for ultrasensitive near-infrared phototransistors. Adv. Mater. 29, 1700439 (2017).
Zhang, W., Netsu, S., Kanazawa, T., Amemiya, T. & Miyamoto, Y. Effect of increasing gate capacitance on the performance of a p-MoS2/HfS2 van der Waals heterostructure tunneling field-effect transistor. Japn. J. Appl. Phys. 58, SBBH02 (2019).
Kang, J., Sahin, H. & Peeters, F. M. Mechanical properties of monolayer sulphides: a comparative study between MoS2, HfS2, and TiS3. Phys. Chem. Chem. Phys. 17, 27742–27749 (2015).
Singh, D., Gupta, S. K., Sonvane, Y. & Kumar, A. 2D-HfS2 as an efficient photocatalyst for water splitting. Catal. Sci. and Technol. 6, 6605–6614 (2016).
Wang, D. et al. Selective direct growth of atomic layered HfS2 on hexagonal boron nitride for high performance photodetectors. Chem. Mater. 30, 3819–3826 (2018).
Wu, N. et al. Strain effect on the electronic properties of 1T-HfS2 monolayer. Physica E 93, 1–5 (2017).
Berwanger, M., Ahuja, R. & Piquini, P. C. HfS2 and TiS2 monolayers with adsorbed C, N, P atoms: a first principles study. Catalysts 10, catal10010094 (2020).
Zheng, B. et al. Vertically oriented few-layered HfS2 nanosheets: growth mechanism and optical properties. 2D Mater. 3, 035024 (2016).
Lei, C. et al. Broken-gap type-III band alignment in WTe2/HfS2 van der Waals heterostructure. J. Phys. Chem. C 123, 23089–23095 (2019).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Ghosez, P., Michenaud, J.-P. & Gonze, X. The physics of dynamical atomic charges: the case of ABO3 compounds. Phys. Rev. B 58, 6224–6240 (1998).
Baroni, S., De Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Cingolani, A., Lugará, M., Scamarcio, G. & Lévy, F. The Raman scattering in hafnium disulfide. Solid State Commun. 62, 121–123 (1987).
Wooten, F. Optical Properties of Solids (Academic Press, 1972).
Sun, Q. C. et al. Spectroscopic determination of phonon lifetimes in rhenium-doped MoS2 nanoparticles. Nano Lett. 13, 2803–2808 (2013).
Letcher, J., Kang, K., Cahill, D. G. & Diott, D. D. Effects of high carrier densities on phonon and carrier lifetimes in Si by time-resolved anti-Stokes Raman scattering. Appl. Phys. Lett. 90, 252104 (2007).
Sun, Q. C., Xu, X., Vergara, L. I., Rosentsveig, R. & Musfeldt, J. L. Dynamical charge and structural starin in inorganic fullerenelike MoS2 nanoparticles. Phys. Rev. B 79, 205405 (2009).
Wieting, T. J. & Verble, J. L. Infrared and Raman studies of long-wavelength optical phonons in hexagonal MoS2. Phys. Rev. B 3, 4286–4292 (1971).
Carr, G. L., Perkowitz, S., & Tanner, D. B. Infrared and millimeter waves, vol. 13 (Academic Press, 1985).
Kittel, C. Introduction to Solid State Physics. 8th ed. (Wiley, 2004).
Sun, Q. C., Xu, X., Baker, S. N., Christianson, A. D. & Musfeldt, J. L. Experimental determination of ionicity in MnO nanoparticles. Chem. Mater. 23, 2956–2960 (2011).
Ashcroft, N. W. & Mermin, D. Solid State Physics (Thomson Learning, 1976).
Ghosez, P. & Gonze, X. Band-by-band decompositions of the Born effective charges. J. Condens. Matter Phys. 12, 9179–9188 (2000).
Pike, N. A. et al. Origin of the counterintuitive dynamic charge in the transition metal dichalcogenides. Phys. Rev. B 95, 201106 (2017).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Nunes, R. W. & Gonze, X. Berry-phase treatment of the homogeneous electric field perturbation in insulators. Phys. Rev. B 63, 155107 (2001).
Souza, I., Íñiguez, J. & Vanderbilt, D. First-principles approach to insulators in finite electric fields. Phys. Rev. Lett. 89, 117602 (2002).
Baroni, S., Giannozzi, P. & Testa, A. Green’s-function approach to linear response in solids. Phys. Rev. Lett. 58, 1861–1864 (1987).
Csonka, G. I. et al. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 79, 155107 (2009).
Sun, J. et al. Self-consistent meta-generalized gradient approximation within the projector-augmented-wave method. Phys. Rev. B 84, 035117 (2011).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Greenaway, D. L. & Nitsche, R. Preparation and optical properties of group IV–VI2 chalcogenides having the CdI2 structure. J. Phys. Chem. Solids 26, 1445–1458 (1965).
Yagmurcukardes, M., Ozen, S., Iyikanat, F., Peeters, F. M. & Sahin, H. Raman fingerprint of stacking order in HfS2- Ca(OH)2 heterobilayer. Phys. Rev. B 99, 205405 (2019).
Marzari, N. & Vanderbilt, D. Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 56, 12847–12865 (1997).
Marzari, N., Fe, P., Yates, J. R. & Vanderbilt, D. Maximally localized Wannier functions: theory and applications. Rev. Mod. Phys. 84, 1419–1475 (2012).
Mostofi, A. A. et al. An updated version of wannier90: a tool for obtaining maximally-localised wannier functions. Comput. Phys. Commun. 185, 2309–2310 (2014).
Acknowledgements
Research at the University of Tennessee is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Materials Science Division under award DE-FG02-01ER45885. The work at the University of Minnesota is supported primarily by the National Science Foundation through the University of Minnesota MRSEC under Award Number DMR-2011401. We acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the research results reported within this paper. We thank S. Najmaei and I. Boulares at the U. S. Army Research Lab for the 1T-HfS2 crystal and useful conversations.
Author information
Authors and Affiliations
Contributions
This project was conceived by J.L.M. and S.N.N. Raman scattering and infrared absorption measurements were conducted by S.N.N., and data were analyzed by S.N.N. and J.L.M. Theoretical studies and density functional calculations were carried out by S.L. and T.B. All authors discussed the data. The paper was written by S.N.N., S.L., T.B., and J.L.M. All authors read and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neal, S.N., Li, S., Birol, T. et al. Chemical bonding and Born charge in 1T-HfS2. npj 2D Mater Appl 5, 45 (2021). https://doi.org/10.1038/s41699-021-00226-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41699-021-00226-z
This article is cited by
-
Vibrational fingerprints of ferroelectric HfO2
npj Quantum Materials (2022)
