
Abstract
Recent studies on pediatric acute myeloid leukemia (pAML) have revealed pediatric-specific driver alterations, many of which are underrepresented in the current classification schemas. To comprehensively define the genomic landscape of pAML, we systematically categorized 887 pAML into 23 mutually distinct molecular categories, including new major entities such as UBTF or BCL11B, covering 91.4% of the cohort. These molecular categories were associated with unique expression profiles and mutational patterns. For instance, molecular categories characterized by specific HOXA or HOXB expression signatures showed distinct mutation patterns of RAS pathway genes, FLT3 or WT1, suggesting shared biological mechanisms. We show that molecular categories were strongly associated with clinical outcomes using two independent cohorts, leading to the establishment of a new prognostic framework for pAML based on these updated molecular categories and minimal residual disease. Together, this comprehensive diagnostic and prognostic framework forms the basis for future classification of pAML and treatment strategies.
Similar content being viewed by others


Main
Acute myeloid leukemia (AML) is characterized by aberrant clonal expansion of hematopoietic progenitors with differentiation defects1,2,3. Although pAML shares many clinical and pathological characteristics with adult AML, genetic differences have also been appreciated4,5. Notably, t(11;x), resulting in KMT2A rearrangements, are more common in pAML, and adult AML frequently harbors mutations in DNMT3A and splicing factor genes, whereas core binding factor (CBF) AMLs are common across the age spectrum4. In addition, progress in diagnostic technologies has identified cryptic fusions of NUP98 (ref. 6) and GLIS family7 members and UBTF tandem duplications8 enriched in pAML. Recent updates in the World Health Organization classification9 (WHO5th) and the International Consensus Classification10 (ICC) define AMLs with KMT2A and NUP98 rearrangements as distinct disease entities. However, recently discovered recurrent driver alterations in pAML remain categorized as ‘acute myeloid leukemia with other defined genetic alterations’ or ‘AML, not otherwise specified (NOS)’, confirming the need to understand both the biological and clinical features of pAMLs with these driver alterations.
Accumulation of clinical outcomes associated with gene alterations enabled risk stratification of adult AML according to detailed mutational profiling, such as the 2022 European LeukemiaNet risk stratification11. By contrast, risk stratification for pAML is still developing, and various strategies are utilized in clinical trials12,13,14,15. This is partly due to genetic differences between adult and pAML4, the rarity of the disease and a shortage of clinical outcome studies related to genetic alterations. To clarify the genomic landscape of pAML and its association with clinical outcomes, we characterized 887 cases of pAML by transcriptome and genome profiling. These analyses resulted in 23 molecular categories, defined by mutually exclusive gene alterations and specific expression profiles that show unique biological and mutational characteristics. These molecular categories have predictive value regarding clinical outcomes that can be leveraged to establish a framework for diagnosis and outcome prediction.
Results
Comprehensive genetic characterization of pAML
pAML samples were collected from previously published studies4,7,8,16,17,18,19,20,21,22,23,24,25 or at St. Jude Children’s Research Hospital, resulting in a cohort of 887 unique pAMLs either at diagnosis (n = 783, 88.3%) or at relapse (n = 104, 11.7%) (Fig. 1a, Extended Data Fig. 1a and Supplementary Table 1). This pAML cohort showed a wide age distribution at diagnosis (range 0–23.5 years; median 9.3) including young adults, with peaks in infancy and adolescence (Extended Data Fig. 1b). We first assessed the genetic landscape of these AMLs using RNA sequencing (RNA-seq) data to detect fusions, internal or partial tandem duplications (ITD/PTD), copy-number variants (CNV), as well as single nucleotide variants (SNV) and insertions and deletions (indels) (Fig. 1a–e, Extended Data Fig. 1c–e and Supplementary Tables 2–9). For 665 cases (74.9%) with either whole-genome sequencing (WGS, 59.2%) or whole-exome sequencing (WES, 44.0%), we also collected processed data from publications or performed de novo calling for newly included samples, which validated 97.3% of calls from the RNA-seq pipeline8 (Fig. 1a and Extended Data Fig. 1f).

a, Study cohort of pAML (n = 887) and study design. b, Recurrent pathogenic or likely pathogenic in-frame fusions (blue) and SVs (gray) detected in the entire cohort (n ≥ 3). Fusions included only in-frame fusions, and SVs included out-of-frame fusions resulting in loss of the C terminus of the protein and alterations detected from WGS data using CREST. c, Recurrent pathogenic or likely pathogenic somatic mutations (n ≥ 15). Colors represent types of mutations. Bars in b and c represent the total number of alterations in the cohort. d, Results of GISTIC analysis for focal chromosomal events (shorter than 90% of the chromosome arm). The left-hand panel shows the enrichment of focal gains (red) and the right-hand panel shows the enrichment of focal losses (blue). Green lines show a significance threshold for q values (0.25). Representative genes in enriched regions are highlighted. e, Genomic landscape and WHO classification of pAML. Representative genes from GRIN analysis or defining alterations are shown. Colors represent types of mutations. Numbers of gene alterations are shown next to gene names, and the lines of the box plot for VAFs represent the 25% quantile, median and 75% quantile. The upper whisker represents the higher value of maxima or 1.5× the interquartile range (i.q.r.), and the lower whisker represents the lower value of minima or 1.5× i.q.r. f, Summary of the WHO classification (WHO5th) of the entire cohort. A box with solid lines indicates categories with defining driver alterations. Boxes with dashed lines indicate subgroups with specified gene alterations, myelodysplasia-related or other defined genetic alterations. NA, Not available.
Pathogenic fusions or structural variants (SVs) were identified in 627 patients (70.7%). Most of these are recurrent and class-defining in pAML (for example, KMT2Ar, 20.3%; RUNX1::RUNX1T1, 12.4%) (Fig. 1b and Supplementary Table 6), whereas we also found fusions recurrent in other leukemias, such as SET::NUP214 (n = 1) or SFPQ::ZFP36L2 (n = 1). Mutational profiling revealed 1,924 pathogenic or likely pathogenic somatic mutations in 749 (84.4%) patients, including class-defining NPM1 (67 patients, 7.6%) and CEBPA (49 patients, 5.5%) mutations (Fig. 1c and Supplementary Tables 7 and 8). Most mutations were in genes involved in signaling pathways (n = 865), epigenetics (n = 312) and transcription factors (n = 432). RAS pathway mutations were most frequent, with 37.5% (333 of 887) having at least one RAS-related mutation and 21.3% of those (71 of 333) having mutations in multiple RAS pathway genes. Gains of chromosome 8 (7.3%) or chromosome 21 (6.2%) and loss of the long arm of chromosome 5 (5q-: 1.5%) or chromosome 7 (4.8%) were commonly observed (Fig. 1d, Extended Data Fig. 1e and Supplementary Table 9). Enrichment of focal deletions involving RB1 (13q14: 2.9%), ETV6 (12p13: 2.1%), NF1 (17q11: 2.0%) and TP53 (17p13: 2.0%), and focal gains involving AKT3 and FH (1q43: 3.0%) or ABCA transporters (17q24: 1.9%) were also identified. Genomic random interval (GRIN) analysis26 identified 142 altered genes with statistical significance (Fig. 1e and Supplementary Table 10). Consistent with previous reports, RAS-related mutations or FLT3-ITD with variable variant allele frequencies (VAFs) were highly co-occurring with class-defining alterations (Fig. 1e and Extended Data Fig. 2a,b). By contrast, mutations in UBTF or CBFB were exclusively found in cases without a defining alteration, as previously shown8,27, suggesting that these alterations define subgroups with distinct molecular characteristics.
Based on these collective data, we classified pAMLs using current WHO and ICC systems, and the frequencies of major classifications are consistent with cytogenetic profiles of European pAML cohorts28,29 (Fig. 1e,f, Extended Data Fig. 1g and Supplementary Fig. 1). In our pAML cohort, 68.5% of cases had specified genetic alterations in WHO5th, 10.7% of cases were defined as ‘acute myeloid leukemia, myelodysplasia-related’ (AML-MR) and the remaining cases with rare fusions or no defining alteration were classified as ‘acute myeloid leukemia with other defined genetic alterations’ (15.8%) or by differentiation stages (3.4%). By contrast, 95.0% of adult AMLs can be classified either by specific gene alteration (67.1%) or as AML-MR (27.8%)30, emphasizing the need for a more comprehensive classification of pAML based on its unique biology.
Molecular categories defined by unique gene alterations
We and others have shown that class-defining driver alterations are associated with specific expression patterns8,31 or that allele-specific and outlier expression of MECOM32,33, BCL11B (ref. 34) or MNX1 (ref. 35) by SVs can define subtypes. We then integrated the mutational landscape with expression profiling to define granular molecular categories for pAML (Supplementary Table 11). Uniform manifold approximation and projection (UMAP) analysis of transcriptional data revealed tight clustering of classes defined in WHO5th, including RUNX1::RUNX1T1, CBFB::MYH11 and CEBPA mutation, suggesting subtype-specific expression patterns (Fig. 2a and Extended Data Fig. 3a). We noted that clustering is also driven in part by differentiation status represented by marker gene expression, French–American–British (FAB) classification or cellular hierarchy36 (Extended Data Fig. 3c–e), contributing to heterogeneity within large categories such as KMT2Ar or NUP98r (Fig. 2a and Extended Data Figs. 3a and 4a). Diffusion maps37 confirmed similar patterns of clustering and differentiation status (Extended Data Fig. 3a–e). Cases with NPM1 fusions or indels outside the C terminus38 clustered with canonical NPM1 mutations, and we assigned them to the NPM1 category (Extended Data Fig. 4a); similarly, we assigned a RAR family fusion, TBL1XR1::RARB, to the acute promyelocytic leukemia category based on expression similarities with PML::RARA cases. Among the remaining cases without class-defining alterations, we found that the following alterations were also mutually exclusive and thus defined them as independent molecular categories: UBTF tandem duplications8, GLIS family (GLIS2-3) fusions7, fusions of FET and ETS family genes39,40 (for example, FUS::ERG), BCL11B SVs34 (Supplementary Table 12), PICALM::MLLT10, KAT6A rearrangements, MNX1 SVs41, RUNX1 fusion with CBFA2T2-3 (ref. 42) (RUNX1::RUNX1T1-like) and newly reported CBFB insertions (CBFB-GDXY)27 (Fig. 2a–c). GATA1 fusions (for example, MYB::GATA1) or mutations, rearrangements involving HOX cluster genes and KMT2A-PTD could rarely co-occur with the above-mentioned category-defining alterations (Fig. 2b). However, they were still predominantly found in cases without category-defining alterations and assigned to these categories only with consistent expression patterns and without previously explained driver alterations. By contrast, defining mutations of AML-MR in WHO5th were overall rare (range 0.1–2.1%), frequently co-occurred with other defining alterations (for example, EZH2 in PICALM::MLLT10), and could be found in various clusters rather than as a distinct group (Extended Data Fig. 3a,f), leading to its exclusion as a defining category for pAML. In addition to 11 categories defined by WHO5th, this pAML classification system with 12 new molecular categories captures 91.4% of pAML cases, contrasting to 68.5% by WHO5th (Fig. 3).

