
Abstract
Genomic analyses have redefined the molecular subgrouping of pediatric acute lymphoblastic leukemia (ALL). Molecular subgroups guide risk-stratification and targeted therapies, but outcomes of recently identified subtypes are often unclear, owing to limited cases with comprehensive profiling and cross-protocol studies. We developed a machine learning tool (ALLIUM) for the molecular subclassification of ALL in retrospective cohorts as well as for up-front diagnostics. ALLIUM uses DNA methylation and gene expression data from 1131 Nordic ALL patients to predict 17 ALL subtypes with high accuracy. ALLIUM was used to revise and verify the molecular subtype of 281 B-cell precursor ALL (BCP-ALL) cases with previously undefined molecular phenotype, resulting in a single revised subtype for 81.5% of these cases. Our study shows the power of combining DNA methylation and gene expression data for resolving ALL subtypes and provides a comprehensive population-based retrospective cohort study of molecular subtype frequencies in the Nordic countries.
Similar content being viewed by others


Introduction
Pediatric acute lymphoblastic leukemia (ALL) comprises a heterogeneous group of patients who can be stratified into subgroups based on the presence of recurrent cytogenetic aberrations, which are important predictors of clinical outcome1,2. Subtypes of pediatric B-cell precursor ALL (BCP-ALL) are often characterized by large-scale chromosomal aberrations, including abnormal chromosomal numbers, translocations that give rise to expressed fusion genes, or other structural rearrangements. Before next-generation sequencing (NGS)-based methods were introduced into clinical practice, as many as 30% of all BCP-ALL cases either lacked conclusive results from standard cytogenetic analyses (denoted undefined) or were negative for the subtype-defining aberrations (denoted B-other) and therefore subtype information was not available for treatment stratification or disease monitoring in this large group of patients3. Recent application of high-resolution transcriptome sequencing (RNA-seq) has enabled the discovery of new oncogenic subgroups characterized by fusion genes, such as DUX4 (DUX4-r), ZNF384 (ZNF384-r), MEF2D (MEF2D-r) and NUTM1 (NUTM1-r) rearrangements4,5,6,7,8,9,10,11,12,13,14,15, as well as subtype-like signatures, such as BCR::ABL1-like/“Ph-like”16,17 or ETV6::RUNX1-like/”ER-like”7,18, and the PAX5-driven subtypes, PAX5 alteration (PAX5alt) and PAX5 P80R19,20,21,22. The clinical significance of the recently identified subtypes is often unclear, owing to the limited number of cases and differences between protocols and studies7,8,20,23,24. Therefore, retrospective ALL cohort analyses have been particularly powerful for studying rare ALL subtypes due to the large sample sizes available in biobanks and the prolonged period of follow-up to collect sufficient data on rare events.
Most of the recurrent molecular alterations in ALL are strongly associated with gene expression (GEX) profiles24,25. RNA-seq has since emerged as a powerful tool for the identification of both fusion genes and GEX subtype profiling in a single assay26,27, which promises to replace cumbersome standard karyotyping (G-banding), PCR-based and fluorescence in situ hybridization (FISH)-based methods in a clinical diagnostic setting28. Compared to DNA, RNA is prone to degradation, making it challenging to obtain high-quality RNA for retrospective cohort analyses. However, epigenetic profiling of DNA methylation (DNAm) using arrays or next-generation sequencing (NGS) has demonstrated comparable subtype-specific distributions in ALL cells29,30,31,32. DNAm is advantageous as an analyte due to its ability to identify methylation patterns associated with disease in degraded archival samples33. Leveraging biobank samples and retrospective cohort studies can provide valuable insights into long-term disease outcomes that may be challenging to obtain through prospective study designs, particularly for rare ALL subtypes.
In the present investigation, we describe a multimodal machine learning classification tool, ALL subtype Identification Using Machine learning (ALLIUM) that uses DNAm and/or GEX signatures (Fig. 1). We trained and applied ALLIUM to a large cohort of 1131 Nordic patient samples and determined the frequencies of recent genetic subtypes, which led to the revision of molecular subtypes in 81.5% of the unclassified BCP-ALL cases.

DNA methylation (DNAm, 450k arrays), gene expression (GEX, RNA-sequencing), and somatic mutation (WGS, targeted sequencing) data were generated from 1131 patients treated on the Nordic Society for Pediatric Hematology and Oncology (NOPHO) protocols diagnosed between 1996 and 2013. In total, the subtype of 281 of the BCP-ALL patients (24.8% of the entire ALL cohort) was unclassified at diagnosis. Molecular screening was performed based on a combination of cytogenetics, fusion gene screening, mutational analysis, and copy number analysis. Molecular screening resolved the subtype of 127 BCP-ALL patients. The remaining 154 patients were denoted “B-other”. A supervised classification method (ALLIUM) was used to build subtype-specific models based on two modalities (DNAm and GEX) for 17 of the known molecular subtypes of ALL. ALLIUM re-classified the subtype of 102 B-other patients. This study expanded the scope of known subtypes across the entire cohort resulting in 1079 with known subtype (95.4% of the entire ALL cohort). The 52 patients remaining unclassified at the end of the study are referred to as ALLIUM B-other.
Results
Molecular characteristics and data generation
Diagnostic bone marrow aspirates or peripheral blood samples from 1131 Nordic ALL patients (n = 1025 BCP-ALL and n = 106 T-ALL) were obtained from a population based cohort diagnosed between 1996 and 2013, and enrolled in the Nordic Society of Pediatric Hematology and Oncology (NOPHO) −92, −2000, −2008, EsPh-ALL, or Interfant treatment protocols34,35,36,37. Genome-wide CpG methylation levels were analyzed in 1125 DNA samples (1125 patients) using 450k arrays (DNAm dataset) and RNA-sequencing was performed in 328 RNA samples (315 patients, GEX dataset). Molecular subtypes were assigned based on standard cytogenetic analysis at ALL diagnosis38, where a total of 850 patients (75%) had an established molecular subtype and 281 were denoted unclassified BPC-ALL (Supplementary Data 1). We initially screened our cohort for the molecular ALL subtypes outlined by the International Consensus Classification (ICC)2 using a combination of genome-wide CNA detection, fusion gene screening (Supplementary Fig. 1), and targeted mutational assessment for PAX5 p.Pro80Arg, IKZF1 p.Asn159Tyr, and ZEB2 p.His1038Arg (Supplementary Data 2). This analysis, combined with putative revised molecular subtype information from previously published results7,9,30,39,40,41, identified 127 patients from the unclassified BCP-ALL group (45.2%) who belonged to one of the ICC subtypes. In total, this yielded 977 ICC subtype-defined cases (Table 1). Of note, this included 27 patients with established subtypes missed by routine diagnostics: HeH (n = 9), ETV6::RUNX1 (n = 9), KMT2A-r (n = 4), TCF3::PBX1 (n = 3), BCR::ABL1 (n = 1), and iAMP21 (n = 1). One patient (ALL_913) was re-labelled from HeH to DUX4-r, after confirmation of the presence of the IGH-DUX4 fusion gene and a modal number of 46 chromosomes. The 30 patients with dic(9;20) aberrations42 were re-labelled as PAX5alt. The remaining 154 cases were denoted as B-other after the application of the aforementioned genomic techniques failed to resolve their subtype (Table 1).
