
Abstract
High-grade glioma is one of the deadliest primary tumors of the central nervous system. Despite the many novel immunotherapies currently in development, it has been difficult to achieve breakthrough results in clinical studies. The reason may be due to the suppressive tumor microenvironment of gliomas that limits the function of specific immune cells (e.g., T cells) which are currently the primary targets of immunotherapy. However, tumor-associated macrophage, which are enriched in tumors, plays an important role in the development of GBM and is becoming a research hotspot for immunotherapy. This review focuses on current research advances in the use of macrophages as therapeutic targets or therapeutic tools for gliomas, and provides some potential research directions.
Similar content being viewed by others


Introduction
Glioma is the most common CNS malignancy in adults with a global annual incidence of 5–6 per 100,000 people and a highly heterogeneous and aggressive nature1. And glioblastoma, the most lethal glioma, accounts for 70–75% of all diffuse glioma diagnoses. Despite the availability of conventional treatments including surgery, radiotherapy, and chemotherapy, the median survival of patients is only 14–17 months2. As a result, the development of new therapies is very crucial, for example, targeted therapy, immunotherapy and electric field therapy.
Studies over the past two decades have revealed that tumor microenvironment (TME) is a pivotal determinant of tumor behavior, and is responsible for tumor progression and metastasis3. In addition, the discovery of intracranial lymphatic vessels has led to an increased recognition of the importance of immune cells in brain tumors, challenging previous assumptions about brain tolerance and immune privilege4. Glioma is characterized by a highly suppressive and unique “cold” immune microenvironment, which includes tumor cell-derived immunosuppressive factors, exhausted cytotoxic T lymphocytes (CTLs), Treg cells, and downregulated MHC and self-presentation5. Although T-cell associated therapies, such as chimeric antigen receptor T (CAR-T) cell therapy, or immune checkpoint inhibitors (ICI) targeting programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), have shown promise in treating extracerebral tumors6, the weak lymphocyte responsiveness of GBM’s TME limits their efficacy in GBM immunotherapy7,8,9.
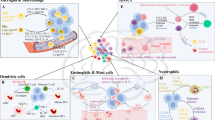
Given the limited success of single lymphocyte-related therapies, investigating other immune cells within the GBM microenvironment may hold the key to effective immunotherapy5. Tumor-associated macrophages (TAMs) comprise a significant portion of the tumor mass, accounting for 30–50% of the cells10, with ~15% derived from intrinsic microglia and 85% recruited from peripheral-derived monocytes by tumor-derived chemokines11. TAMs can be classified into several groups based on their ontogeny, surface protein markers, and transcriptomic data12. Some TAMs promote tumor angiogenesis, immune evasion, and tumor proliferation13,14,15, leading to higher tumor grade, poorer prognosis, and increased treatment resistance, while others display antitumor activity16. In light of these findings, therapeutic strategies targeting pro-tumoral TAMs can be designed, such as TAM depletion, re-educating and suppression of their pro-tumor functions (Fig. 1). In addition, recruited monocyte/macrophages can be manipulated as tools for biological therapies by exploiting their ability to transfer drugs or therapeutic genes (Fig. 2).

TAM mainly originates from microglia in the brain and peripheral-derived monocytes, and its activation types contribute to a suppressive immune microenvironment and thus results in various tumor biological behavior. As a result, TAM-targeted therapy mainly targets its recruitment, polarization, and its multiple function in tumor behavior. TAM tumor-associated macrophage, CCL chemokine ligand, CYP cytochrome P450, CSF colony-stimulating factor, VEGF vascular endothelial growth factor, HIF-1 hypoxia-inducible factors-1 (Created with BioRender.com).

Because of its tumor tropism, macrophages are ideal vehicle to carry therapeutic matters to tumor site. General payloads include conventional chemotherapy drugs, medium of the physical treatments, and genes of immunotherapeutic biomolecules. Latest technology also enables loading of CARs on macrophages, potentially breaking through the limitations of CAR therapy in solid tumors. pCAR chimeric antigen receptor plasmid, PDT photodynamic therapy, CPN conjugated polymer nanoparticles, TMZ NP temozolomide nanoparticle, DOX doxorubicin, PTX paclitaxel, CD cytosine deaminase, IFN interferon, NS nanoshells, EGFRv epidermal growth factor variant, BiTE bispecific T-cell engagers (Created with BioRender.com).
Macrophages as targets in glioma therapy
Therapies targeting TAMs have been performed intending to inhibit their pro-tumor function. Since GBM-derived chemokines such as GDNF (glial cell-derived neurotrophic factor), GM-CSF (granulocyte-macrophage colony-stimulating factor), and CCL2 (chemokine ligand), have been proven to be involved in TAM recruitment and polarization17, classical methods are to deplete TAMs and reshape TAM population by targeting these molecules18 (Table 1). With a deeper understanding of heterogeny and various roles of TAMs in tumor development, therapies targeting specific tumor-promoting mechanisms of TAM are also being developed.
TAM depletion and reduction
CSF-1/CSF-1R plays a central role in microglial and macrophage’s development and maintenance19,20. Unfortunately, depleting TAM by targeting CSF-1/CSF-1R has not been as effective in clinical trials as in preclinical trials21. Quail et al. found that resistance to CSF-1R inhibitors is linked to IGF-1 (insulin-like growth factor) and wound-associated signature from TAMs driven by IL-4/NFAT and Stat6 signaling. IGF-1 causes upregulation of the PI3K (phosphoinositide 3-kinase) pathway in tumor cells by binding to IGF-1R. Co-inhibition of IGF-1/PI3K and CSF-1R using OSI906/Linsitinib +BLZ945 was found to overcome tumor resistance to BLZ945 therapy, and extend median survival of mice from 13 days to 63 days22. In addition, GBM response to CSF-1R inhibition therapy may be related to the dictation of the TME by different tumor subtypes. Wang et al. conducted transcriptomic analyses to investigate the microenvironment of proneural, mesenchymal, and classical-type tumors and observed that mesenchymal GBM with NF1 mutation/loss displayed increased expression of macrophage-associated markers (Iba1, CD11b) and macrophage infiltration. Furthermore, they found that recurrent GBM had a higher tendency to transform into the mesenchymal subtype, which was associated with increased macrophage recruitment23. Rao et al. further investigated the differential response of proneural-like tumor and mesenchymal-like tumor to PLX3397. Single-Cell RNA-Seq reveals that the TAMs of PDGFB-driven proneural tumors is dominated by microglia and PLX3397 treatment can remodels the TAMs in this type of tumor by downregulating pro-tumor gene expression, leading to a favorable response to PLX3397 therapy. In contrast, TAMs from RAS-driven mesenchymal-like tumor exhibit pro-inflammatory and pro-angiogenic signaling, resulting in resistance to PLX3397 monotherapy or combination therapy with anti-PIK3 pathway22. Co-targeting of TAMs and angiogenesis using PLX3397+cediranib (a VEGFR2 inhibitor) could reduce tumor proliferation in mesenchymal-like tumors, but has a negative effect in proneural tumors24. These suggest that treatment targeting TAM needs to be more tailored to specific tumor subtypes.