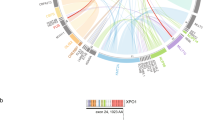
a, UMAP plot of the entire pAML cohort (n = 887) and cord blood CD34+ cells (normal controls: n = 5) using the top 315 variable genes. The colors of each dot denote the molecular categories of the samples. Representative category names are shown, and large clusters enriching specific categories are highlighted in circles (pink: NUP98::NSD1, NPM1, UBTF, DEK::NUP214, KMT2A-PTD; green: KMT2Ar and KAT6Ar; yellow: categories with acute megakaryocytic or erythrocytic expression; blue: MECOM, MNX1, ETS family, PICALM::MLLT10, BCL11B). b, Heatmap showing frequencies of defining gene alterations represented by color. Statistical significance was assessed by two-sided Fisher’s exact test to calculate P values of co-occurrence, followed by Benjamini–Hochberg adjustment for multiple testing to calculate q values (*P < 0.05, **q < 0.05). c, Definition of molecular categories and diagnostic workflow. Molecular categories not defined in WHO5th are highlighted in red. APL, acute promyelocytic leukemia.

The colors of the ribbon plot represent molecular categories of samples in the pAML cohort (n = 887).
Biological characterization of the molecular categories
Establishing updated molecular categories for pAML allowed for the investigation of clinicopathological associations. Categories with acute megakaryoblastic leukemia (AMKL) or acute erythroid leukemia (AEL) phenotypes are clearly enriched in infants, whereas CBF leukemias and mutation-defined leukemias (for example, UBTF, NPM1, CEBPA) were enriched in adolescents and young adults (Fig. 4a and Extended Data Fig. 4b). Notably, among KMT2A fusion partners, MLLT3 and MLLT10 were found in both monocytic AML and AMKL; however, these fusions preferentially show AMKL phenotypes in infants, suggesting that AMKL phenotypes are defined both by driver alterations and by developmental stages as discussed previously43,44. Overall, however, each molecular category showed variable morphological features represented by FAB classification, except categories with acute promyelocytic leukemia (M3) or AMKL (M7) phenotypes. Likewise, complex karyotypes, which also define AML-MR9, were frequently observed in MNX1, HOXr and PICALM::MLLT10 categories. In addition, many of these category-defining alterations are cytogenetically cryptic (for example, NUP98::NSD1 or GLIS family) or somatic mutations (for example, CEBPA, UBTF or GATA1), highlighting the need for sequencing approaches for the appropriate molecular diagnosis of pAML.

a, Clinical background of molecular categories. (Upper) Violin plots showing age distribution within each molecular category. Colors show the molecular categories. Large dots and bars represent the median and the 2.5–97.5 percentile range, respectively. Small dots represent the ages of individuals (n = 887). (Lower) Frequency of FAB classification (blue bars) and karyotype (gray bars) in individual categories. b, Mutational heatmap showing mutation frequencies in each molecular category. The color of each panel represents the frequency of a mutation in each molecular category, and the statistical significance was assessed by two-sided Fisher’s exact test to calculate P values of co-occurrence followed by Benjamini–Hochberg adjustment for multiple testing to calculate q values (*P < 0.05, **q < 0.05 after adjustment). Bars in the upper panel show the frequency of mutations in the entire cohort, and the colors represent mutation types. Molecular categories are clustered according to Ward clustering using the Euclidean distance of the frequency matrix. Genes are grouped according to functional annotations. c, Heatmap showing normalized enrichment scores (NES) and FDR of GSEA for each molecular category. Colors denote NES and asterisks show FDR (*FDR < 0.05, **FDR < 0.01, ***FDR < 0.001). Detailed results are found in Supplementary Table 14. d, Violin plots showing cellular hierarchy scores in each molecular category inferred by CIBERSORT. The colors show molecular categories. The lines of the boxes represent the 25% quantile, median and 75% quantile. The upper whisker represents the higher value of maxima or 1.5× i.q.r., and the lower whisker represents the lower value of minima or 1.5× i.q.r. Dots show outliers. LSPC, leukemic stem and progenitor cell.
We next explored the association between defining alterations and cooperating mutations, because some cooperating mutations co-occur and act synergistically with specific driver events4,45. Signaling alterations were broadly found in 66.3% of patients, although each mutation showed distinct patterns among molecular categories with variable VAFs (Figs. 1e and 4b). Among RAS mutations, NRAS mutations were broadly found and enriched in CBFB::MYH11 and NPM1, whereas KRAS mutations were enriched in KMT2Ar and DEK::NUP214. Similarly, FLT3-ITD showed strong enrichment in NUP98r, NPM1, UBTF, KMT2A-PTD and BCL11B, accounting for 66.2% of FLT3-ITD+ cases, whereas 75.5% of FLT3-TKD (tyrosine kinase domain) were found in KMT2Ar, NPM1 and CBF-AMLs. Similarly, WT1 mutations were specifically enriched in NUP98r, UBTF and BCL11B, and highly co-occurring with FLT3-ITD (Fig. 4b).
We further evaluated gene expression signatures among molecular categories. Top variably expressed genes across the cohort are involved in development, differentiation or inflammation (Extended Data Fig. 5a and Supplementary Table 13), consistent with previous reports that the heterogeneity of AML can be partly attributed to differentiation status3,36,46. Gene set enrichment analysis (GSEA) confirmed known expression profiles of major categories (Fig. 4c and Supplementary Table 14), whereas the new categories proposed in this study show similarities and differences with canonical categories. For example, UBTF showed expression signatures similar to NPM1 and DEK::NUP214, whereas KAT6Ar was similar to KMT2Ar, suggesting shared biological mechanisms. In addition, genes involved in signaling pathways, immunity or drug resistance showed unique enrichment across categories. Weighted gene coexpression network analysis (WGCNA)47 confirmed characteristic patterns of active gene networks associated with specific biological functions in each category (Extended Data Fig. 5b and Supplementary Table 15).
Given recent adult AML-focused studies uncovering the associations of cellular stemness48,49 or hierarchy36,50 with prognosis or drug response, we investigated these features in our pAML dataset. We observed unique patterns of stemness and cellular hierarchy scores in each category. Molecular categories known to have a good prognosis (RUNX1::RUNX1T1, CBFB::MYH11 and CEBPA) tended to have high granulocyte–monocyte progenitor (GMP) scores (median >0.20) (Fig. 4d and Extended Data Fig. 5c), except for the low GMP scores (median 0.078) and mid-high stemness-related scores in NPM1. Also, KMT2Ar, associated with poor prognosis, showed low stemness-related scores and variable differentiation-related scores. Also, various prognostic scores (for example, LSC17 (ref. 48), iScore46) correlated with molecular categories (Extended Data Fig. 5d), collectively demonstrating that molecular categories are associated with unique pathophysiological characteristics.
Superfamilies defined by HOX gene expression profiles
These molecular categories also showed intercategorical similarities, forming large clusters of AMKL/AEL, immature AML, CBF leukemias, CEBPA and two clusters demarcated by HOXA and HOXB gene expression (Fig. 5a,b). The cluster with high HOXA gene expression and low HOXB gene expression consisted mainly of KMT2Ar and KAT6Ar (herein referred to as the HOXA group), and the other cluster characterized by high expression of both HOXA and HOXB genes included NPM1, NUP98r, UBTF, KMT2A-PTD and DEK::NUP214 (HOXB group), which are generally associated with poor prognosis except for NPM1 (Extended Data Fig. 6a). Overall, HOXA and HOXB groups, not including those with AMKL features, account for 18.5% and 23.3% of the cohort, respectively. Differential gene expression analyses revealed that HOXB pAMLs had high expression of stemness-related genes (PRDM16 and NKX2-3) or differentiation genes (CD96 and WT1) (Fig. 5c,d and Supplementary Table 16). By contrast, HOXA group cases showed high expression of monocyte or signaling-related genes. GRIN analysis also revealed striking differences in mutational patterns between HOXA and HOXB groups (Fig. 5e,f and Supplementary Table 17). FLT3 was significantly altered in both groups but with different mutation types; FLT3-TKD was dominant in the HOXA group and FLT3-ITD was prevalent in the HOXB group, accounting for 67.5% of FLT3-ITD+ patients (Fig. 5f and Supplementary Fig. 6b). WT1 mutations were preferentially found in the HOXB group (57.6%). FLT3-ITD (ref. 51 and WT1 mutations16,52 have been associated with poor prognosis; however, these data suggest that FLT3-ITD and WT1 mutations highly confound with specific driver alterations that converge on a common expression signature. KRAS mutations were strongly associated with the HOXA group and were rare in the HOXB group (20.7% and 3.9%, respectively). In comparison, NRAS mutations were prevalent in both HOXA and HOXB groups (20.7% and 17.4%) (Fig. 5f); however, p.G12 or p.G13 mutations were comparable in both categories, whereas p.Q61 mutations were more frequent in the HOXA group (Extended Data Fig. 6b). It is well-established that each RAS mutation has preferential distribution among cancer subtypes53. Expression levels or differences in the downstream signaling of RAS proteins are postulated as the possible mechanisms, and similarly, between FLT3-ITD and TKD54, whereas these genes were homogenously expressed at the RNA level (Extended Data Fig. 6b). Despite varied clinical associations, these molecular category-dependent transcriptional and mutational patterns may reflect shared biology within each HOX group55, and the different signaling dependencies may suggest targeted therapies guided by these biological insights.

a, UMAP plot colored according to groups of molecular categories based on UMAP clustering and HOX cluster gene expression profiles. A gray circle indicates a cluster enriching categories with immature phenotypes (BCL11B, MECOM, MNX1, PICALM::MLLT10, ETS family). b, HOXA9 and HOXB5 expression on UMAP plots. Dot colors represent the relative expression of the genes. c, Volcano plot showing differentially expressed genes between HOXA and HOXB groups. Genes with absolute fold change >2 and FDR < 0.05 are considered differentially expressed. Red or blue dots show genes enriched only in either HOXA or HOXB groups, respectively, and representative gene names are shown. d, Gene Ontology term analyses of genes with significantly high expression in HOXA (red) and HOXB (blue) categories by DAVID (Database for Annotation, Visualization and Integrated Discovery). Bars represent logged FDR. e, Plots showing the results of GRIN analyses in the HOXA group (horizontal axis) and HOXB group (vertical axis). Genes with FDR < 0.05 in either the HOXA or HOXB group are shown. Red or blue dots show genes enriched only in either the HOXA or HOXB group, respectively. Dotted lines represent thresholds for statistical significance (FDR < 0.05). f, Mutational heatmap comparing patterns between the HOXA and HOXB groups. Colors represent mutation types, and molecular categories are annotated on the top. Bar plots on the right show frequencies of mutations in the HOXA and HOXB groups. Statistical significance of GRIN analysis in the HOXA and HOXB groups (*FDR < 0.05) and two-sided Fisher’s exact test between HOXA and HOXB groups (*P < 0.05, **q < 0.05 after Benjamini–Hochberg adjustment) are also shown. GRIN results for FLT3 are for the entire gene, whereas Fisher’s tests were performed separately for ITD, TKD and non-TKD mutations.