ALLIUM is a highly sensitive method for molecular ALL subtype classification
In order to design a DNAm and GEX-based classifiers for ALL, the 977 patients with known ICC molecular subtypes defined based on updated molecular analysis were split into design and hold-out datasets to create and validate the ALLIUM classifier (Table 2). An internally produced replication set (n = 13, GEX) and three external datasets (GEX: GSE16150143, GEX: GSE228632 and DNAm: GSE5660031) were used for additional independent subtype verification. ALLIUM is based on nearest shrunken centroid (NSC)44 models consisting of DNAm and GEX data in a one vs. rest approach. Subtypes with similar molecular profiles, i.e. those characterized by aneuploidies (HeH, low HeH, iAMP21, hypodiploidy), ETV6 gene rearrangements (ETV6::RUNX1, ETV6::RUNX1-like), and the Philadelphia (ph) chromosome (BCR::ABL1, BCR::ABL1-like) were handled in a different manner. For these subtypes, a two-step procedure with initial classification on the group level, followed by a one-vs-one or a multi-class classification within the group was applied (Supplementary Materials and Methods and Supplementary Fig. 2–3). Moreover, to identify misclassification errors due to low blast count, control classifiers for DNAm and RNA were built utilizing data available from ALL patients in remission or healthy blood donors45. As the output contained probability scores for each classifier, multiple subtype classifications could occur. Therefore, we proceeded with a multi- to single-class transformation, assigning the subtype with the highest probability score for each sample.
ALLIUM identified 379 CpGs and 356 genes as most informative for subtype determination (Supplementary Data 3–4). Unsupervised analysis of samples with known subtype revealed clear subtype-driven clustering (Fig. 2a–c, Supplementary Fig. 4). We evaluated the models using hold-out, replication and independent external validation datasets (Table 2, Supplementary Data 5–14). The classifiers were highly predictive overall, with 87.0% concordance between DNAm and true molecular subtype and 94.5% overall concordance between GEX and true molecular subtype (Fig. 2b–d). Both ALLIUM modalities achieved high specificity (>0.99) across the datasets, but the DNAm classifer displayed lower sensitivity (range 0.83 - 0.88) than the GEX classifier (range 0.79–1.00) (Table 2). Both DNAm and GEX classification was performed in samples from 242 patients in our dataset, enabling us to directly compare the two models (Fig. 2e, f). ALLIUM GEX resulted in 95.9% overall concordance with true subtype, 0.96 sensitivity and 0.99 specificity, while ALLIUM DNAm resulted in 93.8% overall concordance, 0.91 sensitivity and 0.99 specificity across these 242 patients (Fig. 2f, Supplementary Data 15). Only 19 cases showed discrepant result between the GEX and/or DNAm classifiers and true subtype. The GEX classifier was more often correct (n = 9), while the DNAm was correct in four cases. A complete mismatch was observed for the remaining six cases, including four cases with no class assignment (Supplementary Data 16). Of the nine patients that were correctly predicted by GEX, the DNAm classifier was unable to return a prediction for five cases (no-class), which supports our previous observation that ALLIUM DNAm is less sensitive than the GEX classifier (Supplementary Data 16). To summarize, the performance of the GEX and DNAm classifiers was largely consistent across the design, hold-out, replication and external validation datasets across the subtypes (Fig. 2g, Supplementary Fig. 4, Supplementary Data 14).

a Unsupervised hierarchical clustering based on the DNA methylation (DNAm) β-values of 379 CpG sites across molecularly defined patients (n = 971) and control samples (n = 139). The heatmap shows the DNA methylation β-value for each CpG (y-axis) and sample (x-axis). The color key is indicated to the right of the panel. b Confusion matrix showing the concordance between ALLIUM DNAm subtype predictions (x-axis) and true molecular subtypes (y-axis) for 971 patients. The numbers indicate the number of patients by subtype. c Unsupervised hierarchical clustering based on gene expression (GEX) levels of 356 genes across molecularly defined patients (n = 248) and control samples (n = 12). The heatmap shows the min-max scaled log2 gene expression levels of the 356 genes (y-axis) by sample (x-axis). The color key for the heatmap is indicated in the right side of the panel. d Concordance between ALLIUM GEX subtype predictions (x-axis) and true molecular subtypes (y-axis) for 248 patients analyzed with ALLIUM GEX. e Barplots showing the degree of concordance between ALLIUM DNAm and GEX predictions for 242 samples with data from both modalities. The subtype is indicated along the y-axis and the number of patients along the x-axis. The light bars represent the overall number of predictions per subtype and the darker bars indicate the number predictions concordant between DNAm and GEX. Patients with “no class” predictions (n = 9) are not shown. f Line plots demonstrating the sensitivity (top) and specificity (bottom) of ALLIUM DNAm (circle) and GEX (square) models overall and by subtype for the 242 patients analyzed with both data modalities. g Bi-directional barplots showing the sensitivity and specificity by subtype for the design, hold-out, replication, DNAm GSE56600, GEX GSE161501 and GEX GSE228632 datasets. The sensitivity is indicated by the left-sided bar, while the specificity is indicated by the right-sided bar for each dataset and subtype. The overall performance is shown on the top of each barplot. The number of patients in each dataset by subtype is indicated to the right of each barplot.
Functional annotation of genes and CpG sites identified by ALLIUM
Next, we explored the relationship between the features (CpG sites and genes) identified by ALLIUM. Initially, we assessed the covariance between the datasets using cross-decomposition analysis, focusing patients in the design cohort with overlapping DNAm and GEX data (n = 201). We analyzed all the available features, totaling 19,774 genes and 167,353 CpG sites, as well as the 356 genes and 379 CpG sites selected by ALLIUM. Notably, the features chosen by ALLIUM exhibited substantial correlation for components 1 (88%) and 2 (96%), while the correlation among all features was consistently below 10% (Fig. 3a, b). The strong correlation highlights a potentially biologically relevant association between the features selected by the two ALLIUM modalities.