CD73 is a promising target for GBM treatment, as this enzyme favors cancer progression and immune evasion by converting extracellular immuno-activating ATP into adenosine. To suppress CD73 expression, researchers have developed siRNA CD73-loaded cationic-Nano emulsion (NE-siRNA CD73R), which effectively reduced the population of Tregs (regulatory T cells), microglia, and macrophages in TME, and suppressed tumor growth25. This nanotechnology holds great potential for the targeted delivery of siRNA and improved GBM treatment. However, further investigations are needed to determine the optimal dosing and maintenance of siRNA in tumor tissues for gene therapy.
Targeting the CCR2/CCR2 axis can be an effective strategy for inhibiting the recruitment of TAMs. The chemokine CCL2, which is derived from the TME, is known to plays a crucial role in the migration and recruitment of blood-derived monocytes that contribute to the immunosuppressive TME and tumor progression26,27. Genetically interrupting CCL2 could prolonged the survival of mice bearing glioblastoma11. In addition, celecoxib has been found to reduce microglia and macrophages by inhibiting the expression of CCL2 and CXCL10 and28, in addition to its direct antitumor effects through the promotion of tumor cell apoptosis and regulation of cell cycle. CCR2 antagonists, such as CCX872, have also been shown to decrease the intratumoral macrophages population. When used in combination with PD-1 antagonists, CCX872 has been found to reduces myeloid-derived suppressive cells (MDSC) in tumor, enhance the activation of IFN axis and T cells, reduce exhaustion of T cells and improve tumor-killing ability, thereby benefiting the survival of mice bearing Kr15829. However, conflicting results have been reported in another research, where GL261 inoculation on Ccr2-deficient strain led to a 30% reduction of TAM, but also augmented tumor volumes30. Moreover, the clinical outcomes of this type of treatment are also not as good as in preclinical experiments suggest31. Such discrepancies may be attributed to the activation of other recruitment signaling pathways when CCR2 is deficient as the monocyte chemoattractant protein family forms a complex regulatory network. In addition, CCR2 expression in different tumor tissues may result in varying responses to CCR2 axis-targeted therapy30. Therefore, more preclinical studies are needed to characterize the chemotaxis of monocytes and verify the potential of this therapeutic target.
Some other therapeutic targets that inhibit TAM recruitment have been initially investigated. M6A demethylase ALKBH5 (AlkB homolog 5) highly expresses in GBM stem-like cells32. ALKBH5 expression promotes tumor proliferation, affects lymphocyte activation and infiltration, and is associated with M2-like macrophage infiltration33.
Kynurenine present in tumor-conditioned media activate AHR (aryl hydrocarbon receptor) in macrophages, resulting in increased expression of CCR2 as well as recruitment of TAM. AHR also drives CD39 expression, which can impair T-cell immune response. This suggests that they are both potential therapeutic targets for the TME34.
PI3Kγ plays a crucial role in promoting microglia chemotaxis and IL-11 secretion, which can in turn lead to tumorigenesis and resistance to TMZ by activating the STAT3-MYC axis in tumor cells. Targeting microglia by PI3Kγ inhibitor TG100–115 can suppress tumor genesis and therapy resistance, leading to a 6-day increase in the survival of mice bearing GL26135. Zheng et al. constructed honokiol (HNK), which can inhibit PIK3/mTOR (mammalian target of rapamycin), and the antitumor agent disulfiram/copper (DSF/Cu) into exosomes (CDX-LIPO) and targeted tumor capillary-specific Nicotinic acetylcholine receptors via DCDX. This approach induces autophagy in tumor cells while promoting pro-inflammatory polarization of macrophages, activating multiple immune cells within the TME, regulating cellular metabolism and inhibiting the production of immunosuppressive lactate36. These findings suggest that PI3Kγ may be a promising therapeutic target for treating gliomas.
p38/MAPK pathway is associated with the recruitment of macrophage/microglia as well as higher PD-L1 expression in both tumor cells and TAMs. Inhibition of this pathway in combination with anti-PD-L1 antibody treatment has been shown to reduce infiltration of blood-derived CD45high/CD11b+ macrophages and decrease PD-L1 protein expression in microglia, leading to improved survival in GBM-bearing mice resistant to TMZ37. These findings suggest that targeting the p38/MAPK pathway in conjunction with PD-L1 blockade may be a promising therapeutic strategy for GBM.
Clodronate can be uptake by macrophages and induce TAM apoptosis in solid tissues in vivo and reduce macrophage-derived VEGF, thereby inhibiting tumor angiogenesis and tumor proliferation. Combining it with exosomes can increase tissue infiltration and reduce systemic toxicity, making it a potential immunotherapeutic tool38,39.
TAM re-educating and repolarizing
Macrophages are highly plastic cells which can be activated and polarized by various factors including glioma cell-derived soluble molecules, non-coding RNAs, and factors induced by radiotherapy and chemotherapy40. Re-educating TAM to increase the proportion of pro-inflammatory subtype helps to remodel the immunosuppressive TME of gliomas, thereby potentially improving the efficacy of glioma treatment.
PD-L1 blockades has been widely used as a therapeutic strategy for many types of cancer, including GBM41,42. However, the exact mechanisms underlying therapeutic effects of PD-L1 blockade are not fully elucidated. Sydney R. Gordon has shed some light on the role of PD-1 in the recruitment and polarization of M2-like monocyte/macrophages showing that PD-1+ TAMs exhibit inferior phagocytic function, which can be reverted by PD-L1 blockade43. PD-L1 knockdown has also been found to upregulate M1-like and downregulate M2-like populations, which can prevent tumor cell invasion and migration44,45. Furthermore, combination therapy with anti-PD-L1 and radiation has been shown to increase the recruitment of immunosuppressive PD-L1+ monocytes in both radiated and non-radiated brain regions46. Additionally, anti-PD-1 antibodies can directly activate macrophages, resulting in an upregulation of cell cycle genes and cytokine production, a pro-inflammatory phenotype, and enhanced phagocytosis of tumor cells46.
In addition to its application in TAM depletion, CSF-1/CSF-1R also serves as a therapeutic target for TAM remodeling47,48. The aforementioned treatments targeting CSF-1R can modulate the polarization of TAM to some extent22,24. Another potential target for TAM modulation has been identified by Liu et al., who found that DExH-box helicase 9 enhances the expression of CSF-1 in tumor tissues by binding to transcription factor 12(TCF12), promoting the recruitment of TAMs and the expression of M2-related markers. Knockdown of this gene can inhibit macrophage recruitment and polarization and suppress tumor growth49. Thus, in CSF-1/CSF-1R inhibition therapy, TAM re-education and reduction act collectively to remodel the immunosuppressive TME of GBM19,20,24.