Along with the global distinction between HOXA and HOXB groups, we also noted heterogeneity within each HOX cluster. The HOXA cluster consisted of subclusters characterized by MECOM or LAMP5 expression (Extended Data Fig. 7a–c and Supplementary Table 18), harboring most KMT2Ar cases (136 of 180; 75.6%). Notably, the largest subcluster expressed XAGE1 family genes specifically (Extended Data Fig. 7b,c), which encode testis-specific proteins postulated as therapeutic targets in various tumors56. Also, the remaining KMT2Ar cases were clustered with other categories with HOXB expression or AMKL less frequently. These clustering patterns were associated with age or fusion partners (for example, KMT2A::ELL in the HOXB cluster), but the associations were not exclusive (Extended Data Fig. 7d,e). Among KMT2Ar, fusion partners and MECOM expression have been reported to be prognostic; however, our data suggest considerable heterogeneity in expression patterns not explained by only fusion partners or MECOM expression. The HOXB cluster showed similar heterogeneity represented by cellular hierarchies (Extended Data Fig. 7f–h). These heterogeneities were occasionally associated with molecular categories or somatic mutations but were not exclusive, with possible factors, including cell-extrinsic factors46 to be investigated.
Molecular basis of AML without defining gene alterations
Seventy-six ‘Unclassified’ cases remained after assignment to these 23 molecular categories. Twenty-one cases had recurrent driver alterations previously reported in the literature (Fig. 6a and Supplementary Table 19), including rare in-frame RUNX1 fusions (n = 2: USP42; n = 1: EVX1 and ZEB2) and MLLT10 fusions (n = 1: DDX3X, TEC and MAP2K2), which require a larger cohort for further categorization. Also, in addition to high-allelic burden JAK2 p.V617F mutation (n = 1), we found candidate driver somatic mutations of MLLT1 p.C119SPAR (n = 1) and H3F3A p.K28M (n = 1) in cases in HOX clusters (Fig. 6a and Extended Data Fig. 8a). These mutations resemble recurrent mutations in other pediatric cancer types with HOX gene expression and immature phenotypes (MLLT1 p.C118QPPG in Wilms tumor57 or H3F3A p.K28M in high-grade glioma58), postulating a shared mechanism of tumorigenesis among these pediatric neoplasms.

a, UMAP plot showing cases without category-defining alterations. Red dots represent cases with rare recurrent gene alterations, blue dots represent cases for which no pathogenic alteration was found and black dots represent cases with at least one gene alteration not defining the phenotype. Gray dots represent cases with classified categories. b, Plot showing the FDR of GRIN analysis for the Unclassified category (horizontal axis) and relative enrichment of the alteration in the unclassified category (vertical axis). Dot sizes and colors denote the Unclassified category’s frequency, which included fusions, mutations, copy-number loss and gain, and copy-neutral heterozygosity. c, Mutational heatmap of the Unclassified cases, including complex karyotypes and monosomy 7. Patients’ clinical and demographic data are shown on the top. Colors represent mutation types. Defining alterations for AML-MR are marked by asterisks. d, UMAP plots showing CD34, CD3D and DNTT expression (left), FAB classification (middle) and cases with ETV6 alterations and RUNX1 alteration (right). For ETV6 and RUNX1 alteration plots, cases with classified categories are shown as gray dots. e, Patterns of alteration in ETV6 (left) and RUNX1 (right). Category-defining fusions are shown in the top row, alterations co-occurring with category-defining alterations in the middle row, and alterations in the Unclassified category in the bottom row. Bars represent a relative fraction of alteration in each group and colors denote the alteration types. WBC, white blood cell.
Pathogenic alterations were not identified in 9 of the remaining 55 Unclassified cases, partly attributed to the lack of WGS data for 8 of these cases. The rest had at least one pathogenic, but not subtype-defining alteration enriched in ETV6, RUNX1, TP53 and myelodysplasia-related genes in addition to complex karyotypes or monosomy 7 (Fig. 6b,c and Supplementary Tables 19 and 20). Of note, AML-MR defining karyotypes (complex karyotypes or monosomy 7) or somatic mutations were found broadly in various clusters (Extended Data Fig. 8b–d), suggesting that these alterations do not define specific categories. By contrast, ETV6 and RUNX1 alterations not defining established categories were found preferentially in clusters associated with FAB M0/1 or immature or T cell-like signatures (Fig. 6d, Extended Data Fig. 8b–d and Supplementary Table 21), as previously described59. Although various ETV6 or RUNX1 alterations can be class-defining (for example, RUNX1::RUNX1T1) or co-occur with other defining alterations, those in the Unclassified category are commonly loss-of-function (Fig. 6e). Given that germline mutations of RUNX1 or ETV6 are associated with leukemia with incomplete penetrance60,61, these data suggest somatic alterations of these genes also require additional mutations for leukemia development, which may cooperatively define the immature leukemic phenotypes. Further accumulation of genomic data and experimental models will be necessary to understand immature pAML with these mutations.
Clinical association of molecular categories
Although the association between KMT2Ar or NUP98r and poor outcomes is well-appreciated, the clinical associations of new molecular categories have been discussed only in separate studies8,25. To address this deficiency and translate them into a clinical framework, we investigated the outcomes of these molecular categories using the COG AAML1031 study13 (n = 1,034; Supplementary Table 22). Analyses of the AAML1031 RNA-seq data using the same pipeline revealed similar clustering of molecular categories and the overall category frequencies (Fig. 7a,b). The AAML1031 cohort confirmed the association of molecular categories with age and FLT3-ITD status, and showed variable minimal residual disease (MRD) positivity among molecular categories (Fig. 7c). Major categories with favorable outcomes aligned with previous reports (for example, RUNX1::RUNX1T1 (n = 141), CBFB::MYH11 (n = 102) and CEBPA (n = 63); Extended Data Fig. 9a). We also confirmed the known association of GLISr7 (n = 20), MECOM (n = 11), PICALM::MLLT10 (n = 8) and KAT6Ar (n = 7) with poor outcomes, except DEK::NUP214 (n = 17) which showed a favorable outcome in the AAML1031 study29,62. New categories of MNX1 (n = 4), RUNX1::RUNX1T1-like (n = 4) and CBFB-GDXY (n = 4) showed favorable outcomes.

a, UMAP plot of transcriptome data of the AAML1031 cohort (n = 1,034) using top 340 variable genes. Dot colors denote molecular categories assigned to the samples according to genomic profiling using the same pipeline as this study cohort. Representative category names are shown, and large clusters enriching specific categories are highlighted in circles (pink: NUP98::NSD1, NPM1, UBTF, DEK::NUP214, KMT2A-PTD; green: KMT2Ar and KAT6Ar; yellow: categories with acute megakaryocytic or erythrocytic expression; blue: MECOM, MNX1, ETS family, PICALM::MLLT10, BCL11B). b, Frequency of molecular categories in the study cohort (black) and AAML1031 cohort (blue). The statistical significance of the frequency of each category assessed by two-sided Fisher’s exact test followed by Benjamini–Hochberg adjustment (q < 0.05; blue indicates fewer and black indicates more in the AAML1031) is shown. c, Clinical features of molecular categories showing age at diagnosis (left), FLT3-ITD status (middle) and MRD positivity at the end of induction (right). Molecular category names associated with megakaryocytic phenotypes are highlighted in red. The lines of the boxes represent the 25% quantile, median and 75% quantile. The upper whisker represents the higher value of maxima or 1.5× i.q.r., and the lower whisker represents the lower value of minima or 1.5× i.q.r. d, Grouping of molecular categories into low, intermediate and high-risk groups by recursive partitioning (upper) and Kaplan–Meier curves of overall survival of patients in each risk group (lower). e, Kaplan–Meier curves and statistical significance of overall survival of patients with known prognostic factors (FLT3-ITD status (upper left), age (lower left) and MRD positivity at the end of the induction I (upper right)). f, Kaplan–Meier curves of overall survival of patients in six risk strata using risk groups (low–intermediate–high) and MRD positivity. For survival curves in d, e and f, statistical significance was assessed by the log-rank test, and P values are shown in the plot. For survival analysis involving MRD status, patients with available MRD status (MRD+: n = 273; MRD−: n = 703) are included. NS, not significant.
We also investigated the clinical association of molecular heterogeneities within major categories. Among KMT2Ar, fusion partners or MECOM expression63,64 also confounded in the AAML1031 cohort (Extended Data Fig. 9b). Cox hazard models showed that both fusion partners and expression clusters are prognostic (P = 0.00052 and 0.0015, respectively), with fusions with SEPTIN family and MLLT11 or immature expression patterns associated with favorable outcomes (Extended Data Fig. 9c). The association of fusion partners or expression clusters with prognosis did not significantly differ (difference in C-index of 95% bootstrap interval for fusions and clusters: −0.025 to 0.093). Although HOXB categories of NUP98r, NPM1 and UBTF also showed heterogeneity of expression patterns, their outcomes were not associated with UMAP clusters or fusion partners (Supplementary Fig. 2a).
Given these findings, we next applied recursive partitioning models65 for censored event time data of molecular categories and fusion partners of KMT2Ar, which revealed three groups with distinctive prognoses (Fig. 7d and Supplementary Fig. 2b–d). Univariate analyses revealed that age and FLT3-ITD were not prognostic, which could reflect the sorafenib given to patients with high-allelic FLT3-ITD in the AAML1031 study13 (Fig. 7e). Contrarily, MRD positivity and a subset of cellular hierarchy scores were associated with overall survival (Fig. 7e and Extended Data Fig. 9d). A Cox proportional hazards model using risk groups and prognostic factors showed that hierarchy scores did not significantly contribute to prognosis, whereas risk groups and MRD positivity were independently prognostic (Supplementary Table 23). These data led us to establish a simple predictive framework solely based on molecular categories and MRD positivity, resulting in six risk strata with granular outcome prediction (Fig. 7f and Extended Data Fig. 9e). The prognostic values were validated using the separate AML08 trial12 cohort (n = 221; Extended Data Fig. 10a–c and Supplementary Tables 24 and 25). Hematopoietic stem cell transplantation in the first remission showed a benefit for high-risk categories with MRD, whereas that for the remaining groups needs further assessment (Extended Data Fig. 10d). Also, the predictive value of this prognostic framework was comparable or superior to various risk stratifications currently used in clinical trials for pAML13,14,15 or ELN2022 (ref. 11) for adult AML (Supplementary Fig. 3). These data suggest that the proposed framework could be a basis for future risk stratification and clinical decisions.