Cross-decomposition analysis with Partial Least Squares (PLS) Canonical analysis. The UMAP plots indicate components 1 and 2 for a the DNAm (n = 167,353) vs the GEX (n = 19,774) unselected signatures, and b the ALLIUM DNAm (n = 379) and GEX (n = 356) signatures (right). The points indicate the training (67%, blue) and test sets (33%, red). The Pearson’s correlation coefficient for the comparing modalities per component is denoted in the title of each plot. Boxplots demonstrating the c GEX levels for four selected genes across 315 patients grouped by revised molecular subtype. d DNAm levels for four selected CpG sites across 1125 patients by revised molecular subtype. The boxes are color-coded by respective subtype according to the key at the bottom of the panel. The Benjamini-Hochberg (BH) corrected Kruskal-Wallis H-test p value indicates the statistical significance between subtypes (bottom right). Asterisks indicate the subtype(s) for which ALLIUM chose each specific CpG or GEX signature. The lines (whiskers) on the boxplots represent the distribution of residual data points beyond the lower and upper quartiles.
To further explore this association, we considered the genomic distribution, the extent and magnitude of gene expression and DNA methylation changes, as well as their regulation across the features specific for the different subtypes. The features were distributed across all chromosomes, with no significant overall enrichment in the genomic locations of CpG sites or genes (FDR q value > 0.05, Supplementary Fig. 5 and Supplementary Data 17–18). However, the CpGs sites did show a significant enrichment in intergenic “open sea” regions outside of CpG islands (FDR q value < 0.0001, Supplementary Data 19).
We assessed the directionality of the subtype-defining features, and observed they were more frequently hypomethylated at the CpG level (Supplementary Fig. 6) and displayed greater expression levels and variability at the gene expression level (Supplementary Fig. 7). Further investigation of the genomic overlap between CpG sites and genes revealed 21 CpG sites that overlapped with the genomic location of 18 ALLIUM GEX genes (Supplementary Data 20–21). Among these, eight CpG sites located in seven genes were selected for the same subtype. For example, PAX5alt shared four CpG sites in the RAPGEF4, CALN1 and NAV2 genes, the aneuploidy group had one CpG site located within each of the LCN6 and CELSR1 genes, the ETV6 group had one CpG site in FARP1, and TCF3::PBX1 had one CpG site in KANK1. For these genes we observed an inverse relationship between methylation level and gene expression (Pearson’s correlation coefficient = −0.58, Supplementary Fig. 8).
Importantly, ALLIUM consistently selected well-known ALL genes such as NUTM1 for NUTM1-r, PBX1 for TCF3::PBX1, MEF2C for MEF2D-r, CEBPA for ZNF384-r, CpG sites in CBFA2T3 for the ETV6-group, the expression of CDKN2A and CpG sites in AUTS2 for the PAX5alt group, along with CpG sites in ETV6, RUNX2, and IKZF1 for PAX5 P80R (Fig. 3c, d). These findings cumulatively underscore that ALLIUM classifiers consistently selected biologically relevant features for characterizing the subtypes.
Comparisons of model performance
Several GEX-based methods for ALL subtyping have been developed independently. We evaluated the ALLIUM GEX classifier against ALLSorts26 and ALLCatchR27. As both classifiers are trained specifically for BCP-ALL, we removed T-ALL from the comparison. ALLIUM GEX, ALLSorts, and ALLCatchR were evaluated for the 309 BCP-ALL samples of known subtype across all the five GEX datasets included herein. Overall, the three classifiers performed similarly (Fig. 4a–c), although notable differences between ALLIUM GEX, ALLSorts, and ALLCatchR, respectively, were observed for classification of PAX5alt (100%, 71%, 71%), HeH (86%, 71%, 86%), and iAMP21 (100%, 33%, 67%) using our hold-out dataset (Supplementary Figs. 4 and 9–12). Specifically, we noted that ALLSorts and ALLCatchR predicted three (including a multi-class case) and eight out of 14 PAX5alt patients with dic(9;20) as BCR::ABL1-like, respectively. ALLIUM was not trained on BCL2/MYC, IKZF1 N159Y, HLF, CEB and CDX2::UBTF subtypes and ALLSorts and ALLCatchR did not predict any of these rare subtypes in our data sets.

a Concordance between ALLIUM GEX subtype predictions (x-axis) and true molecular subtypes (y-axis) for 309 BCP-ALL samples of known subtype (95.5%, 295/309). b Concordance between ALLSorts subtype predictions (x-axis) and true molecular subtype (y-axis) (83.5%, 258/309). c Concordance between ALLCatchR subtype predictions (x-axis) and true molecular subtype (y-axis) (87.4%, 270/309). ALLCatchR was not trained on low HeH. d Boxplots demonstrating classification performance, including precision, sensitivity, specificity, F1 score and accuracy (balanced) for the three GEX models (n = 309 samples) and ALLIUM DNAm (n = 1104 samples with known subtype). The lines (whiskers) on the boxplots represent the distribution of residual data points beyond the lower and upper quartiles.
Overall, ALLIUM DNAm performed similarly to the GEX models (Fig. 4d). No other model is currently available for subtyping in ALL by DNA methylation, with the exception of a model built by us previously for eight ALL subtypes30. We compared the 379 CpG sites selected by ALLIUM to our previous classifier (n = 232 CpG sites), which resulted in 28.9% (67/232) overlapping sites with ALLIUM (Supplementary Data 22).
Resolved molecular subtypes of unclassified BCP-ALL
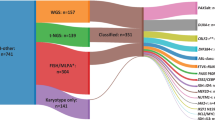
Next, we applied ALLIUM to the 154 remaining B-other cases in our cohort (Supplementary Data 23–24). For 67 cases where both DNAm and GEX data were available, 51 (76.1%) received concordant subtype predictions (Fig. 5a). The highest concordance was observed for the prediction of subtypes with fusion genes, i.e. ZNF384 (4/4, 100%), KMT2A (1/1, 100%), DUX4-r (11/12, 91.7%), ETV6::RUNX1-like (6/7, 85.7%), and PAX5alt (20/24, 83.3%). To establish consensus molecular subtypes for the B-other group, we constructed a 4-tier system to improve the confidence of subtype re-annotation (Fig. 5b). Tier 1 included 28 patients with a high score from the DNAm or GEX classification combined with molecular evidence to support the subtype: expressed fusion gene, CNA, karyotype, or mutation. Tier 2 comprised 34 patients with concordant GEX and DNAm classification, but lacked conclusive molecular evidence. Tier 3 included 40 patients with only DNAm predictions or a discordant prediction with one non-class and one high-score subtype prediction. Lastly, tier 4 included 52 patients where ALLIUM generated low confidence predictions or two conflicting predictions. Figure 5c illustrates the distribution of molecular subtypes within the group of 281 initially unclassified BCP-ALL patients following molecular screening and ALLIUM classification (tier 1-3).