In PTEN-null GBM, the deficiency of PTEN gene leads to an increased secretion of Gal-9 (galectin-9) via AKT-GSK3β-IRF1 pathway. Gal-9 activate Tim-3 on macrophages and induce M2-like polarization and enhanced secretion of vascular endothelial growth factor A, resulting in tumor angiogenesis and tumor growth. Inhibition of different parts of this axis by α-lactose or Tim-3 antibody or LGALS9-targeted shRNAs can regulate TAM polarization and inhibit angiogenesis and tumor proliferation50.
CD40 costimulatory receptors on macrophages promote the effect of IL-12 and IFN and activate T-cell immunity. While IL-6 promotes the transition of macrophages to a pro-tumor phenotype and promotes the secretion of inflammatory suppressors such as IL-10 and TGF-β. However, Yang et al. found that IL-6 induces CD40 expression through Stat3 and HIF-1α. The combination of IL-6 neutralizing antibody and CD40 agonist enhances T-cell activation and ICB (immune checkpoint blockade) efficacy and extends survival from 21 to 37 days in GL261-bearing mice51.
Zhang et al. developed a mRNA based nanocarrier that stably express IRF5 (interferon regulatory factor) and its kinase IKKβ in TAMs by using a poly (β-amino ester) backbone linked to an anionic mRNA and decorated with Di-mannose moieties and polyglutamic acid on the surface. The approach leads to a shift towards M1-like phenotype, resulting in enhanced T-cell activation and recruitment52. Combination therapy with radiation resulted in a significant survival benefit in a preclinical model of glioma (25–52 days). CpG-decorated gold (Au) nanoparticles were also shown to re-educate TME. CpG is a toll-like receptor 9 agonist that can activate macrophages, promote M1-like polarization, and enhance phagocytosis and antigen presenting53. In addition, gold NPs serve as radio enhancers, making them ideal candidates for combination therapy with radiation54. These immunotherapies can promote abscopal response after radiotherapy and remodel the suppressive TAM recruited during radiotherapy55.
MiRNA-155 has been shown to downregulate the expression of anti-inflammatory proteins in microglia and macrophages, resulting in a pro-inflammatory phenotype. In addition, macrophage phagocytosis of viral particles can also promote the conversion to a pro-inflammatory phenotype. Gao et al. developed a viral gel by combining MiR-155 with nanohydrogels that are encapsulated with erythrocyte membranes and M2pep and HA2 peptides, which target tumor-promoting macrophages. This viral gel is stable in vivo and can be efficiently phagocytosed by macrophages, inducing differentiation to a pro-inflammatory phenotype and produce antitumor effects56.
In glioma, the presence of MFG-E8 (Milk fat globule EGF factor) derived from tumor cells promotes the ITGB3/STAT3 pathway, which leads to increased secretion of IL-4, ultimately resulting in the differentiation of microglia into a pro-tumor phenotype and upregulation of molecules such as TGF, IL-10, and CD206. Targeting MFG-E8 and its integrin β3 receptor inhibits TAM infiltration and reduce the expression of anti-inflammatory genes, reduces tumor size and improves tumor sensitivity to drugs57.
Targeting the pro-tumor functionality of TAMs
TAMs perform a multitude of pro-tumor functions in the TME, including promoting therapy resistance, pro-tumor signaling, pro-angiogenesis and regulating energy metabolism16. Due to the diverse functionalities of TAMs, various mechanisms need to be investigated, and different therapies targeting have the potential to be developed58 (Fig. 1).
In the context of drug resistance, TAMs have been shown to contribute to the resistance against temozolomide (TMZ) and PD-L1 checkpoint blockade therapy. This resistance is associated with a more suppressive immune microenvironment and macrophage education35,37,59. Interestingly, propofol, a conventional anesthetic, has been found to downregulate HIF-1α expression, promote a pro-inflammatory phenotype and reduce drug resistance to TMZ3. As for PD-L1 checkpoint blockade, the pro-tumoral phenotype of TAMs is associated with ARG1 type of arginine metabolism60,61. MDSCs expressing ARG1 can alter T-cell activation and enhance tumor invasion62. However, inhibiting ARG1/2 by OAT-1746 unlocks antitumor response in myeloid cells, T cells and NK cells, reduce the expression of tumor-supportive gene in TAMs, and improves the efficacy of the PD-1 checkpoint inhibition63.
Tumor-associated macrophages (TAM) have been shown to play a role in various reactions following radiation therapy. For instance, memory deficits after whole-brain radiotherapy have been linked to post-radiotherapy monocyte recruitment, which can be mitigated by treatment with PLX5622 for 21 days. However, the exact role of macrophages in causing memory loss remains unclear64. In addition, Akkari et al. observed increased recruitment of TAMs as well as an increased proportion of mononuclear-derived macrophages in recurrent tumors after radiotherapy. Transcriptomic data from their study suggest that TAMs develop distinct genetic signatures after radiotherapy compared to untreated tumors. Specifically, both SMAD and RBPJ pathways were upregulated, while signatures associated with both types of TAM ontogeny remained relatively stable65. Targeting microglia and macrophages with BLZ945 not only enhances the efficacy of initial radiotherapy, but also inhibits tumor recurrence66.
As for enhancement of tumor aggression, CCL8 secreted by TAMs binds to CCR5 and CCR1 receptors, activating ERK1/2 phosphorylating signaling pathway and inducing pseudopodia formation of GBM cells. Blocking TAM-secreted CCL8 by neutralized antibody significantly decreases invasion of glioma cells67.
For tumor angiogenesis, M2-like cells have been demonstrated to contribute to glioma angiogenesis, which is prominently driven by the interactions between TGF-β1 and surface integrin (αvβ3) interactions. Tuning cell-adhesion receptors using an integrin (αvβ3)-specific collagen hydrogel can regulate inflammation-driven angiogenesis68. TAM cells also induce angiogenesis via CYP2J2 (cytochrome P450 2J2) and 11,12-EET (epoxyeicosatrienoic acid) expression. Thus, targeting CYP2J2 can reduce tumor angiogenesis and benefit glioma therapy69. Moreover, macrophage-derived VEGFA (vascular endothelial growth factor A) plays a crucial role in tumor angiogenesis in PTEN-null GBM, and α-lactose can attenuated tumor growth by inhibiting angiogenesis in this way50.
Regarding the altered tumor metabolism, tumor metabolites also educate macrophages in the tumor microenvironment to become a pro-tumor phenotype, and the products of TAM in turn lead to tumor growth70. One of the most well-studied metabolites is 2-HG, the product of mIDH (mutant isocitrate dehydrogenase). It causes downregulation of leukocyte chemotaxis, resulting in repression of the tumor-associated immune system71. In addition, IDH-dependent macrophage education, which is associated with decreased antigen presenting and CCL2 expressing, is related to a complex re-orchestration of tryptophan metabolism. Inhibition of AhR cellular chemoreceptor, a high regulated pathway receptor after TAM exposure to 2-HG, or tryptophan metabolism can reverse immunosuppression, enhance the immune function of macrophages, and prolong the survival of IDH1-mutant glioma-bearing mice in combination with PD-L1 blockade70,72,73.