Discussion
In addition to known enrichment of chromosomal events like t(11,x) in pAML, sequencing technologies have identified additional pediatric-enriched driver alterations7,8,27. This prompted us to comprehensively investigate the increasingly complex genomic landscape of pAML in the context of the latest classification systems for hematological malignancies (WHO5th (ref. 9) and ICC10) and to develop a pAML-focused categorization. In this study, we systematically categorized our pAML cohort of 887 patients using an approach based on RNA-seq, resulting in 23 molecular categories defined by mutually exclusive driver alterations, covering 91.4% of the entire cohort. Of these 23 categories, 12 are not currently defined by WHO5th. These include common categories like UBTF, GLISr and GATA1, otherwise categorized as ‘AML-MR’ or ‘acute myeloid leukemia with other defined gene alterations’ in the current WHO classification. Notably, myelodysplasia-related mutations or chromosomal alterations often co-occur with many pAML category-defining alterations and override them in WHO5th or do not drive consistent gene expression patterns even without category-defining alterations. Considering that the current classification systems are mainly based on evidence from adult AML, we propose an alternative framework for pAML to better reflect its biology.
These molecular categories show unique expression and mutational profiles, whereas some categories also show critical similarities, which can suggest common molecular mechanisms and potential therapeutics. In particular, we noticed two large clusters characterized by HOXA-B expression profiles. Molecular categories with HOXB signatures were strongly associated with FLT3-ITD and WT1 mutations, whereas those with HOXA signatures were associated with KRAS mutations. Considering that AMLs with KMT2Ar, NUP98r and NPM1 are dependent on KMT2A/Menin66,67,68 and that several Menin inhibitors targeting KMT2Ar and NPM1 AML are in clinical trials69,70, our data suggest that other subtypes marked by HOX expression may also be candidates for Menin inhibitors. This is supported by our recent study showing that UBTF AMLs are sensitive to Menin inhibitors71. Also, the high frequency of FLT3-ITD in categories with HOXB expression implies that FLT3 signaling is closely related to biology and that treatment with FLT3 inhibitors for FLT3-ITD+ HOXB subtypes independent of the allelic ratio may be effective.
Some cases without category-defining alterations could be characterized by rare fusion or mutations, which need further evidence to establish as a disease entity, including MLLT1 and H3F3A mutations that are frequent and class-defining in Wilms tumor57 and glioma58, respectively. Considering that AML and Ewing sarcoma also share ETS family fusions40 (for example, EWSR1::ERG), it would be intriguing to incorporate knowledge of these solid tumors to understand the biology behind pAML with these rare alterations. Also, enrichment of RUNX1 or ETV6 loss-of-function alterations in immature AML implies that these can be class-defining in the absence of other defining alterations and likely with specific cooperating mutations. These findings further suggest a continuum with other immature leukemias, such as early T cell precursor-ALL and mixed phenotype acute leukemias (T/My) with similar mutational features72,73.
We further investigated the clinical outcomes of these molecular categories using two independent cohorts: the COG AAML1031 study and the AML08 study. Using both cohorts, we show a strong association of new molecular categories with outcomes (for example, PICALM::MLLT10, UBTF and KAT6Ar as high risk, and CBFB-GDXY as low risk). These analyses also revealed that molecular categories and known prognostic factors, such as FLT3-ITD status or cellular hierarchy scores, are confounding. With this comprehensive profiling recognizing new pAML subtypes, we established a simple risk stratification using molecular categories and MRD. This strategy, however, heavily relies on the analysis of next-generation sequencing data. Although the WHO classification requires targeted sequencing or WGS, we propose a diagnostic pipeline utilizing RNA-seq, which is highly sensitive for canonical and cryptic fusion calling, allows for categorization based on gene expression signatures, including outlier and allele-specific expression (MECOM, BCL11B and MNX1), and provides limited but sufficiently sensitive mutation calling to enable our comprehensive molecular categorization strategy to newly diagnosed pAML. This approach is favored over current commercial panels commonly used for pAML, which either lack coverage of all the defining genes (for example, UBTF) or are unsuitable for detecting complex structural variations that drive aberrant expression of MECOM or BCL11B. Given that clinical sequencing is not readily available globally and these molecular analyses require substantial expertise, robust and easy pipelines are needed for future and broad application of this framework for pAML in the general clinical setting.
Methods
Subject cohorts and sample details
Tumor samples from patients with AML from the St. Jude Children’s Research Hospital tissue biorepository were obtained with written informed consent from patient, parents or guardians using a protocol approved by the St. Jude Children’s Research Hospital institutional review board. Studies were conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects. No patient received compensation for the enrollment to this study. Samples for RNA-seq (n = 221), WGS (n = 58) and WES (n = 7) are newly sequenced in this study, and the rest of the data were obtained from previous publications4,7,8,16,17,18,19,20,21,22,23,24,25 or public databases (see details in ‘Data availability’ and Supplementary Table 1). For samples with multiple available data points, we included one representative time point with a high tumor purity and good RNA-seq data quality. Cases were assigned to current WHO5th (ref. 9) and ICC10 by board-certified hematopathologists (P.K. and J.M.K.).
Genotype fingerprints
To make sure that the study cohort cases represent unique individuals, we performed a pairwise genotype concordance comparison among all the study cases using the estimated genotype from single nucleotide polymorphisms (SNPs) with ≥20 coverage in RNA-seq Binary Alignment Map (BAM) files. We set genotype concordance percentage cutoff at ≥90% of SNPs shared between two individuals to identify potential duplicates, confirming the uniqueness of the 887 patients in the study cohort.
Sample processing, library preparation and sequencing
For newly sequenced samples with low tumor purity (<60%), the leukemic cell population was enriched either by flow cytometric sorting or T cell depletion by magnetic beads (EasySep Human CD3 Positive Selection Kit II; StemCell Technologies, catalog no. 17851). For flow cytometric sorting, CD45dimCD33dim positive population was sorted using anti-CD45 PerCP-Cyanine5.5 (eBioscience, catalog no. 8045-9459-120; 1:20 dilution), anti-CD33 APC (eBioscience, catalog no. 17-0338-42; 1:20 dilution) and DAPI (BD Biosciences, catalog no. 564907) using FACS Aria III instrument and FACS Diva v.9.0 (both BD Biosciences) (Extended Data Fig. 1c). CD34 gating using anti-CD34 PE (phycoerythrin) (Beckman, catalog no. IM1459U; 1:5 dilution) was added depending on the positivity of each patient sample. Enrichment of the tumor population was confirmed by flow cytometric analysis of the postsorting samples (generally >90%). Libraries were constructed using the TruSeq Stranded Total RNA Kit, with Ribozero Gold (Illumina, catalog no. 20020598) for RNA-seq, the TruSeq DNA PCR-Free Library Prep Kit (Illumina, catalog no. 20015963) for WGS and the TruSeq Exome Kit v.1 (Illumina, catalog no. 20020614) for WES according to the manufacturer’s instructions. After library quality and quantity assessment, samples were sequenced on HiSeq2000 or 2500 (Illumina, RRID:SCR_020132, RRID:SCR_016383) instruments with paired-end (2 × 101 bp, 2 × 126 bp or 2 × 151 bp) sequencing using TruSeq SBS Kit v3-HS (Illumina, catalog no. FC-401-3001) or TruSeq Rapid SBS Kit (Illumina, catalog no. FC-402-4023) and HiSeq Control Software with most recent version at the time of sequencing.
RNA-seq mapping, fusion detection and large-scale CNV calling
RNA reads from newly sequenced samples and from publications were mapped to the GENCODE (RRID:SCR_014966) human genome assembly release 19 gene annotation (GRCh37/hg19) using the StrongARM pipeline74. Chimeric fusion detection was carried out using CICERO75 (v.0.3.0). For the cases with only RNA-seq data, RNAseqCNV76 (v.1.2.1) was used to call large-scale CNV.
Somatic mutation calling from RNA-seq
To detect SNV and indel from RNA-seq data, we applied the following approach to simultaneously account for germline polymorphisms (without germline control) and sequencing artifacts specific to RNA-seq on a panel of 87 predefined genes previously reported to be significantly mutated in pAML4 and myelodysplastic syndrome (Supplementary Table 5). Briefly, candidate SNVs/indels were called by Bambino77 (v.1.07) or RNAindel78,79 (v.3.0.4), annotated by VEP80 (v.95), filtered by excluding variants with gnomAD (v.2.1.1, RRID:SCR_014964)81 population allele frequency >0.1% as possible germline variants, and in turn, classified for putative pathogenicity with PeCanPie/MedalCeremony82 (not versioned). Candidate variants with putative pathogenicity were considered germline or artifacts if present in >5% of the cases. Candidate variants were further filtered if the number of supporting reads was ≤5 or if the VAF was ≤5%. UBTF tandem duplications were detected by CICERO focusing ITD or PTD with supporting reads ≥3 within exon 13 of UBTF gene or adjacent introns and CICERO score <10, detection of indels on exon 13 of the UBTF gene, and counting reads with 10 or more soft-clipped nucleotide sequences and total reads on the 3′-end of exon 13 that contains a hotspot of ITD and PTD (GRCh37-lite, chr17:42288162-42288192; GRCh38, chr17: 44210794-44210824)8.
WGS and WES data analysis
The previous genomic lesion calls for the cases (WGS: n = 394; WES: n = 284) from published studies4,7,8,16,18,19,20,23,25 were collected from their respective publications. For the unpublished cases with DNA data (WGS: n = 136; WES: n = 107), DNA reads were mapped using BWA83,84 (WGS: v0.7.15-r1140 and v0.5.9-r26-dev; WES: v0.5.9-r26-dev and v0.5.9, RRID:SCR_010910) to the GRCh37/hg19 human genome assembly. Aligned files were merged, sorted and de-duplicated using Picard tools 1.65 (broadinstitute.github.io/picard/). SNVs and indels were called using Bambino. For cases paired with matched germline controls, germline variants were filtered out if present in the matched germline sample. For unpaired cases, possible germline variants were filtered and classified as for somatic mutation calling from RNA-seq. The counting of somatic mutations included all the pathogenic or likely pathogenic mutations detected by WGS, whereas mutation detection from cases with only RNA-seq data is limited to the 87 preselected genes. SVs were analyzed using CREST (Clipping REveals STructure)85 (v.1.0), and CNVs were analyzed using CONSERTING86 on the WGS data. CNVs were also called on cases with only WES DNA data using the following methods. Briefly, Samtools87 (v.1.16) mpileup command was used to generate a mpileup file from matched germline and tumor BAM files with duplicates removed. If a matched germline was not available, a high-quality normal sample was used to pair with the tumor sample. VarScan88 (v.2.3.5) was then used to take the mpileup file to call somatic CNVs after adjusting for normal/tumor sample read coverage depth and GC content. Circular Binary Segmentation algorithm89 implemented in the DNAcopy R package (v.1.52.0) was used to identify the candidate CNVs for each sample. B-allele frequency information was also used to assess allelic imbalance.