a Concordance of ALLIUM subtype estimation for 67 B-other patients with both DNA methylation (DNAm, x-axis) and gene expression (GEX, y-axis) data. b ALLIUM stratification into subtype and tier group for the 154 B-other patients. c Subtype distribution after molecular and ALLIUM re-classification for the complete set of 281 patients with unclassified subtype at the start of the study. d Unsupervised dimensionality reduction (UMAP) based on the DNAm levels of 379 CpG sites across the 971 samples with molecularly defined subtype and 139 controls used to train ALLIUM DNAm and the 102 B-other samples reclassified by ALLIUM DNAm. e UMAP based on 356 genes across 248 samples with molecularly defined subtype and 12 controls used to design ALLIUM GEX and the 56 B-other samples reclassified by ALLIUM GEX. f Flow chart of molecular subtype revision in the study. g Distribution of subtypes across entire BCP-ALL cohort (n = 1025) color-coded by subtype determined at ALL diagnosis (start of study) and h distribution after molecular screening and ALLIUM re-classification (end of study).
The reclassified samples clustered with samples of known subtype (Fig. 5d, e, Supplementary Fig. 13). Sub-clusters were observed for KMT2A-r, DUX4-r, and T-ALL. In concordance with previous reports46,47, these included two putative KMT2A-r and DUX4-r subclusters based on GEX data and two T-ALL clusters in the DNAm data. Notably, the KMT2A-r and DUX4-r clusters were not visible in the DNAm visualization, while the T-ALL cluster was not visible in the GEX data, but this may be due to few (n = 19) patients with RNA-seq data. We examined fusion gene usage and found that the KMT2A-r cluster 1 (C1, n = 5) was characterized by USP2 (n = 3) and USP8 (n = 1) fusions, while C2 (n = 7) primarily contained patients with KMT2A::AFF1 fusions (n = 4), indicating sub-clustering associated with the fusion partner. The patients in DUX4-r cluster 1 (C1, n = 15) expressed DUX4::IGH (n = 10), alongside a diverse array of other fusions, including CRLF2::IRF1, PAX5::FLI1, ELL::KLF2, ATAD2::NPM1 and PAX5::FOXP1. In DUX4-r cluster 2 (C2, n = 16), DUX4::IGH (n = 9) was the most prevalent fusion. Seven of the 19 T-ALL patients with RNA-seq data carried fusion genes, but no apparent clustering by fusion partner was observed. Additional information about the fusion genes detected by group can be found in the Supplementary Fig. 14. In summary, by employing molecular screening and the ALLIUM classification method, we successfully elucidated the molecular subtype for 229 out of 281 BCP-ALL patients who had not been previously characterized (Fig. 5f). Consequently, our study expanded the scope of known subtypes across the entire cohort, encompassing a total of 973 out of 1025 BCP-ALL patients (94.9%) (Fig. 5g, h).
Clinical characteristics of molecular subtypes
We obtained complete clinical data from 1124 out of the 1131 patients in our study cohort. The median follow-up period for surviving patients was 16.0 years (interquartile range, IQR, 13.0–19.0). As anticipated, the basic clinical variables exhibited significant variation across different subgroups and these closely aligned with well-established patterns observed in other well-described ALL cohorts (Supplementary Data 25). The distribution of ALL subtypes based on patient age at the time of diagnosis is similar to other cohorts (Fig. 6a)1. The white blood cell (WBC) count also demonstrated significant variability among subtypes. High-risk subtypes, including T-ALL, KMT2A-r, BCR::ABL1, and BCR::ABL1-like ALL, were associated with elevated WBC counts, whereas patients with established low-risk subtypes, such as HeH and ETV6::RUNX1, exhibited the lowest WBC levels. Notably, the DUX4 group consistently displayed the lowest WBC counts across all subgroups (Fig. 6b).

a Histogram of subtype distribution by age. The age distribution color coded by subtype determined at ALL diagnosis is indicated in the top panel. The distribution of the originally unclassified patients color coded by revised molecular subtype is indicated in the lower panel. b Boxplots of the white blood cell count (WBC) at ALL diagnosis by revised molecular subtype. c Boxplot of minimal residual disease (MRD) levels at day 29 of treatment, for 368 patients with MRD information available. d Stacked barplots showing sex, treatment protocol, risk groups, primary event, and cause of death per subtype by reclassified subtype. CR1: complete remission, DCR1: death in complete remission, smn: secondary malignant neoplasm. The plots are color-coded based on their respective subtypes. The lines (whiskers) on the boxplots represent the distribution of residual data points beyond the lower and upper quartiles.
Minimal residual disease (MRD) status at the end of the induction phase (day 29) exhibited notable variation among the subtypes (Kruskal-Wallis p-value = 1.36e-08, Fig. 6c). For instance, the majority of patients with MEF2D-r (n = 5/5, 100%), TCF3::PBX1 (n = 13/13, 100%), and PAX5alt (n = 30/32, 93.8%) were MRD-negative at day 29. In contrast, all eight ZNF384-r cases were MRD-positive. Intriguingly, despite the high MRD levels observed in the ZNF384-r analyzed by MRD, only six relapses and one death were recorded in this subgroup. Patients belonging to the ETV6::RUNX1-like and PAX5-alt groups also demonstrated favorable overall outcomes, with >75% achieving complete remission without events. In line with expectations, emerging high-risk subtypes, such as BCR::ABL1-like, presented poor outcomes.
Discussion
Recent developments in integrated large-scale genomic analyses have greatly improved our knowledge of the genetic basis of ALL, identification of new subtypes and disrupted pathways that can be targeted therapeutically20,24,46,48. Accurate detection of the subtype-defining alterations in the clinical setting is crucial to guide risk and treatment stratification, monitor treatment response, and is very important for future implementation of tailored or precision therapy2. Given the low frequencies of rare subtypes in ALL and the long follow-up data needed to evaluate their clinical relevance, it is imperative to have methods that allow for retrospective analysis of biobank material, in addition to robust diagnostics in prospective cases. Herein, we designed and implemented a multimodal classification approach for ALL (ALLIUM) that captures epigenomic and transcriptomic alterations left as a detectable footprint in ALL cells. We demonstrate the utility of ALLIUM by retrospectively evaluating the frequency and clinical impact of emerging molecular cytogenetic subtypes in a large cohort of patients treated uniformly on NOPHO protocols between 1996 and 2013 and in external datasets.