Non-coding RNA and exosomes are crucial mediators of intercellular communication between tumor cells and TAMs74,75,76,77. They have been found to play important roles in shaping the tumor microenvironment. Moreover, they have been identified as promising targets for novel cancer therapies. For instance, non-coding RNAs can also be utilized for immunotherapy, such as controlling target gene expression and replication of oncolytic viruses78. Exosomes, on the other hand, can be used as vehicles for the delivery of therapeutic agents to target cells. Understanding the complex interplay between non-coding RNA, exosomes, and tumor-associated macrophages will provide valuable insights for the development of more effective cancer treatments.
With a deeper understanding of the role of TAM in tumor development, we can develop therapies that target more specified pro-tumor functions22,79. This will help to optimize treatment efficacy while minimizing potential side effects, ultimately improving patient outcomes.
Macrophages as tools in glioma therapy
To achieve sufficient intratumoral accumulation, researchers exploit tumor-associated macrophages within the special tumor microenvironment to carry drugs or express genes80,81 (Fig. 2), for example, immune molecules and CARs (chimeric antigen receptor)82. Some details and features of these studies are presented as follows in Table 2.
Engineered macrophages as therapeutic gene vector
Macrophages have been applied for the delivery and expression of the genes of biotherapeutic substances, of which one of the most classic is IFN (interferon). IFN has been used in tumor therapy since 1986 to modulate immunity and inhibit angiogenesis. Unfortunately, its clinical application is limited due to its short half-life and high toxicity. Thus, lentiviral vector transduced IFN-α monocytes, which selectively express IFN-α under the control of GBM-specific angiopoietin receptor Tie2 promoter/enhancer elements and accumulate to tumor, were used as a vehicle for targeted delivery83. Because of the preferential activation of the Tie2 promoter in the TME, continuous, low-dose IFN-α would be released at the tumor site without inducing counterregulatory responses and systemic toxicity. IFN-α then stimulates and activates immune cells (e.g., macrophages, DCs, and T cells), inhibits angiogenesis, and suppresses tumor growth and metastasis. And a clinical study based on this technology is also underway (NCT03866109).
As for IFN-β, to exploit tumor stoma cells including TAMs in situ to secret antitumor agent IFN-β(interferon), AAV (adeno-associated virus) was injected intravenously with exosomes (exo-AAV) enhancing the ability to infect cells. The AAV encoding IFN-β was mediated by glioma stoma-specific promoter (GFAP for astrocyte and 5-NF for macrophages/microglia)84. Both types of cells can then secrete IFN-β, but the therapeutic effect of modified TAMs is weaker than that of modified astrocytes. The dilution of AAV vectors due to tumor growth may result in less effective gene expression in TAMs than in the more stable astrocytes, though AAV does effectively infect cells in CNS85. Besides, this study also mentioned the narrow therapeutic window of IFN-β. Therefore, the application of in situ genetic engineering requires the selection of a more persistent virus and a more refined TAM-specific promoter.
Macrophages have also been engineered to express BiTEs (bispecific T-cell engager) to facilitate the interactions of T cells and tumor cells via binding of a CD3ε and GBM-specific EGFRvIII (epidermal growth factor variant)86. Human monocyte-derived macrophages were transduced with lentivirus and secreted BiTEs in EGFRvIII expressing tumor site. The method resulted in the enduring expression of BiTEs, upregulated genes expression involved in T-cell activation, survival, cytokine signaling and T-cell toxicity (e.g., IL2RA, IL2RB, PRDM1, ICOS, CD40), and prevent tumor growth for 36 days87,88. Besides, immunomodulating and antigen-presenting function of engineered macrophages also help with T-cell activation in TME.
In another research, engineered microglia as the source of IL-15 was recruited to the tumor site. Researchers used rAAV2 (recombinant AAV serotype 2) carrying IL-15 to modify microglia. IL-15 (interleukin) promotes a pro-inflammatory phenotype of microglia and the cytotoxic activity of natural killer (NK) cells in TME, which also promote the production of IFN-γ, and counteracted tumor growth89. IL-12 has a similar therapeutic effect as an immunomodulator, but requires local administration to reduce systemic toxicity90. Expression of IL-12 by macrophages at subcutaneous tumor sites can improve the function of IFN cascade and activate T cells, slow tumor growth and prolong survival91. The therapeutic effect of immunomodulatory factors is confirmed. The use of macrophage carriers can improve the targeting and persistence of such therapies.
In the context of suicide gene/prodrug therapy, a novel non-viral gene vector technique based on light treatment was used to mediate the transfection of CD (cytosine deaminase) gene to macrophages in vitro. Transfected NR8383 cells could express CD with F98 glioma cells in the presence of 5-FC (5-fluorocytosine), a nontoxic precursor to 5-FU (5-fluorouracil). Because of the multi-drug resistance of NR8383 macrophages, transformed 5-FU is significantly more toxic to tumor cells than macrophages, allowing them to survive and consistently express CD92,93. This study shows high potential; however, further research is required to construct human-derived drug-resistant macrophage vectors for suicide gene therapy.
Macrophages as therapeutic drug carrier
In spite of the emergence of new chemotherapy or immunotherapy agents, passive delivered free drugs show limited efficacy because of poor diffusion into brain tumor tissue81,94. The transport of free drug is affected by blood–brain barrier, uneven tumor vasculature and the pH of the tumor microenvironment95. Tumor-targeted cell-based delivery system exploit neural stem cells, mesenchymal stem cells, and monocyte/macrophages96, among which monocytes have the widest source, and this makes monocytes/macrophages an ideal vehicle for drug delivery97. In this way, the pro-tumor microenvironment was exploited as cellular “Trojan Horses” against malignances94.
To deliver chemotherapy drugs by macrophages, the most significant aspect is to avoid the toxicity of chemotherapy drugs to the carrier. Wang et al. built ND-PG-RGD-DOX (doxorubicin) with good aqueous solubility in physiological media, and it binds to the integrin receptor avβ3 that is overexpressed on the surface of multiple cells. Nano-DOX was sequestered in the lysosomal compartment which may mechanistically contribute to monocytes’ tolerance to the drug. Monocyte took up the Nano-DOX and maintained good viability for at least 48 h. Upon recruitment to the tumor microenvironment, monocytes are induced by GBM cells to differentiate and release more Nano-DOX than in peripheral blood. The drug delivery and tumor-killing efficacy of this method has been demonstrated in orthotopic GBM xenografts95. Notably, Monocytes release Nano-DOX in the periphery no slower than in the tumor, but the exocytosis of Nano-DOX from monocytes is calcium channel dependent. Perhaps the combination with calcium channel blockers (e.g., verapamil) may limit the non-specific release of the drug in the periphery95.