Validation of somatic alterations called by the RNA-seq pipeline
We focused on 243 cases (27.4%) with data from all three platforms (matched WGS, WES and RNA-seq) to cross-validate the accuracy of our RNA-seq based pipeline8. Of 374 SNV/indel variant calls from RNA-seq data, 329 variants (88%) were called from either WGS or WES, whose VAFs showed significant correlation with those of RNA-seq calls (Extended Data Fig. 1f). Of the remaining 45 calls, 35 have supporting reads in DNA data, which were not called, likely because of sequence noises and low VAF, validating in total 97.3% of the RNA-seq calls.
GRIN analysis for significantly mutated genes
For the 887 AML cases, the GRIN (v.2.0) model26 was used to evaluate the statistical significance of the number of subjects with each type of lesion: fusions, CNVs (amplifications and deletions), copy-neutral loss of heterozygosity, SNV/indels and tandem duplications in each gene. For each type of lesion, robust false discovery estimates were computed from P values using Storey’s q value90 with the Pounds–Cheng estimator of the proportion of hypothesis tests with a true null hypothesis91. A false discovery rate (FDR) cutoff of <0.05 was used to obtain significantly mutated genes, where we focused on protein-coding genes and genes that are known or likely to be pathogenic in leukemia. We also excluded genes that are part of a large chromosomal gain, loss or copy-neutral loss of heterozygosity but not the target of the CNVs based on Genomic Identification of Significant Targets in Cancer (GISTIC) analysis. Subgroup GRIN analyses for HOXA categories (n = 164), HOXB categories (n = 207) categories and the Unclassified category (n = 76) were similarly performed.
GISTIC analysis for significant recurring copy-number alterations
We used GISTIC (v.2.0.23, RRID:SCR_000151)92,93 to identify genomic regions that are significantly amplified or deleted across our 895 samples. Each aberration was assigned a G-score that considered the amplitude of the aberration as well as the frequency of its occurrence across samples. FDR q values were then calculated for the aberrant regions, and regions with q values ≤0.25 were considered significant. A ‘peak region’ was identified for each significant region with the greatest amplitude and frequency of alteration. In addition, a ‘wide peak’ was determined using a leave-one-out algorithm to allow for errors in the boundaries in a single sample. Each significantly aberrant region was also tested to determine whether it resulted primarily from broad or focal events (a broad event was set as >90% of the chromosome arm, whereas a focal event was ≤90%).
Allele-specific expression estimation for MNX1, BCL11B and MECOM categories
For cases with both WGS and RNA-seq available, SNP markers in the respective gene locus with ≥10x coverage that are heterozygous (defined as 0.2 ≤ VAF ≤ 0.8) in WGS and present in RNA-seq were extracted, and a two-sided binomial test (with probability of success P = 0.5) was performed on each marker for allelic imbalance in RNA expression. The median of binomial P values was used to assess allele-specific expression. For RNA-seq only cases, SNP markers in the respective gene locus with ≥10x coverage and allelic imbalance (VAF ≤ 0.2 or VAF ≥ 0.8) support allele-specific expression.
Germline variant curation methods
We focused on 15 candidate genes relevant to AML that define specific categories in WHO5th (Supplementary Table 26) and scanned for germline mutations in the cases with WGS or WES germline BAM files available (WGS: n = 367; WES: n = 354). For cases with germline mutation called in previously published studies8,21, we collected calls from the studies. For the remaining cases, the putative germline variants were called using Bambino, annotated by VEP, and classified for putative pathogenicity with PeCanPie/MedalCeremony. We then used the following criteria to obtain the candidate germline variants: gnomAD population allele frequency ≤0.001; read coverage SNV ≥ 20 and indel ≥ 15; for SNV, VAF between 0.2 and 0.8; for indel, ≥3 reads supporting the alternative allele. All candidate germline variants were comprehensively reviewed and classified based on recommendations from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology94 and the Clinical Genome Resource95,96,97,98 by a variant scientist (J.L.M.).
Inference of genetic ancestry
For each individual, the admixture fraction was estimated using the iAdmix program99 and allele frequencies from the 1000 Genomes Project reference populations (European (EUR), African (AFR), Native American (NA), East Asian (EAS), South Asian (SAS)) were used as a reference100. Overall, the genetic ancestral composition for each single individual was derived based on a comparison of allele frequencies between each individual and reference genome. The sum of coefficients from the five populations was assumed to sum to 100%. An RNA-seq BAM file was used as input directly to iAdmix program, where allele frequencies for the coding SNPs from the 656,129 SNPs were used in the ancestry estimation. The categorization of individuals into ancestral groups was performed based on the composition of genetic ancestry estimated from iAdmix program (Black: AFR > 70%; East Asian: EAS > 90%; Hispanic: NA > 10% and NA greater than AFR; South Asian: SAS > 70%; White: EUR > 90%). The remaining patients with majority EAS or SAS were categorized into ‘Other-Asian’, and the rest of patients with majority EUR or AFR or NA > 10% with NA less than AFR, were categorized into ‘Other-US’101 (Supplementary Table 1).
Gene expression data summarization, batch correction, dimension reduction and clustering
Reads from aligned BAM files were assigned to genes and counted using HTSeq102 (v.0.11.2, RRID:SCR_005514) with the GRCh37/hg19 GTF file. For a gene to be considered as expressed, we required that at least five samples should have ≥10 read counts per million (cpm) reads sequenced. The count data were transformed to log2(cpm) using Voom103 available from R package Limma104 (v.3.50.3, RRID:SCR_010943). We corrected for library strand (stranded total RNA versus unstranded messenger RNA) and batch effect between the TARGET and the rest of cohorts using the ComBat method available from R package SVA105 (v.3.42.0, RRID:SCR_012836). The R package Seurat106,107,108,109 (v.4.1.0, RRID:SCR_016341) was used for dimension reduction and sample clustering. Briefly, the top 315 variable genes were selected using the ‘vst’ method. The expression data were then scaled and used for principal component analysis, and the top 100 principal components were used for dimension reduction using UMAP110,111 (RRID:SCR_018217) (n_neighbors = 12 and min_dist = 0.2). Samples were clustered using the top 100 principal components by first constructing a K nearest-neighbor graph and then iteratively optimizing the modularity using Louvain algorithm (resolution = 3.5). Dimension reduction was also performed by Diffusion maps37,112 algorithm available in the R package destiny113 (v.3.10.0) using the same 315 genes with the default setting except for number of principal components (n_pcs = 50).
Differential gene expression analysis was performed by Limma104, and we set log2(cpm) = 0 if it is <0 based on the log2(cpm) data distribution. P values were adjusted by the Benjamini–Hochberg method to calculate the FDR using the R function p.adjust. Genes with absolute fold change >2 and FDR <0.05 were regarded as significantly differentially expressed. GSEA114 was performed by GSEA v.4.2.3 (RRID:SCR_003199) using MSigDB gene sets c2.all (v.7.5.1), comparing each category with the rest of the categories. Permutations were done 1,000 times among gene sets with sizes between 15 and 1,500 genes. Normalized enrichment scores and FDR for arbitrary gene sets representing hematopoiesis, leukemia phenotype, biological processes and drug responses were shown. WGCNA was carried out by R package WGCNA47 (v.1.70-3, RRID:SCR_003302) using the top 2,000 variable genes and default setting with the exception of block-wide module calculation with reassignThreshold = 0 and mergeCutHeight = 0.25. Functional annotations of the top 315 variable genes, differentially expressed genes and genes in WGCNA modules were performed with DAVID115 (v.6.8), and results for the Gene Ontology term, biological process (GOTERM_BP_DIRECT) were exported. Inference of cellular hierarchy by CIBERSORT116 (RRID:SCR_016955) was performed by the web interface of CIBERSORTx in absolute mode with S-mode batch correction without a permutation36. Transcript per million values and Malignant Signature Matrix and Malignant Single Cell Reference Samples from a publication36 were used as input files, and the malignant cell populations were normalized to 1 to calculate the relative fraction scores, which were shown in UMAP space or violin plots. Prognostic scores of LSC1748, pLSC6 (ref. 49), ADE-RS117 and iScore46 were calculated as reported. Hierarchical clustering (RRID:SCR_014673) of expression data, mutual-exclusivity matrix and GSEA scores was performed using the Euclidian distance and Ward method with pheatmap (v.1.0.12, RRID:SCR_016418).
Statistics and reproducibility
No sample size, power calculation or randomization of patients was performed in this study utilizing retrospective profiling of patients with available materials or sequence data. No analysis depending on patient background was performed in this study. No blinding was performed in the enrollment of patients or data collection of public data, and blinding in group allocation was not possible because the grouping is based on the molecular characteristics of individual patients. For discrete values of the molecular category and the mutation frequency in cohorts, statistical significance and mutual exclusivity were assessed by two-sided Fisher’s exact test and Pearson’s correlation. Adjustment of multiple testing was performed by the Benjamini–Hochberg method using the p.adjust function in R when appropriate. For survival data, decision trees were established by a recursive partitioning method using R library rpart65 (v.4.1.19, RRID:SCR_021777). Kaplan–Meier curves for the probability of overall survival and event-free survival were constructed using the R package survival (v.3.3-1, RRID:SCR_021137). Events in the probability of event-free survival calculations were defined as relapse, death in remission by any cause and nonresponse, which was included as an event at the date of diagnosis. The Cox proportional hazards model was used to calculate the hazard ratio. The log-rank test (two-sided) was used to calculate the statistical significance of individual prognostic factors by univariate analyses first, and significant factors were included in a multivariate analysis. Clinical association of the molecular categories was first assessed using the AAML1031 study (NCT01371981, n = 1,034), and the results were validated using the AML08 cohort (NCT00703820, n = 221, independent from the AAML1031, a part of this study cohort). We quantified the predictiveness of recursive partitioning survival tree models and risk classification systems with Harrel’s concordance index for Cox models118 using a bootstrap procedure. We generated 1,000 bootstrap datasets by sampling patients with replacement and computed concordance index values for each bootstrap dataset. The 2.5 and 97.5 percentiles were used to define the bootstrap confidence interval endpoints. Concordance index values of a pair of risk classification systems were similarly computed similarly. Regression tree models were refit to each bootstrap dataset in the model development analysis on the AAML1031 cohort. For all other analyses, the risk classification was defined externally from the cohort and thus risk-group definitions for individual patients remained constant across bootstrap datasets. R statistical environment (R v.4.0.2, RRID:SCR_001905) was used for statistical tests.