Machine learning (ML) has the potential to improve clinical diagnostics by enabling automated and accurate diagnostics, with reduced cost49. A unique feature of ALLIUM, over other ML-based subtype algorithms26,27 is that it can use multiple modalities (DNA methylation and/or gene expression) for subtype determination. We demonstrate that a DNAm-based classifier can achieve a comparable performance to GEX-based methods. A specific strength of DNAm as an analyte is its ability to identify disease-related methylation patterns and potential biomarkers in archived samples33. By using biobank samples and retrospective cohort studies, insight into long-term disease outcome can be gained, which would be difficult to obtain through prospective study designs, especially for rare subtypes. One limitation of the DNAm classifier was its reduced sensitivity, which can be ascribed to the inherent constraint of DNAm data, which ranges from 0% to 100% methylation per CpG site. This limited range renders DNAm data more susceptible to confounding effects stemming from lower blast percentages. In contrast, the dynamic range inherent in gene expression (GEX) may offer greater flexibility, enabling compensation or correction in scenarios involving low blast percentages27. The ability of RNA-seq to detect fusion genes and coding mutations that can provide clear molecular evidence for subtype decision making, gives additional value to the GEX approach26,27 for prospective clinical diagnostics. However, RNA is not as readily available from historical material in biobanks, limiting the usefulness of GEX classifiers for retrospective interrogations. On the other hand, immerging studies demonstrate how DNA methylation holds significant potential for prognostication across a spectrum of hematological malignancies47,50,51,52,53, which will be an interesting avenue to pursue in future studies. Array-based DNAm assays also have the added advantage of concurrently generating comprehensive CNA profiles54. These CNA profiles help distinguishing subtypes characterized by large-scale copy number changes, such as HeH, low HeH, hypodiploidy and iAMP21. In centers where DNAm subtyping for brain cancer is already established55,56, the incorporation of ALLIUM DNAm subtyping could serve as a complementary diagnostic modality.
Using ALLIUM as a tool, we were able to accurately detect molecular ALL subtypes for up to 81.5% of previously unclassified BCP-ALL cases in our population-based Nordic cohort spanning three NOPHO protocols (1992, 2000, 2008). We found that the molecular composition of BCP-ALL cases in the Nordics is comparable to studies from Europe57,58, USA20,46,59, and Asia60, and others61. In order of prevalence, these include PAX5alt (with a frequency of 8% compared to a range of 4–10% in the aforementioned studies), BCR::ABL1-like (3% vs 3–13%), DUX4-r (4% vs 4–7%), ETV6::RUNX1-like (2% vs 1–3%), MEFD2-r (<1% vs 1–2%), NUTM1-r (<1%, vs < 1–1%), PAX5 P80R (<1% vs 1–2%). ALLIUM performed comparably to two other GEX subtyping models26,27, with two differences. Due to lack of training samples from rare subtypes, including t(5;14)(q31.1;q32.3)/IL3::IGH, IKZF1 N159Y, and CDX2::UBTF62 in the Nordic training set, ALLIUM cannot differentiate these subtypes. A second notable difference between ALLIUM and the other models is their performance distinguishing PAX5alt from BCR::ABL1-like. This may be due to differences in how these subtypes were defined in the training sets. PAX5 alterations have been carefully curated in the Nordics, based on a combination of PCR and high-resolution genome-wide analyses21,42,63,64. For example, the PAX5-driven aberration dic(9;20) was included as an obligatory risk-stratifying subgroup in the NOPHO-2008 protocol, and thus this aberration has been studied in detail38,65. The low-risk clinical indicators (low WBC, MRD-, 76% EFS) in the PAX5alt group in combination with high-risk indicators (high WBC, MRD + , 37% EFS) in the BCR::ABL1-like group would indicate that the groups defined by ALLIUM distinguish potentially clinically relevant groups. Further clarification will be needed in future efforts to refine subtype-decision models and highlights the need for large international collaborations to achieve this.
An additional strength of our study is our ability to assess the added value of MRD risk stratification in light of new molecular subtypes37. Although MRD remains as one of the best prognostic markers for treatment outcome in ALL, our results further underscores that MRD stratification in the new subtypes may not be uniformly applicable59,60,66. Furthermore, early monocytic lineage switching, which includes loss of the B-cell immunophenotype, has been described in DUX4-r, ZNF394-r and PAX5 P80R subtypes66, potentially leading to an underestimation of MRD levels in these groups. However, questions still remain if MRD is a clinically relevant measure for treatment decisions in these new groups. Although we do not know what the MRD levels were of the patients included herein treated prior to 2008, our confirmatory observations further support that slow clearance of MRD specifically in the ZNF384-r may not accurately measure future outcome.
Our study primarily focused on molecular subtype classification. While our study provides valuable insights into molecular subtypes, we did not comprehensively analyze correlations with clinical data, treatment regimens, or long-term follow-up information. This lack of comprehensive clinical investigation limits our ability to draw conclusions about the impact of specific subtypes on treatment responses and long-term outcomes, which will be important to address in future studies. Finally, our study is based on the current state of knowledge and the 17 of the 22 know ICC subtypes that were present in our Nordic training cohort. The field is rapidly evolving, with new subtypes, biomarkers, and machine learning models adapted for this purpose26,27,67,68. The development of multiple ALL classification methods ensures that solutions are well-tested, adaptable, and capable of addressing a wide array of samples and cohorts from diverse genetic backgrounds. Although ALLIUM was not trained on all ICC subtypes, it remains the only model available for predicting ALL subtypes from DNA methylation data.
In summary, by implementing ALLIUM for retrospective analysis of a large retrospective ALL cohort, we were able to accurately assess molecular subtype in previously undefined Nordic BCP-ALL cases. ALLIUM is freely available on GitHub and can be applied to determine molecular subtype membership of patients with either DNA methylation array data or RNA-seq data for research, or to support future precision diagnostics in pediatric ALL.
Methods
Patients
Bone marrow aspirates or peripheral blood samples collected at diagnosis from 1131 unique population.-based pediatric ALL patients were obtained from children diagnosed in the Nordic countries during 1996–2013 and enrolled on the Nordic Society of Pediatric Hematology and Oncology (NOPHO) NOPHO-92 (n = 201), NOPHO-2000 (n = 493), NOPHO-2008 (n = 380), EsPh-ALL (n = 17), or Interfant (n = 40) treatment protocols34,35,36,37. Molecular diagnosis of ALL was established by analysis of leukemic cells at the time of diagnosis with respect to morphology, immunophenotype and cytogenetics. The guardians and/or the patients provided written consent. The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Regional Ethical Review Authority in Uppsala, Sweden and by the NOPHO Scientific Committee (Study #56).
DNA and RNA extraction
DNA and RNA were extracted from primary ALL cells after Ficoll gradient separation using reagents from the AllPrep DNA/RNA/miRNA Universal Kit (Qiagen) or the AllPrep DNA/RNA Kit (Qiagen), including a DNase treatment step (Qiagen). DNA and RNA were quantified using the reagents from the double-stranded DNA Broad Range Kit or the RNA Broad Range kit on a Qubit instrument (Life Technologies). RNA quality was determined using the RNA Integrity Number (RIN) assessed by the Bioanalyzer or TapeStation system (Agilent).