Liposomes are also employed to isolate the drug and reduce toxicity to carrier cells. Using dipalmitoyl phosphatidylserine (DPPS) as a “eat me” signal, paclitaxel (PTX)-loaded liposomes were phagocyted by BV2 microglia. Microglia then cross the blood–brain barrier and deliver the drug to tumor cells via extracellular vesicles and microtubules. Owing to its high targeting performance and natural accumulation in gliomas, this cell remedy requires far less dose of PTX, and has superior antitumor effect than sole PTX-liposome or PTX therapy98. In addition, the increase in CD86/CD206, TNF-α/IL-10, and CD8/FoxP3 ratios of TAM after administration also suggested that this regime could modulate the tumor microenvironment toward a pro-inflammatory phenotype.
Conventional chemotherapy drug TMZ also faces under-delivery for GBM therapy. Mia et al. have developed a noninvasive gut-to-brain oral drug delivery system dependent on macrophages. TMZ prodrug was encapsulated in nanoparticle (NP) with β-glucans using a GSH-responsive disulfide-containing linker and were phagocytosed in situ by resident macrophages in the intestinal tract, and then delivered to brain tumor site via the lymphatic and circulatory system. Bisulfide bonds within the prodrug NPs make sure that the drugs are only released in GSH (glutathione)-overexpressing tumor microenvironment99. The TMZ that can be delivered to the intracerebral tumor tissue using prodrug NPs is five times more than that using free TMZ with some present in the liver and minimal amounts in other major organs. This treatment improved survival and reduced weight loss in mice.
Some non-chemotherapy approaches require targeted delivery of therapeutic agents. Photothermal therapy (PTT) is to induce rapid heating in tumors via gold–silica nanoshells (AuNS) mediator, which are loaded into NR8383 macrophages for delivery to the tumor site and absorbs near-infrared light to produce a therapeutic effect100,101. Photodynamic therapy (PDT) has been implemented for GBM therapy. Its effect depends on photosensitizer (PS), light and oxygen in the irradiated tumor position. Conjugated polymer nanoparticles (CPNs), as a photosensitizer, were transported by macrophages infiltrated GBM in the U87 and GL261 bearing mice. CPNs were not found to affect monocyte viability, and the using of macrophage vehicle shows superior delivery efficacy to using sole CPNs102. However, this study did not provide in vivo antitumor experimental results.
Chimeric antigen receptor-macrophage therapy
Although CAR T-cell therapy has demonstrated effectiveness and enhanced targeting for hematologic tumors, the recruitment of T cells to GBM tumor sites is limited by multiple mechanisms, including the blood–brain barrier, T-cell deletion103, and T-cell sequestration104. While several approaches have been tried to increase the infiltration of CAR-T cells in solid tumors, including GBM105,106, the suppressive tumor microenvironment could also render T-cell anergy107 and dysfunction108. As a result, creating CAR-T cells suitable for GBM treatment remains a challenge109. However, macrophages, as a crucial part of innate immune system, efficiently infiltrate into tumors, phagocyte and deplete abnormal cells, and ingest and present antigens to T cells110. These properties of monocytes/macrophages suggest expressing CAR on macrophages platform can enhance targeting and serves as a potential method of immunotherapy.
CAR-macrophage therapy is a promising area of research that has shown potential in treating various types of tumors. While very few studies have targeted GBM, one impressive method for extracranial tumors was developed by Klichinsky et al. Their CAR is based on a replication-incompetent chimeric adenoviral vector (Ad5f35), which persistently expressed CARs in macrophages and did not affect other functions. The receptor structure consists of a HER2 antigen-binding domain and an intracellular CD3ζ base domain which activate the phagocytosis of cells111. CAR-macrophages became pro-inflammatory (classically activated) phenotype after the stimulation of Ad5f35 vector, which could remodel suppressive TME. What’s more, as professional antigen-presenting cells, CAR-macrophages cross-present tumor antigens and activate T cells. Their CAR-macrophages extended the median survival time of SKOV3-burdened mice from 63 days to 88.5 days112 and CAR-M constructed using their strategy is in clinical trials (NCT04660929). CAR-macrophage therapy introduces a new means of exploiting adenovirus for tumor treatment and to some extent reshapes the immune microenvironment of tumors, suggesting that this therapy is of great significance.
Except for conventional CAR cell treatment that requires isolation, genetic modification and then reinfusion, there are new techniques to genetically engineering the intracavitary macrophages in situ to express CAR. Chen et al. constructed CD68 promoter-driven anti-CD133 CAR plasmids (pCARs) encoding the CD3ζ intracellular costimulatory domain, and used nanoporter (NP)–hydrogel superstructure for locoregional induction of CD133-specific CAR-MΦs in tumor resection cavity. Surrounding macrophages can be effectively transfected to express CARs, and then phagocytizes CD133 marked glioma stem cells and suppresses tumor growth and recurrence113. Locally engineered CAR-M cells exhibit a pro-inflammatory phenotype with only minor systemic side effects. This research provided us a new stand in CAR-macrophage therapy.
Conclusion and future perspectives
TAMs play a crucial role in the tumor immune response due to their phenotypic diversity, which can result in either tumor-promoting or tumor-suppressing effects. However, the success of therapies targeting TAMs relies on our comprehensive understanding of their properties. A deeper understanding of TAMs' role in tumor development can facilitate the identification of more precise therapeutic targets and reduce tumor resistance to treatment. Conventional treatments and immunotherapy can be evaded by tumor cells through evolution114 and immune editing115. Studies have shown that TME components, particularly TAMs, co-evolve with tumors, making it challenging to target all macrophages crudely, leading to therapeutic resistance116. Potential strategy to overcome this challenge is to design more precise treatments based on the various TAM components' functions. In addition, combining TAM clearance with engineered macrophage introduction can utilize the competitive effect between cell populations to evade treatment resistance112,117,118. These strategies highlight the need to comprehend TAMs' mechanism of action and drugs.
During TAM polarization, macrophages undergo a complex process that involves metabolic changes and changes in the expression level of human leukocyte antigen (HLA) and CCL molecules. However, our understanding of this process is not yet complete. A study has identified the temporal changes of some M2-like polarization-associated molecules (such as MEK/ERK, peroxisome proliferator-activated receptor (PPARγ)) after treatment with IL-4119, and therapies have been designed to target these signaling pathways. Gradient changes in several immune molecules (e.g., chemokine and major histocompatibility antigen) during the development of GBM have also been identified120. As a result, a deeper understanding of macrophage-related temporal changes in both intrinsic and engineered macrophages will help us gain a more specific understand of the detailed mechanisms of TAM-related therapy and develop better treatment methods.