Visualization
Mutational heatmaps and mutations on individual genes were visualized using ProteinPaint (proteinpaint.stjude.org/). Heatmaps of expression data, mutual-exclusivity matrix and GSEA scores were created by pheatmap function. Other data visualizations were performed by ggplot function of R library ggplot2 (v.3.3.6, RRID:SCR_014601), survminer (v.0.4.9) and base plot function in R statistical environment. Figures are incorporated and edited using Adobe Illustrator (2021, RRID:SCR_010279). Annotation of genes in mutational heatmaps depends on common knowledge, and the definition of RAS pathway genes included causative genes of Noonan or Noonan-like syndrome119.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genomic analyses in this study are based on the GENCODE GRCh37/hg19, and gnomAD v.2.1.1 was used for classification for germline and somatic mutations. The genomic data and expression data newly generated in this study (RNA-seq: n = 221, WGS: n = 58, WES: n = 7) have been deposited in the European Genome-Phenome Archive (EGA, RRID:SCR_004944), which is hosted by the European Bioinformatics Institute (EBI), under accession EGAS00001005760. Subsets of the new data (RNA-seq: n = 221, WGS: n = 53, WES: n = 5) have been also deposited to St. Jude Cloud under Pan-AML study (https://permalinks.stjude.cloud/panaml). Details are found in Supplementary Table 1. For previously published RNA-seq data (n = 393), 266 are available either on EGA or St. Jude Cloud7,8,17,19,20,21,22,23,25 or from the original publication24. For the other 127 published cases18, we downloaded the BAM files from EGA (EGAS00001004701). For previously published WGS data (n = 198), 106 from the original publications7,8,19,20,23,25 are available on either EGA or St. Jude Cloud, and the other 92 published BAM files18 were downloaded from EGA (EGAS00001004701). For the previously published WES data (n = 273), 153 with data from the original publications7,8,17,19,20,21,22,23,25 are available either on St. Jude Cloud or EGA, and the BAM files for the other 120 published cases18 were downloaded from EGA (EGAS00001004701). We also downloaded data for publicly available but previously unpublished RNA-seq data (n = 86) on St. Jude Cloud under the PCGP study (https://permalinks.stjude.cloud/permalinks/PCGP, n = 8) and the RTCG study (https://platform.stjude.cloud/data/cohorts?dataset_accession=SJC-DS-1007, n = 78). Similarly, we obtained unpublished WGS data (n = 82: RTCG) and WES data (n = 2: PCGP, n = 99: RTCG study). The data generated by the TARGET initiative4,16 (n = 187), including additional samples from the AAML1031 trial13 (n = 1,034), are also available under accession phs000218 (TARGET-AML) and phs000465 (TARGET substudy, data is available as a part of phs000218), managed by the NCI, and were obtained through GDC Portal managed by NCI under the TARGET-AML study (https://portal.gdc.cancer.gov/projects/TARGET-AML). Information about TARGET can be found at http://ocg.cancer.gov/programs/target. These sequencing data are available through controlled access as part of the NIH Genomic Data Sharing Policy (https://grants.nih.gov/grants/guide/notice-files/NOT-OD-14-124.html) and data access is restricted for academic use. Source data are provided with this paper.
Code availability
We did not use custom code or software for this study.
References
Tenen, D. G. Disruption of differentiation in human cancer: AML shows the way. Nat. Rev. Cancer 3, 89–101 (2003).
Klco, J. M. et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 25, 379–392 (2014).
Miles, L. A. et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 587, 477–482 (2020).
Bolouri, H. et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 24, 103–112 (2018).
Cancer Genome Atlas Research Network; Ley T. J.et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
Jaju, R. J. et al. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35;p15.5) in de novo childhood acute myeloid leukemia. Blood 98, 1264–1267 (2001).
Gruber, T. A. et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3–GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 22, 683–697 (2012).
Umeda, M. et al. Integrated genomic analysis identifies UBTF tandem duplications as a recurrent lesion in pediatric acute myeloid leukemia. Blood Cancer Discov. 3, 194–207 (2022).
Khoury, J. D. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 36, 1703–1719 (2022).
Arber, D. A. et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood 140, 1200–1228 (2022).
Mrozek, K. et al. Outcome prediction by the 2022 European LeukemiaNet genetic-risk classification for adults with acute myeloid leukemia: an Alliance study. Leukemia 37, 788–798 (2023).
Rubnitz, J. E. et al. Clofarabine can replace anthracyclines and etoposide in remission induction therapy for childhood acute myeloid leukemia: the AML08 Multicenter, Randomized Phase III Trial. J. Clin. Oncol. 37, 2072–2081 (2019).
Pollard, J. A. et al. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ acute myeloid leukemia: a report from the children’s oncology group protocol AAML1031. J. Clin. Oncol. 40, 2023–2035 (2022).
Reinhardt, D., Antoniou, E. & Waack, K. Pediatric acute myeloid leukemia – past, present, and future. J. Clin. Med. 11, 504 (2022).
Tomizawa, D. et al. A phase III clinical trial evaluating efficacy and safety of minimal residual disease-based risk stratification for children with acute myeloid leukemia, incorporating a randomized study of gemtuzumab ozogamicin in combination with post-induction chemotherapy for non-low-risk patients (JPLSG-AML-20). Jpn. J. Clin. Oncol. 52, 1225–1231 (2022).
McNeer, N. A. et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia 33, 1934–1943 (2019).
Iacobucci, I. et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat. Genet. 51, 694–704 (2019).
Fornerod, M. et al. Integrative genomic analysis of pediatric myeloid-related acute leukemias identifies novel subtypes and prognostic indicators. Blood Cancer Discov. 2, 586–599 (2021).
Newman, S. et al. Genomes for kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 11, 3008–3027 (2021).
Rusch, M. et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat. Commun. 9, 3962 (2018).
Schwartz, J. R. et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat. Commun. 8, 1557 (2017).
Andersson, A. K. et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 47, 330–337 (2015).
Faber, Z. J. et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 48, 1551–1556 (2016).
Buelow, D. R. et al. Uncovering the genomic landscape in newly diagnosed and relapsed pediatric cytogenetically normal FLT3-ITD AML. Clin. Transl. Sci. 12, 641–647 (2019).
de Rooij, J. D. et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 49, 451–456 (2017).
Pounds, S. et al. A genomic random interval model for statistical analysis of genomic lesion data. Bioinformatics 29, 2088–2095 (2013).
Ryland, G. L. et al. Description of a novel subtype of acute myeloid leukemia defined by recurrent CBFB insertions. Blood 141, 800–805 (2023).
von Neuhoff, C. et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J. Clin. Oncol. 28, 2682–2689 (2010).
Harrison, C. J. et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J. Clin. Oncol. 28, 2674–2681 (2010).
Huber, S. et al. AML classification in the year 2023: how to avoid a Babylonian confusion of languages. Leukemia 37, 1413–1420 (2023).
Ross, M. E. et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 104, 3679–3687 (2004).
Groschel, S. et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381 (2014).
Schwartz, J. R. et al. The acquisition of molecular drivers in pediatric therapy-related myeloid neoplasms. Nat. Commun. 12, 985 (2021).
Montefiori, L. E. et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage-ambiguous stem cell leukemia. Cancer Discov. 11, 2846–2867 (2021).
Tosi, S. et al. Paediatric acute myeloid leukaemia with the t(7;12)(q36;p13) rearrangement: a review of the biological and clinical management aspects. Biomark. Res. 3, 21 (2015).
Zeng, A. G. X. et al. A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia. Nat. Med. 28, 1212–1223 (2022).
Haghverdi, L., Buettner, F. & Theis, F. J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 31, 2989–2998 (2015).
Martelli, M. P. et al. Novel NPM1 exon 5 mutations and gene fusions leading to aberrant cytoplasmic nucleophosmin in AML. Blood 138, 2696–2701 (2021).
Panagopoulos, I. et al. Fusion of the FUS gene with ERG in acute myeloid leukemia with t(16;21)(p11;q22). Genes Chromosomes Cancer 11, 256–262 (1994).
Thomsen, C., Grundevik, P., Elias, P., Stahlberg, A. & Aman, P. A conserved N-terminal motif is required for complex formation between FUS, EWSR1, TAF15 and their oncogenic fusion proteins. FASEB J. 27, 4965–4974 (2013).
von Bergh, A. R. et al. High incidence of t(7;12)(q36;p13) in infant AML but not in infant ALL, with a dismal outcome and ectopic expression of HLXB9. Genes Chromosomes Cancer 45, 731–739 (2006).
Gamou, T. et al. The partner gene of AML1 in t(16;21) myeloid malignancies is a novel member of the MTG8(ETO) family. Blood 91, 4028–4037 (1998).
Li, Z. et al. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat. Genet. 37, 613–619 (2005).
Lopez, C. K. et al. Ontogenic changes in hematopoietic hierarchy determine pediatric specificity and disease phenotype in fusion oncogene-driven myeloid leukemia. Cancer Discov. 9, 1736–1753 (2019).
Yun, H. et al. Mutational synergy during leukemia induction remodels chromatin accessibility, histone modifications and three-dimensional DNA topology to alter gene expression. Nat. Genet. 53, 1443–1455 (2021).
Lasry, A. et al. An inflammatory state remodels the immune microenvironment and improves risk stratification in acute myeloid leukemia. Nat. Cancer 4, 27–42 (2023).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Ng, S. W. et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 540, 433–437 (2016).
Elsayed, A. H. et al. A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia 34, 735–745 (2020).
Bottomly, D. et al. Integrative analysis of drug response and clinical outcome in acute myeloid leukemia. Cancer Cell 40, 850–864 e9 (2022).
Meshinchi, S. et al. Clinical implications of FLT3 mutations in pediatric AML. Blood 108, 3654–3661 (2006).
Ho, P. A. et al. Prevalence and prognostic implications of WT1 mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood 116, 702–710 (2010).
Prior, I. A., Lewis, P. D. & Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 (2012).
Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J. Hematol. Oncol. 4, 13 (2011).
Spencer, D. H. et al. Epigenomic analysis of the HOX gene loci reveals mechanisms that may control canonical expression patterns in AML and normal hematopoietic cells. Leukemia 29, 1279–1289 (2015).
Mahmoud, A. M. Cancer testis antigens as immunogenic and oncogenic targets in breast cancer. Immunotherapy 10, 769–778 (2018).
Perlman, E. J. et al. MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat. Commun. 6, 10013 (2015).
Schwartzentruber, J. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226–231 (2012).
Gutierrez, A. & Kentsis, A. Acute myeloid/T-lymphoblastic leukaemia (AMTL): a distinct category of acute leukaemias with common pathogenesis in need of improved therapy. Br. J. Haematol. 180, 919–924 (2018).
Brown, A. L. et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 4, 1131–1144 (2020).
Feurstein, S. & Godley, L. A. Germline ETV6 mutations and predisposition to hematological malignancies. Int. J. Hematol. 106, 189–195 (2017).
Tarlock, K. et al. Significant improvements in survival for patients with t(6;9)(p23;q34)/DEK-NUP214 in contemporary trials with intensification of therapy: a report from the Children’s Oncology Group. Blood 138, 519 (2021).
Groschel, S. et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German–Austrian Acute Myeloid Leukemia Study Group and the Dutch–Belgian–Swiss HOVON/SAKK Cooperative Group. J. Clin. Oncol. 31, 95–103 (2013).