DNA methylation arrays
Genome-wide DNA methylation levels were determined using the Infinium HumMeth450K BeadChip assay (450k array, Illumina). DNAm data were generated using 250 ng input DNA from 384 newly collected BCP-ALL samples on the 450k array. Data from 741 patients were retrieved from Gene Expression Omnibus (GEO) entry GSE4903145. Batch correction was not applied to the DNAm data (Supplementary Fig. 15). The complete DNAm dataset (1125 patients) was firstly filtered according to a previous study45 resulting in 435,941 CpG sites and then to include probes present on the MethylationEPIC v.1.0. B5 manifest file (https://emea.support.illumina.com/downloads/infinium-methylationepic-v1-0-product-files.html), resulting in 406,542 CpG sites. After variance-based filtering (variance < 0.01), 167,353 CpGs remained for downstream analysis. CNAs were detected using intensity levels from the 450k arrays using the R package “CopyNumber450kCancer”69. Data from 50 normal blood cell samples (CD3 + and CD19 + , GSE49031) was used as control data for normalization and transformation of probe intensities (log2 ratio, LogR).
RNA sequencing
A total of 328 samples from 315 patients were subjected to RNA sequencing (Supplementary Data 2). RNA sequencing libraries were prepared from 132 samples with RIN > 7 using the Illumina TruSeq stranded Total RNA (RiboZero human/mouse/rat) kit with 300 ng of total input RNA. The libraries were paired-end (PE) sequenced (150 bp) on an Illumina HiSeq2500 or NovaSeq 6000 instrument to an average of 49.8 M (range 30.2-113.2 M) PE 150 bp reads per sample. Samples with RIN < 7 or with less than 300 ng input RNA available were prepared with the Illumina TruSeq RNA Access library preparation kit (n = 28 samples) and sequenced on an Illumina HiSeq 2500 instrument PE 150 bp to an average 34.0 M (range 12.6-55.1 M). RNA-seq data from 162 samples, generated with 1000 ng input RNA using the Script-Seq kit (EpiCentre)9,39,70 and 6 samples prepared with Illumina RNA access protocol39,41 were collected from previous studies. The raw sequencing data for each of the 328 libraries included in the study were processed together using the nextflow-based (21.02.0.edge) nf-core/rnaseq (3.0) pipeline, which includes trimming of the paired-end reads by trimgalore (0.6.6), alignment to GRCh38.103 with STAR (2.6.1d). The aligned reads were quantified at the transcript level using Salmon (1.4.0) and the transcript level expression values were subsequently summarized to the gene level using the bioconductor package tximeta (1.8.0). The gene count matrix was corrected for batch effects with ComBat-Seq. The genes were subsequently filtered to remove Y chromosome, scaffold, mitochondrial (MT), and ribosomal (RPS and RPL) genes, as well as non-protein coding genes resulting in 19,774 protein-coding genes for downstream analysis. Data were normalized using Gene Length corrected trimmed mean of M-values (GeTMM), adjusting the data for both gene length and library size and finally log2 transformed. Technical (n = 5, repeated RNA-seq library construction from same RNA sample) and biological replicates (n = 8, sample taken at relapse) from 11 patients were used to validate merging the different library types (Supplementary Fig. 15).
Fusion genes were detected using a combination of FusionCatcher 0.99.7d71 and targeted screening of 22 genes known ALL fusions (Supplementary Data 2). Fusion gene status for 61 patients in the study were described previously7. Candidate fusion genes were validated by supporting karyotype data, copy number analysis and/or by experimental validation using Sanger sequencing as previously described9.
Mutational analysis
Somatic single nucleotide variants (SNVs) were retrieved from a 872-cancer gene Haloplex panel for 144 patients in our study cohort72,73 and from whole genome sequencing performed on 41 patients40,41,72. Variant alleles PAX5 p.Pro80Arg, IKZF1 p.Asn159Tyr, and ZEB2 p.His1038Arg were screened for in the 328 samples with RNA-seq data using alleleCount/3.2.2 (https://github.com/cancerit/alleleCount) on bam files.
ALL subtype classification
ALLIUM was built using the scikit-learn package, based on the Nearest Shrunken Centroid (NSC) method44,74. Classifiers were built for each of 17 established molecular ALL subtypes present in our cohort and for healthy controls in a supervised manner. Models for DNAm and GEX datasets were designed separately. First, the data were split into design (known subtypes), hold-out (known subtypes) and discovery (B-other) sets. The models were trained, optimized and features were selected on the design set and then their performance was evaluated on hold-out and internal replication datasets. The models were further validated in independent external validation datasets: RNA-seq data from 65 Finnish patients from GSE228632 for which detailed information can be found in the Supplementary Materials and Methods, and published datasets from RNA-seq of 19 BCP-ALL patients from GSE16150143 and 450k DNAm from 227 BCP-ALL patients GSE5660031. Additional details can be found in the Supplementary Materials and Methods. ALLSorts and ALLCatchR were run on the corrected count matrix (n genes = 60,666) according to the instructions (https://github.com/Oshlack/ALLSorts/wiki/1.-Installation, https://github.com/ThomasBeder/ALLCatchR)26,27.
Cross-decomposition and enrichment analysis
Cross decomposition was performed with the Partial Least Squares (PLS) Canonical analysis using the scikit-learn package (sklearn.cross_decomposition) in Python to quantify the covariance between the DNA methylation and GEX datasets. PLS analysis used data from 201 patients with known molecular subtype with both data modalities (DNAm and RNA) available. The analysis was performed on the unselected set of CpG sites (n = 167,353) and genes (19,774), as well as the CpG sites (n = 379) and genes (n = 356) selected by ALLIUM. The patients were randomly split into a train (n = 134) and test data set (n = 67). Two components for the PLS Canonical transformer were chosen. Pearson’s correlation coefficient was used to measure the correlation between the two modalities for each component. One proportion z-tests followed by Benjamini-Hochberg correction were performed to measure the enrichment of the genomic locations of CpG sites and genes selected by ALLIUM in comparison to the unselected set of CpG sites and genes.
Clinical data
Outcome data for the 1131 patients in the study was retrieved from the NOPHO leukemia database in February 2022. In total, 1124 patients had complete follow-up data available and the average time since diagnosis was 16.5 years (range 9-26). OS was calculated as the time from the date of diagnosis to the date of last follow-up or death of any cause. Kruskal-Wallis H-test from the Python library scipy.stats assessed the significance of subtype-stratified MRD distributions. A p value < 0.05 (2-tailed) was considered statistically significant.
Data availability
The public data were available from their original studies and available at the Gene Expression Omnibus (GEO) under accession numbers GSE49031, GSE56600, GSE228632, and GSE161501. The GEX data are available under GSE227832. The 450k DNA methylation data are available under controlled access via https://doi.org/10.17044/scilifelab.22303531 (https://figshare.scilifelab.se/). Requests for data sharing may be submitted to Jessica Nordlund (jessica.nordlund@medsci.uu.se).
Code availability
All scripts and environment requirements to reproduce the analyses, as well as the ALLIUM model are available at GitHub https://github.com/Molmed/Krali_2023.
References
Inaba, H. & Mullighan, C. G. Pediatric acute lymphoblastic leukemia. Haematologica 105, 2524–2539 (2020).