Genetically engineered macrophage-based platforms can reduce the impact of the unique GBM tumor microenvironment on exogenous gene vectors121. However, conventional methods for modifying immune cells, such as T cells and NK cells, are not effective for monocytes/macrophages and their progenitors. Some intrinsic mechanisms of macrophages, such as restriction factors122 and the lack of corresponding receptors on the surface123, limit the function of commonly used viral vectors. Elaborate adenovirus may provide us with a way to stably express the desired gene in macrophages. Modified adenovirus recognizes a wider range of cellular markers than the commonly used coxsackie and adenovirus receptors124,125. For example, the Ad5/F35 chimeric virus has been used in preclinical and clinical studies for viral therapy of hematologic diseases as well as CAR-macrophage therapy because it recognizes the CD46 marker on macrophages and can effectively transduce them126. In addition to modified adenoviruses, modified lentiviral vectors which resist to certain restriction factors are also capable of expressing exogenous genes in monocytes and macrophages127. As for in vivo macrophage modifying, some AAV vector for gene therapy (such as AAV9) are able to cross the blood–brain barrier and can be used in combination with exosomes to enhance infection of TAM84. Several nano- or physical methods for transfection of macrophages using non-viral vectors are also under investigation93,113, although the persistence of these vectors still needs to be improved. To increase the density of expression vectors in the tumor microenvironment, we can also use oncolytic viruses that can replicate specifically in the tumor128. It is interesting to note that the vector used to modify macrophages can also affect their phenotype. Adenovirus and certain non-viral vectors can cause a shift in macrophages towards a pro-inflammatory phenotype112,113, while lentiviruses may not alter macrophage phenotype or lead to a pro-tumor phenotype91,129. The mechanism underlying these effects requires further investigation. Ultimately, the choice of vector depends on factors such as safety, efficiency, impact on macrophages, and potential toxicity of the gene being expressed.
The appropriate cell sources are crucial for macrophage therapy. However, monocytes, the primary source of macrophages, are scarce in peripheral blood, which makes it challenging to harvest enough macrophages for therapy. In such cases, induced pluripotent stem cells (iPSCs) can be used to produce macrophages with therapeutic effects, such as CAR-macrophages129,130. Moreover, self-renewing hematopoietic stem/progenitor cell (HSPC) are also potential sources of macrophages. Under certain conditions, such as bone marrow transplantation with CSF-1R blockade treatment, circulation-derived myeloid cells (CDMC) can replace microglia in brain tissue and potentially serve as a source of macrophages for therapy117. However, further validation of this approach is needed under tumor conditions. Although processing the human hematopoietic system is complex, based on the tumor tropism of macrophages, it is hypothesized that delivering HSC-derived macrophages to tumor tissue may not be as demanding as delivery to brain tissue83.
In terms of CAR therapy, several targets have been explored for GBM therapy, including interleukin-13 receptor alpha 2 (IL13Rα2), EGFRvIII, HER2, CD70, B7-H3, and others86,131,132,133, although many of them do not produce decisive outcomes in CAR-T therapy due to immunosuppressive TME, antigen drift or downregulation and heterogeneity of solid tumors. Various approaches, such as the introduction of immunosuppressants, chemokines, and increased types of CARs or CAR-T cells, have been tried to overcome these challenges105,133,134. CAR-macrophage therapy shows promise in overcoming several challenges that have hindered the application of CAR-T cells in GBM. Unlike CAR-T cells, CAR-macrophages have superior tumor infiltration capabilities and work by not only directly killing tumor cells but also stimulating the immune system, remodeling the TME, and presenting antigens113,134. As a result, CAR-macrophages may be less affected by tumor heterogeneity and downregulated CAR targets, which can hinder the effectiveness of CAR-T therapy135. However, more research is needed to fully understand the mechanisms underlying CAR-macrophage therapy. In addition, compared to CAR-T therapy, CAR-macrophage therapy has shown relatively low systemic toxicity in preclinical studies. It is hypothesized that macrophages downregulate migration-associated receptors (CCL2, CCL5) in the hypoxic TME, which may trap recruited CAR-macrophages in the tumor site and reduce systemic toxicity136. Moreover, local treatment strategies following routine surgeries for GBM may also help to reduce systemic toxicity113.
Despite the potential benefits of CAR-macrophage therapy, there are unique challenges that need to be addressed. One major challenge is maintaining the pro-inflammatory phenotype of macrophages while avoiding their pro-tumor functions. In addition, many of the current CAR-M treatments utilize the same CAR structure as in T cells, which may not be optimal for achieving both tumor cell killing and TME regulation. To overcome these challenges, it may be necessary to design CARs that can more effectively activate multiple functions of macrophages137. In conclusion, further research is needed to explore the specific mechanism of CAR-M therapy, investigate the temporal changes in the immune microenvironment after administration, and develop the optimal design of CARs suitable for macrophages.
References
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro-Oncol. 20, iv1–iv86 (2018).
Molinaro, A. M., Taylor, J. W., Wiencke, J. K. & Wrensch, M. R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 15, 405–417 (2019).
Zhao, W. & Yun, K. Propofol enhances the sensitivity of glioblastoma cells to temozolomide by inhibiting macrophage activation in tumor microenvironment to down-regulate HIF-1α expression. Exp. Cell Res. 418, 113277 (2022).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
Lim, M., Xia, Y., Bettegowda, C. & Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 15, 422–442 (2018).
Chuntova, P. et al. Unique challenges for glioblastoma immunotherapy—discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro-Oncol. 23, 356–375 (2021).
O’Rourke, D. M. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017).
Koyama, S. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 10501 (2016).
Weulersse, M. et al. Eomes-dependent loss of the co-activating receptor CD226 restrains CD8+ T cell anti-tumor functions and limits the efficacy of cancer immunotherapy. Immunity 53, 824–839.e10 (2020).
Hambardzumyan, D., Gutmann, D. H. & Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27 (2016).
Chen, Z. et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res 77, 2266–2278 (2017).
Pittet, M. J., Michielin, O. & Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 19, 402–421 (2022).
Najafi, M. et al. Macrophage polarity in cancer: a review. J. Cell. Biochem. 120, 2756–2765 (2019).
Komohara, Y., Ohnishi, K., Kuratsu, J. & Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 216, 15–24 (2008).
Qian, B.-Z. & Pollard, J. W. Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 (2010).
Engblom, C., Pfirschke, C. & Pittet, M. J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462 (2016).
Wei, J. et al. Immune biology of glioma associated macrophages and microglia: functional and therapeutic implications. Neuro-Oncol. 22, 180–194 (2020).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272 (2013).
Stanley, E. R. & Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 6, a021857–a021857 (2014).
Chitu, V., Gokhan, Ş., Nandi, S., Mehler, M. F. & Stanley, E. R. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 39, 378–393 (2016).
Butowski, N. et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncol. 18, 557–564 (2016).
Quail, D. F. et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018 (2016).
Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e6 (2017).
Rao, R. et al. Glioblastoma genetic drivers dictate the function of tumor-associated macrophages/microglia and responses to CSF1R inhibition. Neuro-Oncol. 24, 584–597 (2022).
Azambuja, J. H. et al. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol. Immunother. 69, 1801–1812 (2020).
Vakilian, A., Khorramdelazad, H., Heidari, P., Sheikh Rezaei, Z. & Hassanshahi, G. CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 103, 1–7 (2017).
Chang, A. L. et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 76, 5671–5682 (2016).