Bill, M. et al. Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A. Proc. Natl Acad. Sci. USA 117, 26340–26346 (2020).
Breiman, L., Friedman, J. H. & Olshen, R. A. Classification and Regression Trees (Chapman and Hall, 1984).
Krivtsov, A. V. et al. A menin–MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell 36, 660–673 e11 (2019).
Uckelmann, H. J. et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 367, 586–590 (2020).
Heikamp, E. B. et al. The menin–MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood 139, 894–906 (2022).
Issa, G. C. et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature 615, 920–924 (2023).
Swaminathan, M., Bourgeois, W., Armstrong, S. A. & Wang, E. S. Menin inhibitors in acute myeloid leukemia – what does the future hold? Cancer J. 28, 62–66 (2022).
Barajas, J. M. et al. Acute myeloid leukemias with UBTF tandem duplications are sensitive to Menin inhibitors. Blood https://doi.org/10.1182/blood.2023021359 (2023).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Alexander, T. B. et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 562, 373–379 (2018).
Wu, G. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 46, 444–450 (2014).
Tian, L. et al. CICERO: a versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol. 21, 126 (2020).
Jakubek, Y. A. et al. Large-scale analysis of acquired chromosomal alterations in non-tumor samples from patients with cancer. Nat. Biotechnol. 38, 90–96 (2020).
Edmonson, M. N. et al. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics 27, 865–866 (2011).
Hagiwara, K. et al. RNAIndel: discovering somatic coding indels from tumor RNA-seq data. Bioinformatics 36, 1382–1390 (2020).
Hagiwara, K., Edmonson, M. N., Wheeler, D. A. & Zhang, J. indelPost: harmonizing ambiguities in simple and complex indel alignments. Bioinformatics 38, 549–551 (2022).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Edmonson, M. N. et al. Pediatric Cancer Variant Pathogenicity Information Exchange (PeCanPIE): a cloud-based platform for curating and classifying germline variants. Genome Res. 29, 1555–1565 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
Wang, J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 8, 652–654 (2011).
Chen, X. et al. CONSERTING: integrating copy-number analysis with structural-variation detection. Nat. Methods 12, 527–530 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Olshen, A. B., Venkatraman, E. S., Lucito, R. & Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004).
Storey, J. D. A direct approach to false discovery rates. J. R. Stat. Soc. Series B Stat. Methodol. 64, 479–498 (2002).
Pounds, S. & Cheng, C. Robust estimation of the false discovery rate. Bioinformatics 22, 1979–1987 (2006).
Beroukhim, R. et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl Acad. Sci. USA 104, 20007–20012 (2007).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Lee, K. et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 39, 1553–1568 (2018).
Luo, X. et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel recommendations for germline RUNX1 variants. Blood Adv. 3, 2962–2979 (2019).
Gelb, B. D. et al. ClinGen’s RASopathy expert panel consensus methods for variant interpretation. Genet. Med. 20, 1334–1345 (2018).
Bansal, V. & Libiger, O. Fast individual ancestry inference from DNA sequence data leveraging allele frequencies for multiple populations. BMC Bioinformatics 16, 4 (2015).
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Lee, S. H. R. et al. Association of genetic ancestry with the molecular subtypes and prognosis of childhood acute lymphoblastic leukemia. JAMA Oncol. 8, 354–363 (2022).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 e21 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e29 (2021).
McInnes L, H. J. & Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at arXiv https://doi.org/10.48550/arXiv.1802.03426 (2018).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019).
Haghverdi, L., Buttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Steen, C. B., Liu, C. L., Alizadeh, A. A. & Newman, A. M. Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. Methods Mol. Biol. 2117, 135–157 (2020).
Elsayed, A. H. et al. A 5-Gene Ara-C, Daunorubicin and Etoposide (ADE) drug response score as a prognostic tool to predict AML treatment outcome. Blood 134, 1429 (2019).
Harrell, F. E. Jr, Califf, R. M., Pryor, D. B., Lee, K. L. & Rosati, R. A. Evaluating the yield of medical tests. JAMA 247, 2543–2546 (1982).
Tartaglia, M., Gelb, B. D. & Zenker, M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 25, 161–179 (2011).
Acknowledgements
We thank all the patients and their families at St. Jude Children’s Research Hospital (SJCRH) for their contribution to the biological specimens used in this study. We also thank the Biorepository, the Flow Cytometry and Cell Sorting Core, and the Hartwell Center for Bioinformatics and Biotechnology at SJCRH for their essential services. This work was funded by the American Lebanese and Syrian Associated Charities of SJCRH and grants from the National Institutes of Health (grant no. P30 CA021765, Cancer Center Support Grant and a Developmental Fund Award to J.M.K. and X.M., and grant no. U54CA243124, Fusion Oncoproteins in Childhood Cancers (FusOnC2) Consortium to J.M.K. (co-principal investigator). The content, however, does not necessarily represent the official views of the National Institutes of Health and is solely the responsibility of the authors. This work was also supported in part by the Fund for Innovation in Cancer Informatics (the-ici-fund.org, to X.M. and J.M.K.). J.M.K. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a previous recipient of the V Foundation Scholar Award (Pediatric). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.M.K., J.M., T.W. and M.U. conceptualized and managed the entire project. J.M.K., J.M., M.U., G.S. and M.P.W. performed mutational analyses. S.P., Y.N., T.A.A. and M.U. performed clinical outcome analyses. J.M.K. and J.L.M. reviewed and classified germline mutations. J.M.K. and P.K. reviewed mutational data and performed classification of the WHO and ICC. M.R., D.R., S.F., Y.L., W.Y., Y.F., G.W., X.M., B.J.H. and S.P. provided resources and software for data analysis. S.D.B., L.W., T.A.A. and J.E.R. provided data. M.U., J.M. and Y.N. prepared figures. M.U., J.M. and J.M.K. wrote the original draft of the manuscript. All authors reviewed and edited the manuscript. J.M.K. and S.P. supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Rachel Rau and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Cohort details.
a. Data source of each patient with acute myeloid leukemia (AML), including publications and clinical trials. b. Age distribution of patients at diagnosis (red: age<3, blue: 3<age<10, gray: 10<age). Lines of the box represent 25% quantile, median, and 75% quantile. The upper whisker represents the higher value of maxima or 1.5 x interquartile range (IQR), and the lower whisker represents the lower value of minima or 1.5 x IQR. NA: not available. c. Representative gating strategy for sorting of the myeloid cell population. Vertical and horizontal axes are linear for FSC (forward scatter) and SSC (side scatter) and log-scaled for fluorescence-conjugated antibodies. CD34 gating was adjusted for individual patients depending on the positivity. d. A Venn diagram showing data platforms available for each patient. WGS: whole-genome sequencing, WES: whole-exome sequencing, RNA-Seq: RNA-sequencing. e. Results of GISTIC (Genomic Identification of Significant Targets in Cancer) analysis for arm-level chromosomal events. The left panel shows the enrichment of chromosomal gains (red), and the right panel shows the enrichment of chromosomal losses (blue). Green lines show a significance threshold for q values (0.25). f. Cross-validation of single nucleotide variant (SNV) and insertion/deletion (indel) calls from the RNA pipeline using whole-genome/exome sequencing (WGS/WES) data. The bar graph shows mutation calls and the validation status. For those also called from DNA data, a comparison of variant allele frequency (VAF) and Pearson’s correlation are shown in the bottom left, and the statistical test was performed as two-sided. A regression line is shown in red. For unvalidated calls, details are shown in the bottom right. g. A comparison of major classes of the World Health Organization (WHO) classification in the study cohort with karyotyping in previous large pediatric AML cohorts.
Extended Data Fig. 2 Mutational correlation.
Pair-wise correlation among most frequent 97 genetic alterations (n ≥ 5 in the entire cohort) from GRIN analysis and chromosomal changes (complex karyotype and monosomy 7) (a) and category-defining gene alterations (b). KMT2A-PTD (partial tandem duplication) is independently included from other KMT2A alterations, and FLT3 alterations are classified into ITD (internal tandem duplication), TKD (tyrosine kinase domain) mutations, and non-TKD mutations due to the known functional difference. Colors correspond to Pearson correlation. Statistical significance was assessed by two-sided Fisher’s exact test to calculate P values followed by the Benjamini-Hochberg adjustment for multiple testing to calculate q values (*P < 0.05, **q < 0.05).
Extended Data Fig. 3 Transcriptional and mutational characterization of the study cohort.
a. UMAP (Uniform Manifold Approximation and Projection) plots and diffusion maps colored according to the WHO classification. b. A diffusion map colored according to molecular categories of the samples. DC: diffusion component, APL: acute promyelocytic leukemia. c. Expression of marker genes on UMAP plots and diffusion maps. Colors represent scaled expression levels. d. Cellular hierarchy scores inferred by CIBERSORT on UMAP plots and diffusion maps. Colors represent scaled scores. LSPC: leukemia stem and progenitor cell, GMP: granulocyte and macrophage projenitor, cDC: classic dendritic cell, ProMono: promonocyte, Mono: monocyte. e. UMAP plots and diffusion maps colored according to the French-American-British (FAB) classification f. A heatmap showing frequencies of defining gene alterations of AML, myelodysplasia-related in the WHO classification in each category. Colors denote the frequencies. Statistical significance was assessed by two-sided Fisher’s exact test to calculate P values of co-occurrence followed by the Benjamini-Hochberg adjustment for multiple testing to calculate q values. No pair remained significant (q < 0.05) after adjustment, and P values (<0.05) are shown instead.
Extended Data Fig. 4 Details of molecular categories.
a. Details of molecular categories with multiple category-defining alterations. The distribution on the UMAP plot according to fusion partners (KMT2Ar, NUP98r, ETS family, and APL categories) or mutation and fusions (NPM1 and GATA1 categories) are shown with colors representing the types of alterations. Age distributions according to fusion partners are also shown for KMT2Ar and NUP98r. Acute megakaryocytic/erythoid leukemia (AMKL/AEL) cases are shown separately in red. b. Proportion of molecular categories among different age groups (left: age<3, middle: 3<age<10, right: 10<age). Each column is colored according to the molecular categories, and categories associated with AMKL/AEL phenotypes are highlighted in a red square. Representative category names are shown in the columns.
Extended Data Fig. 5 Transcriptional analysis of the study cohort.
a. Plots showing averaged log2 CPM (count per million) values and standardized variance in the entire cohort (left). The top 315 variable genes used for the UMAP analysis were colored red, and representative variable gene names are shown in the right enlarged plot. The top results of the Gene Ontology (GO) term analysis by DAVID (Database for Annotation, Visualization and Integrated Discovery) are shown in the right panel. Bars represent logged FDR (false discovery rate<0.1). b. A heatmap colored according to scaled module intensities of WGCNA (weighed-gene correlation network analysis) in each molecular category. Representative genes and results of GO term analysis of genes in each module are shown on the right. Blue module enriched no GO term with FDR < 0.1. c. Distribution of differentiated cell-related hierarchy scores inferred by CIBERSORT among molecular categories. d. Distribution of prognostic scores among molecular categories. LSC17: leukemia stem cell 17 score, pLSC6: pediatric leukemia stem cell 6 score, iScore: inflammation-associated gene score, ADE-RS: Ara-C, Daunorubicin and Etoposide Drug Response Score. In c and d, lines of the box represent 25% quantile, median, and 75% quantile. The upper whisker represents the higher value of maxima or 1.5 x IQR, and the lower whisker represents the lower value of minima or 1.5 x IQR. Dots represent outliers. The colors of plots show molecular categories.