Arber, D. A. et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood 140, 1200–1228 (2022).
Lilljebjörn, H. & Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 130, 1395–1401 (2017).
den Boer, M. L. et al. Outcomes of paediatric patients with B-cell acute lymphocytic leukaemia with ABL-class fusion in the pre-tyrosine-kinase inhibitor era: a multicentre, retrospective, cohort study. Lancet Haematol. 8, e55–e66 (2021).
Gu, Z. et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 7, 13331 (2016).
Hirabayashi, S. et al. ZNF384 -related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102, 118–129 (2017).
Lilljebjörn, H. et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 7, 11790 (2016).
Liu, Y.-F. et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 8, 173–183 (2016).
Marincevic-Zuniga, Y. et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J. Hematol. Oncol. J. Hematol. Oncol. 10, 148 (2017).
Ohki, K. et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 104, 128–137 (2019).
Shago, M., Abla, O., Hitzler, J., Weitzman, S. & Abdelhaleem, M. Frequency and outcome of pediatric acute lymphoblastic leukemia with ZNF384 gene rearrangements including a novel translocation resulting in an ARID1B/ZNF384 gene fusion. Pediatr. Blood Cancer 63, 1915–1921 (2016).
Yasuda, T. et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 48, 569–574 (2016).
Boer, J. M. et al. Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia 35, 2978–2982 (2021).
Hormann, F. M. et al. NUTM1 is a recurrent fusion gene partner in B-cell precursor acute lymphoblastic leukemia associated with increased expression of genes on chromosome band 10p12.31-12.2. Haematologica 104, e455–e459 (2019).
Dickerson, K. M. et al. ZNF384 Fusion Oncoproteins Drive Lineage Aberrancy in Acute Leukemia. Blood Cancer Discov. 3, 240–263 (2022).
Cario, G. et al. BCR-ABL1-like acute lymphoblastic leukemia in childhood and targeted therapy. Haematologica 105, 2200–2204 (2020).
Tasian, S. K., Loh, M. L. & Hunger, S. P. Philadelphia chromosome–like acute lymphoblastic leukemia. Blood 130, 2064–2072 (2017).
Zaliova, M. et al. ETV6/RUNX1 -like acute lymphoblastic leukemia: A novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype: ZALIOVA et al. Genes. Chromosomes Cancer 56, 608–616 (2017).
Bastian, L. et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 33, 1895–1909 (2019).
Gu, Z. et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 51, 296–307 (2019).
Marincevic-Zuniga, Y. et al. PAX5-ESRRB is a recurrent fusion gene in B-cell precursor pediatric acute lymphoblastic leukemia. Haematologica 101, e20–e23 (2016).
Passet, M. et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 133, 280–284 (2019).
Zhang, J. et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 48, 1481–1489 (2016).
Li, J.-F. et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc. Natl. Acad. Sci. 115, (2018).
Hetzel, S. et al. Acute lymphoblastic leukemia displays a distinct highly methylated genome. Nat. Cancer 3, 768–782 (2022).
Schmidt, B. et al. ALLSorts: an RNA-Seq subtype classifier for B-cell acute lymphoblastic leukemia. Blood Adv. 6, 4093–4097 (2022).
Beder, T. et al. The Gene Expression Classifier ALLCatchR Identifies B-cell Precursor ALL Subtypes and Underlying Developmental Trajectories Across Age. HemaSphere 7, e939 (2023).
Berglund, E. et al. A Study Protocol for Validation and Implementation of Whole-Genome and -Transcriptome Sequencing as a Comprehensive Precision Diagnostic Test in Acute Leukemias. Front. Med. 9, 842507 (2022).
Milani, L. et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 115, 1214–1225 (2010).
Nordlund, J. et al. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin. Epigenetics 7, 11 (2015).
Lee, S.-T. et al. Epigenetic remodeling in B-cell acute lymphoblastic leukemia occurs in two tracks and employs embryonic stem cell-like signatures. Nucleic Acids Res. 43, 2590–2602 (2015).
Chatterton, Z. et al. Epigenetic deregulation in pediatric acute lymphoblastic leukemia. Epigenetics 9, 459–467 (2014).
Dirks, R. A. M., Stunnenberg, H. G. & Marks, H. Genome-wide epigenomic profiling for biomarker discovery. Clin. Epigenetics 8, 122 (2016).
Biondi, A. et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol. 13, 936–945 (2012).
Pieters, R. et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370, 240–250 (2007).
Schmiegelow, K. et al. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 24, 345–354 (2010).
Toft, N. et al. Results of NOPHO ALL2008 treatment for patients aged 1–45 years with acute lymphoblastic leukemia. Leukemia 32, 606–615 (2018).
Norén-Nyström, U. et al. Genetic Subtypes and Outcome of Patients Aged 1 to 45 Years Old With Acute Lymphoblastic Leukemia in the NOPHO ALL2008 Trial. HemaSphere 7, e883 (2023).
Ivanov Öfverholm, I. et al. Overexpression of chromatin remodeling and tyrosine kinase genes in iAMP21-positive acute lymphoblastic leukemia. Leuk. Lymphoma 61, 604–613 (2020).
Nordlund, J. et al. Refined detection and phasing of structural aberrations in pediatric acute lymphoblastic leukemia by linked-read whole-genome sequencing. Sci. Rep. 10, 2512 (2020).
Sayyab, S. et al. Mutational patterns and clonal evolution from diagnosis to relapse in pediatric acute lymphoblastic leukemia. Sci. Rep. 11, 15988 (2021).
Zachariadis, V. et al. The frequency and prognostic impact of dic(9;20)(p13.2;q11.2) in childhood B-cell precursor acute lymphoblastic leukemia: results from the NOPHO ALL-2000 trial. Leukemia 25, 622–628 (2011).
Diedrich, J. D. et al. Profiling chromatin accessibility in pediatric acute lymphoblastic leukemia identifies subtype-specific chromatin landscapes and gene regulatory networks. Leukemia 35, 3078–3091 (2021).
Tibshirani, R., Hastie, T., Narasimhan, B. & Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl Acad. Sci. 99, 6567–6572 (2002).
Nordlund, J. et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 14, r105 (2013).
Brady, S. W. et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 54, 1376–1389 (2022).
Haider, Z. et al. DNA methylation and copy number variation profiling of T-cell lymphoblastic leukemia and lymphoma. Blood Cancer J. 10, 45 (2020).
Roberts, K. G. et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 371, 1005–1015 (2014).
Kann, B. H., Hosny, A. & Aerts, H. J. W. L. Artificial intelligence for clinical oncology. Cancer Cell 39, 916–927 (2021).
Duran-Ferrer, M. et al. The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome. Nat. Cancer 1, 1066–1081 (2020).
Yamato, G. et al. Genome-wide DNA methylation analysis in pediatric acute myeloid leukemia. Blood Adv. 6, 3207–3219 (2022).