Shono, K. et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 axes contributes to antitumor effects in a mouse model of malignant glioma. Sci. Rep. 10, 15286 (2020).
Flores-Toro, J. A. et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 117, 1129–1138 (2020).
Felsenstein, M. et al. CCR2 of tumor microenvironmental cells is a relevant modulator of glioma biology. Cancers 12, 1882 (2020).
Lim, S. Y., Yuzhalin, A. E., Gordon-Weeks, A. N. & Muschel, R. J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 7, 28697–28710 (2016).
Zhang, S. et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606.e6 (2017).
Wei, C. et al. Pan-cancer analysis shows that ALKBH5 is a potential prognostic and immunotherapeutic biomarker for multiple cancer types including gliomas. Front. Immunol. 13, 849592 (2022).
Takenaka, M. C. et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 22, 729–740 (2019).
Li, J. et al. PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc. Natl. Acad. Sci. USA 118, e2009290118 (2021).
Zheng, Z. et al. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J. Immunother. Cancer 8, e000207 (2020).
Dang, W. et al. Combination of p38 MAPK inhibitor with PD-L1 antibody effectively prolongs survivals of temozolomide-resistant glioma-bearing mice via reduction of infiltrating glioma-associated macrophages and PD-L1 expression on resident glioma-associated microglia. Brain Tumor Pathol. 38, 189–200 (2021).
Zeisberger, S. M. et al. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br. J. Cancer 95, 272–281 (2006).
Zhang, H. et al. Doxorubicin-liposome combined with clodronate-liposome inhibits hepatocellular carcinoma through the depletion of macrophages and tumor cells. Int. J. Pharm. 629, 122346 (2022).
Xu, C. et al. Origin, activation, and targeted therapy of glioma-associated macrophages. Front. Immunol. 13, 974996 (2022).
Cloughesy, T. F. et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 25, 477–486 (2019).
Reardon, D. A. et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. 6, 1003–1010 (2020).
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Zhu, Z. et al. PD-L1-mediated immunosuppression in glioblastoma is associated with the infiltration and M2-polarization of tumor-associated macrophages. Front. Immunol. 11, 588552 (2020).
Fierro, J. et al. Dual-sgRNA CRISPR/Cas9 knockout of PD-L1 in human U87 glioblastoma tumor cells inhibits proliferation, invasion, and tumor-associated macrophage polarization. Sci. Rep. 12, 2417 (2022).
Ene, C. I. et al. Anti-PD-L1 antibody direct activation of macrophages contributes to a radiation-induced abscopal response in glioblastoma. Neuro-Oncol. 22, 639–651 (2020).
Stafford, J. H. et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-Oncol. 18, 797–806 (2016).
Barca, C. et al. The colony stimulating factor-1 receptor (CSF-1R)-mediated regulation of microglia/macrophages as a target for neurological disorders (glioma, stroke). Front. Immunol. 12, 787307 (2021).
Liu, L. et al. RNA ‐binding protein DHX9 promotes glioma growth and tumor‐associated macrophages infiltration via TCF12. CNS Neurosci. Ther. 29, 988–999 (2023).
Ni, X. et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN -null glioblastoma. Sci. Adv. 8, eabl5165 (2022).
Yang, F. et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat. Commun. 12, 3424 (2021).
Zhang, F. et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 10, 3974 (2019).
Sylvestre, M., Crane, C. A. & Pun, S. H. Progress on modulating tumor-associated macrophages with biomaterials. Adv. Mater. 32, e1902007 (2020).
Cao, Y. et al. Reeducating tumor-associated macrophages using CpG@Au nanocomposites to modulate immunosuppressive microenvironment for improved radio-immunotherapy. ACS Appl. Mater. Interfaces 13, 53504–53518 (2021).
Frank, M. J. et al. In situ vaccination with a TLR9 agonist and local low-dose radiation induces systemic responses in untreated indolent lymphoma. Cancer Discov. 8, 1258–1269 (2018).
Gao, X. et al. A virus‐mimicking nucleic acid nanogel reprograms microglia and macrophages for glioblastoma therapy. Adv. Mater. 33, 2006116 (2021).
Wu, J. et al. Knockdown of milk‐fat globule EGF factor‐8 suppresses glioma progression in GL261 glioma cells by repressing microglial M2 polarization. J. Cell. Physiol. 235, 8679–8690 (2020).
Chen, Y. et al. Tumor-associated macrophages: an accomplice in solid tumor progression. J. Biomed. Sci. 26, 78 (2019).
Azambuja, J. H. et al. Glioma sensitive or chemoresistant to temozolomide differentially modulate macrophage protumor activities. Biochim. Biophys. Acta BBA - Gen. Subj. 1861, 2652–2662 (2017).
Hernández, A., Domènech, M., Muñoz-Mármol, A. M., Carrato, C. & Balana, C. Glioblastoma: relationship between metabolism and immunosuppressive microenvironment. Cells 10, 3529 (2021).
Lisi, L. et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 645, 106–112 (2017).
Rodriguez, P. C., Ochoa, A. C. & Al-Khami, A. A. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 8, 93 (2017).
Pilanc, P. et al. A novel oral arginase 1/2 inhibitor enhances the antitumor effect of PD-1 inhibition in murine experimental gliomas by altering the immunosuppressive environment. Front. Oncol. 11, 703465 (2021).
Feng, X. et al. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuroinflammation 13, 215 (2016).
Akkari, L. et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 12, eaaw7843 (2020).
Almahariq, M. F. et al. Inhibition of colony-stimulating factor-1 receptor enhances the efficacy of radiotherapy and reduces immune suppression in glioblastoma. Vivo 35, 119–129 (2021).
Zhang, X. et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 100, 619–629 (2020).
Cui, X. et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 161, 164–178 (2018).
Lei, X., Chen, X., Quan, Y., Tao, Y. & Li, J. Targeting CYP2J2 to enhance the anti-glioma efficacy of cannabinoid receptor 2 stimulation by inhibiting the pro-angiogenesis function of M2 microglia. Front. Oncol. 10, 574277 (2020).
Friedrich, M. et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2, 723–740 (2021).
Amankulor, N. M. et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 31, 774–786 (2017).
Shinde, R. et al. Apoptotic cell–induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat. Immunol. 19, 571–582 (2018).
Halaby, M. J. & McGaha, T. L. 2-HG modulates glioma macrophages via Trp metabolism. Nat. Cancer 2, 677–679 (2021).
Xu, J. et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 12, 373 (2021).
Li, M. et al. Tumor-derived exosomes deliver the tumor suppressor miR-3591-3p to induce M2 macrophage polarization and promote glioma progression. Oncogene 41, 4618–4632 (2022).
Qian, M. et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene 39, 428–442 (2020).
Adamus, T. et al. Glioma-targeted delivery of exosome-encapsulated antisense oligonucleotides using neural stem cells. Mol. Ther. Nucleic Acids 27, 611–620 (2022).
Huang, H. et al. Oncolytic adenovirus programmed by synthetic gene circuit for cancer immunotherapy. Nat. Commun. 10, 4801 (2019).