Extended Data Fig. 6 Transcriptional and mutational characterization of clusters demarcated by HOXA-B expression.
a. A heatmap showing expression patterns of HOXA and HOXB cluster genes among molecular categories. Each panel color shows the expression level (log2CPM) of genes. Molecular categories are clustered using the Euclidean distance of the expression levels and the Ward method. b. Expression (left) and ProteinPaint of mutation patterns (right) of FLT3 (top), NRAS (middle), and KRAS (bottom) in the HOXA-B categories. The distribution of log2CPM values among molecular categories is shown for the expression level, and the colors represent molecular categories. For the mutation plots, mutation types and frequencies in the HOXA and HOXB categories are shown separately, and the colors represent mutations types. Statistical significances of mutation distribution and frequency of each mutation were assessed by two-sided Fisher’s exact test (P value), and no adjustment for multiple testing was applied. For each type of NRAS and KRAS mutations, variant allele frequencies (VAFs) are also shown. The lines of the box represent 25% quantile, median, and 75% quantile. The upper whisker represents the higher value of maxima or 1.5 x IQR, and the lower whisker represents the lower value of minima or 1.5 x IQR. Dots represent outliers.
Extended Data Fig. 7 Molecular heterogeneity among HOXA and HOXB groups.
a. UMAP plot showing the distribution of fusion partners of KMT2Ar among different clusters. The dot colors denote fusion partners. b. A volcano plot showing differentially expressed genes (DEG) between the HOXA-main1-2 clusters. Genes with absolute fold change > 2 and FDR < 0.05 are considered DEGs (red: HOXA-main2 cluster high, blue: HOXA-main1 cluster high). Representative gene names are shown. c. Expression of representative DEGs on UMAP plot. The dot colors represent the relative expression of the genes. d. The association of fusion partners of KMT2Ar among different clusters. The statistical significance of the enrichment and exclusivity were assessed by two-sided Fisher’s exact test followed by the Benjamini-Hochberg adjustment (*P < 0.05, **q < 0.05, blue: exclusive, red: enriched). e. Distribution of age at diagnosis among KMT2Ar different clusters. The colors of violin plots represent clusters and lines of the box represent 25% quantile, median, and 75% quantile. The upper whisker represents the higher value of maxima or 1.5 x IQR, and the lower whisker represents the lower value of minima or 1.5 x IQR. Dots represent outliers. f. UMAP plot highlighting molecular categories in the HOXB cluster. The dot colors denote molecular categories. g. Cellular hierarchy scores represented by the color (top) and patterns of frequent mutations (bottom) in the HOXB cluster. Circles in the top highlight clusters with high hierarchy scores. Blue and red dots in the bottom show mutational status. HSPC: hematopoietic stem and progenitor cell. h. The association of molecular categories and HOXB subclusters. The statistical significance of the enrichment and exclusivity were calculated and shown as in d.
Extended Data Fig. 8 Characterization of cases without category-defining alterations.
a. ProteinPaint of rare somatic mutation in the study cohort. As comparisons, data from the COSMIC (Catalogue of Somatic Mutations in Cancer) database. Wilms tumor cohort for MLLT1 mutation and glioma cohort for H3F3A are shown at the bottom. The colors represent mutation types. b. Design of GSEA (gene set enrichment analysis) comparing immature clusters with cluster membership 6, 9, and 16 with the rest of AML samples (left) and representative results for gene sets involved in hematopoietic stem cells or lymphocytes (right). Colors of dots of UMAP show clusters. Representative enrichment score plots are also shown. c. Distribution of the WHO classification (left) and myelodysplasia-related karyotypes and genetic alterations (right) in the Unclassified cases on UMAP plots. The dot colors of the right panel represent mutational status (red-positive, blue-negative), while black dots represent excluded Unclassified cases with recurrent alterations and gray dots represents other categories. The statistical significance of the enrichment and exclusivity of WHO classification and clusters were assessed by two-sided Fisher’s exact test, and P values of cluster-wise comparison and overall distribution are shown in a table (bottom). d. Distribution of other recurrent genetic alterations in the Unclassified cases on UMAP plots. The dots are colored as in c.
Extended Data Fig. 9 Clinical association of molecular categories and known prognostic factors in the AAML1031 cohort.
a. Kaplan-Meier curves of overall survival of patients in each molecular category. Category names and curves are colored according to outcomes (blue: favorable, black: intermediate, red: unfavorable). b. Details of KMT2Ar category in the AAML1031 cohort showing the distribution of KMT2Ar cases among transcriptional clusters colored by fusion partners (left) and by XAGE1A and MECOM expression (top-right) on UMAP plot, and the association of fusion partners of KMT2Ar among different clusters (bottom-right). Circles on the UMAP highlight clusters (white: XAGE1A high, orange: MECOM high, purple: both low, pink: HOXB, yellow: AMKL, blue: immature). The statistical significance of the enrichment and exclusivity were assessed by two-sided Fisher’s exact test followed by the Benjamini-Hochberg adjustment (*P < 0.05, **q < 0.05, blue: exclusive, red: enriched). c. Kaplan-Meier curves of overall survival of patients of KMT2Ar with each fusion (left), in each cluster (middle), and Low and High-risk fusion groups by recursive partitioning (right). For the validity of prediction by KMT2Ar fusion partners and clusters, c-index scores assessed by bootstrapping (1,000 times) were shown below the plots. d. Cellular hierarchy scores on UMAP plots (top) and Kaplan-Meier curves and statistical significance of overall survival (bottom). Significant scores in univariate analysis are highlighted with asterisks (Cycling, GMP-like, and cDC-like scores). For survival curves in c-d, statistical significance was assessed by the log-rank test, and P values are shown in the plots. e. Frequency of risk assignment by bootstrapping (1,000 times). Molecular categories are sorted according to the frequency within each risk group.
Extended Data Fig. 10 Validation of the prognostic model.
a. Grouping of molecular categories into Low, Intermediate, and High-risk groups (top) and Kaplan-Meier curves of overall survival of patients in each risk group (bottom) in the AML08 cohort. b. Kaplan-Meier curves and statistical significance of overall survival of patients with known prognostic factors (FLT3-ITD status: top-left, age: bottom-left, MRD (minimal residual disease) positivity at the end of the induction I: top-right) in the AML08 cohort. C. Kaplan-Meier curves of overall survival of patients in six risk strata using risk groups (Low-Intermediate-High) and MRD (measurable residual disease) positivity in the AML08 cohort. d. Outcomes in each risk group depending on MRD and HSCT (hematopoietic stem cell transplant) status in the AAML1031 cohort. left-Hazard ratio (dot) and 95% confidence intervals (lines) in each group. right-Kaplan-Meier curves of overall survival. Survival curves start from the earliest transplant day within the cohort (day 96) and exclude patients who died before that timepoint. For survival curves in a-c, statistical significance was assessed by the log-rank test, and P values are shown in the plots. For d, the statistical significance of HSCT in each risk group was assessed by incorporating HSCT status as time-dependent variables and shown next to the hazard ratio plot. For survival analysis involving MRD status, patients with available MRD status (MRD+:n = 273, MRD-: n = 703) are included.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3.
Supplementary Tables
Supplementary Table 1. Patient characteristics of the study cohort (n = 887). Supplementary Table 2. Quality control data for the RNA-seq samples newly deposited at EGA (EGAS00001005760) in this study (n = 221). Supplementary Table 3. Quality control data for the whole-genome sequencing samples newly deposited at EGA (EGAS00001005760) in this study (paired: n = 38, unpaired: n = 20). Supplementary Table 4. Quality control data for the whole-exome sequencing samples newly deposited in this study (n = 7 for tumor samples and germline controls). Supplementary Table 5. Candidate genes (n = 87) for somatic mutation calls (RNA indels and Bambino) from RNA-seq BAM files. Supplementary Table 6. Fusions called from RNA-seq data by CICERO and structural variant (SVs) called from DNA data by CREST. Supplementary Table 7. Somatic and germline mutations (SNV, single nucleotide variants; indels, insertions and deletions) called from RNA-seq data and DNA data. Supplementary Table 8. Internal and partial tandem duplications (ITD/PTD) called by CICERO from RNA-seq data. Supplementary Table 9. Copy-number variant (CNV) data and analysis by GISTIC (Genomic Identification of Significant Targets in Cancer). Supplementary Table 10. (Genomic random interval) analysis of the entire cohort (n = 887). Supplementary Table 11. Summary of the RNA-seq cohort (887 AMLs + 5 cord blood CD34+ cell controls). Supplementary Table 12. Summary of categories defined by oncogenic genes (MNX1, BCL11B and MECOM). Supplementary Table 13. Top 315 variable genes in the entire cohort and characterization by GO term analysis using DAVID. Supplementary Table 14. GSEA (gene set enrichment analysis) of the individual categories comparing the rest of AML data using MSigDB gene sets (c2.all). Supplementary Table 15. Module–category correlations and gene–module correlations from WGCNA (weighed gene correlation network analysis). Supplementary Table 16. Differentially expressed gene (DEG) analysis between HOXA and HOXB groups and functional annotation of DEGs by GO term analysis using DAVID. Supplementary Table 17. (Genome random interval) analysis of significantly altered genes in HOXA and HOXB groups. Supplementary Table 18. Differentially expressed gene (DEG) analysis between the main clusters 1 and 2. Supplementary Table 19. Summary of 76 cases without category-defining alterations (Unclassified). Supplementary Table 20. GRIN (genome random interval) analysis of significantly altered genes in Unclassified category. Supplementary Table 21. GSEA (gene set enrichment analysis) of the immature clusters comparing the rest of AML data using MSigDB gene sets (c2.all). Supplementary Table 22. Patient characteristics and outcome data for the AAML1031 cohort (n = 1,034). Supplementary Table 23. Univariate and multivariate analyses of the AAML1031 cohort (n = 1,034). Supplementary Table 24. AML08 cohort for validation (n = 221). Supplementary Table 25. Cohort summary, univariate analyses, and multivariate analysis of the AML08 cohort (n = 221). Supplementary Table 26. List of candidate genes for germline mutations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Umeda, M., Ma, J., Westover, T. et al. A new genomic framework to categorize pediatric acute myeloid leukemia. Nat Genet 56, 281–293 (2024). https://doi.org/10.1038/s41588-023-01640-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-023-01640-3