Blecua, P., Martinez-Verbo, L. & Esteller, M. The DNA methylation landscape of hematological malignancies: an update. Mol. Oncol. 14, 1616–1639 (2020).
Krali, O. et al. DNA Methylation Signatures Predict Cytogenetic Subtype and Outcome in Pediatric Acute Myeloid Leukemia (AML). Genes 12, 895 (2021).
Galbraith, K. & Snuderl, M. DNA methylation as a diagnostic tool. Acta Neuropathol. Commun. 10, 71 (2022).
Pickles, J. C. et al. DNA methylation-based profiling for paediatric CNS tumour diagnosis and treatment: a population-based study. Lancet Child Adolesc. Health 4, 121–130 (2020).
Wu, Z. et al. Impact of the methylation classifier and ancillary methods on CNS tumor diagnostics. Neuro-Oncol. 24, 571–581 (2022).
Zaliova, M. et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 104, 1396–1406 (2019).
Ryan, S. L. et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia 37, 518–528 (2023).
Jeha, S. et al. Clinical Significance of Novel Subtypes of Acute Lymphoblastic Leukemia in the Context of Minimal Residual Disease–Directed Therapy. Blood Cancer Discov. 2, 326–337 (2021).
Li, Z. et al. Distinct clinical characteristics of DUX4- and PAX5 -altered childhood B-lymphoblastic leukemia. Blood Adv. 5, 5226–5238 (2021).
Lee, S. H. R. et al. Association of Genetic Ancestry With the Molecular Subtypes and Prognosis of Childhood Acute Lymphoblastic Leukemia. JAMA Oncol. 8, 354–363 (2022).
Duffield, A. S., Mullighan, C. G. & Borowitz, M. J. International Consensus Classification of acute lymphoblastic leukemia/lymphoma. Virchows Arch. Int. J. Pathol. https://doi.org/10.1007/s00428-022-03448-8 (2022).
Rezayee, F. et al. Feasibility to use whole-genome sequencing as a sole diagnostic method to detect genomic aberrations in pediatric B-cell acute lymphoblastic leukemia. Front. Oncol. 13, 1217712 (2023).
Öfverholm, I. et al. The prognostic impact of IKZF1 deletions and UKALL genetic classifiers in paediatric B-cell precursor acute lymphoblastic leukaemia treated according to NOPHO 2008 protocols. Br. J. Haematol. 202, 384–392 (2023).
Toft, N. et al. Risk group assignment differs for children and adults 1-45 yr with acute lymphoblastic leukemia treated by the NOPHO ALL-2008 protocol. Eur. J. Haematol. 90, 404–412 (2013).
Novakova, M. et al. DUX4r, ZNF384r and PAX5-P80R mutated B-cell precursor acute lymphoblastic leukemia frequently undergo monocytic switch. Haematologica 106, 2066–2075 (2021).
Hu, Z. et al. MD-ALL: an integrative platform for molecular diagnosis of B-acute lymphoblastic leukemia. Haematologica https://doi.org/10.3324/haematol.2023.283706 [Early view].
Mäkinen, V.-P., Rehn, J., Breen, J., Yeung, D. & White, D. L. Multi-Cohort Transcriptomic Subtyping of B-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 23, 4574 (2022).
Marzouka, N. et al. CopyNumber450kCancer: baseline correction for accurate copy number calling from the 450k methylation array. Bioinformatics 32, 1080–1082 (2016).
Marincevic-Zuniga, Y. et al. The Heterogeneous Fusion Gene Landscape in Pediatric Acute Lymphoblastic Leukemia. Blood 128, 4081–4081 (2016).
Nicorici, D. et al. FusionCatcher - a tool for finding somatic fusion genes in paired-end RNA-sequencing data. https://doi.org/10.1101/011650 (2014).
Lindqvist, C. M. et al. The Mutational Landscape in Pediatric Acute Lymphoblastic Leukemia Deciphered by Whole Genome Sequencing. Hum. Mutat. 36, 118–128 (2015).
Lindqvist, C. M. et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget 7, 64071–64088 (2016).
Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Acknowledgements
This work was supported by grants from the Swedish Research Council (2019-01976 to JN), the Swedish Cancer Society (CAN2018-623 to ACS and CAN2022-2395 to JN), the Swedish Childhood Cancer Foundation (PR2017-0023 to ACS and PR2019-0046 to JN), the Göran Gustafsons Foundation (to JN), the Jane and Aatos Erkko Foundation and the Academy of Finland #321550 (to OL and MH). DNA methylation array analysis and RNA-sequencing was performed with assistance from the SciLifeLab National Genomics Infrastructure, SNP&SEQ Technology Platform, which is funded by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. Computational resources were provided by the Swedish National Infrastructure for Computing (SNIC), National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Finnish IT Centre of Science (CSC) and University of Eastern Finland Bioinformatics Center. SNIC and NAISS are partially funded by the Swedish Research Council. We thank Sara Nystedt and Sara Nilsson for technical assistance, Jonas Carlsson Almlöf and Christofer Bäcklin for input on DNA methylation classification, and our colleagues from NOPHO LL Biology Group, Lucia Cavelier, Anna Bremer and Tatjana Pandzic for valuable input on the study design. We especially thank the ALL patients who contributed samples to this study. Figures 1, 5f and Supplementary Figs. 2–3 were made with Biorender.com.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
Y.M.Z., G.L., A.C.S., and J.N. conceived the study. O.K., Y.M.Z., G.A., A.P.E., A.L., S.S., V.Z., I.I.Ö., and L.O. analyzed the data. O.K. designed ALLIUM and made the figures. Y.M.Z., A.L., V.Z., L.O., and K.V. performed experiments. V.Z., M.H., J.S., I.I.Ö., G.B., A.N., L.O., K.V., H.L., T.Fi., and O.L. provided RNA-seq data. V.Z., I.I.Ö., G.B., A.N., and U.N.N. provided expertise on clinical diagnostics. H.O.M. and H.V.M. provided the MRD data. T.F.l., E.F., O.G.J., J.K., O.L., U.N.N., K.S., A.H., M.H., and G.L. provided clinical material and data. M.H. provided information from the NOPHO registry. E.F. and U.N.N. provided karyotyping expertise and data. M.H., O.L., G.L., A.C.S., and J.N. secured funding. O.K., Y.M.Z., A.P.E., G.A., G.L., and J.N. wrote the manuscript. All authors read and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krali, O., Marincevic-Zuniga, Y., Arvidsson, G. et al. Multimodal classification of molecular subtypes in pediatric acute lymphoblastic leukemia. npj Precis. Onc. 7, 131 (2023). https://doi.org/10.1038/s41698-023-00479-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00479-5
This article is cited by
-
Refining risk prediction in pediatric acute lymphoblastic leukemia through DNA methylation profiling
Clinical Epigenetics (2024)