Espinoza, F. I. & Walker, P. R. Untangling macrophage/microglia complexity in glioblastoma subtypes to elucidate the impact of CSF1R inhibition. Neuro-Oncol. 24, 598–600 (2022).
Riley, R. S., June, C. H., Langer, R. & Mitchell, M. J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196 (2019).
Madsen, S. J. & Hirschberg, H. Macrophages as delivery vehicles for anticancer agents. Ther. Deliv. 10, 189–201 (2019).
Chen, Y. et al. CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 139, 111605 (2021).
De Palma, M. et al. Tumor-targeted interferon-α delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 14, 299–311 (2008).
Volak, A. et al. Virus vector-mediated genetic modification of brain tumor stromal cells after intravenous delivery. J. Neurooncol. 139, 293–305 (2018).
Dashkoff, J. et al. Tailored transgene expression to specific cell types in the central nervous system after peripheral injection with AAV9. Mol. Ther. Methods Clin. Dev. 3, 16081 (2016).
Choi, B. D. et al. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc. Natl. Acad. Sci. USA 110, 270–275 (2013).
Gardell, J. L. et al. Human macrophages engineered to secrete a bispecific T cell engager support antigen-dependent T cell responses to glioblastoma. J. Immunother. Cancer 8, e001202 (2020).
Choi, B. D. et al. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol. Res 1, 163–167 (2013).
Mormino, A. et al. Enriched environment cues suggest a new strategy to counteract glioma: engineered rAAV2-IL-15 microglia modulate the tumor microenvironment. Front. Immunol. 12, 730128 (2021).
Chiocca, E. A. et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: results of a phase 1 trial. Sci. Transl. Med. 11, eaaw5680 (2019).
Brempelis, K. J. et al. Genetically engineered macrophages persist in solid tumors and locally deliver therapeutic proteins to activate immune responses. J. Immunother. Cancer 8, e001356 (2020).
Wang, J. L., Scheitler, K. M., Wenger, N. M. & Elder, J. B. Viral therapies for glioblastoma and high-grade gliomas in adults: a systematic review. Neurosurg. Focus 50, E2 (2021).
Romena, G., Nguyen, L., Berg, K., Madsen, S. J. & Hirschberg, H. Enhanced gene transfection of macrophages by photochemical internalization: potential for gene-directed enzyme prodrug therapy of gliomas. Photodiagnosis Photodyn. Ther. 33, 102098 (2021).
Ibarra, L. E. Cellular Trojan horses for delivery of nanomedicines to brain tumors: where do we stand and what is next? Nanomedicine 16, 517–522 (2021).
Wang, C. et al. Monocyte-mediated chemotherapy drug delivery in glioblastoma. Nanomedicine 13, 157–178 (2018).
Hosseinalizadeh, H., Mahmoodpour, M., Razaghi Bahabadi, Z., Hamblin, M. R. & Mirzaei, H. Neutrophil mediated drug delivery for targeted glioblastoma therapy: a comprehensive review. Biomed. Pharmacother. 156, 113841 (2022).
Parker Kerrigan, B. C., Hossain, A., Yamashita, S. & Lang, F. F. Stem cell therapy of gliomas. In Progress in Neurological Surgery (eds Chernov, M. F. et al.) Vol. 32, 124–151 (S. Karger AG, 2018).
Du, Y. et al. Engineered microglia potentiate the action of drugs against glioma through extracellular vesicles and tunneling nanotubes. Adv. Healthc. Mater. 10, 2002200 (2021).
Miao, Y. et al. A noninvasive gut‐to‐brain oral drug delivery system for treating brain tumors. Adv. Mater. 33, 2100701 (2021).
Madsen, S. J. et al. Nanoparticle-loaded macrophage-mediated photothermal therapy: potential for glioma treatment. Lasers Med. Sci. 30, 1357–1365 (2015).
Christie, C., Madsen, S. J., Peng, Q. & Hirschberg, H. Photothermal therapy employing gold nanoparticle- loaded macrophages as delivery vehicles: comparing the efficiency of nanoshells versus nanorods. J. Environ. Pathol. Toxicol. Oncol. 36, 229–235 (2017).
Ibarra, L. E. et al. Trojan horse monocyte-mediated delivery of conjugated polymer nanoparticles for improved photodynamic therapy of glioblastoma. Nanomed 15, 1687–1707 (2020).
Walker, D. G., Chuah, T., Rist, M. J. & Pender, M. P. T-cell apoptosis in human glioblastoma multiforme: implications for immunotherapy. J. Neuroimmunol. 175, 59–68 (2006).
Chongsathidkiet, P. et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 24, 1459–1468 (2018).
Wang, G. et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 31, 134–153 (2023).
Wang, D. et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 12, eaaw2672 (2020).
Schwartz, R. H. T cell anergy. Annu. Rev. Immunol. 21, 305–334 (2003).
Thommen, D. S. & Schumacher, T. N. T cell dysfunction in cancer. Cancer Cell 33, 547–562 (2018).
Choe, J. H. et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 13, eabe7378 (2021).
Gatto, L., Nunno, V. D., Franceschi, E. & Brandes, A. A. Chimeric antigen receptor macrophage for glioblastoma immunotherapy: the way forward. Immunotherapy 13, 879–883 (2021).
Niu, Z. et al. Chimeric antigen receptor-modified macrophages trigger systemic anti-tumour immunity. J. Pathol. 253, 247–257 (2021).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Chen, C. et al. Intracavity generation of glioma stem cell–specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci. Transl. Med. 14, eabn1128 (2022).
Bhang, H. C. et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 21, 440–448 (2015).
Tsai, C.-H. et al. Immunoediting instructs tumor metabolic reprogramming to support immune evasion. Cell Metab. 35, 118–133.e7 (2023).
Kloosterman, D. J. & Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 186, 1627–1651 (2023).
Shibuya, Y. et al. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci. Transl. Med. 14, eabl9945 (2022).
Muthana, M. et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 71, 1805–1815 (2011).
He, L. et al. Global characterization of macrophage polarization mechanisms and identification of M2-type polarization inhibitors. Cell Rep. 37, 109955 (2021).
Virtuoso, A. et al. Tumor microenvironment and immune escape in the time course of glioblastoma. Mol. Neurobiol. 59, 6857–6873 (2022).
Moyes, K. W. et al. Genetically engineered macrophages: a potential platform for cancer immunotherapy. Hum. Gene Ther. 28, 200–215 (2017).
Lahouassa, H. et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228 (2012).
Nilsson, M., Karlsson, S. & Fan, X. Functionally distinct subpopulations of cord blood CD34+ cells are transduced by adenoviral vectors with serotype 5 or 35 tropism. Mol. Ther. J. Am. Soc. Gene Ther. 9, 377–388 (2004).
Katayama, K. et al. Enhanced in vivo gene transfer into the placenta using RGD fiber-mutant adenovirus vector. Biomaterials 32, 4185–4193 (2011).
Collins, L. T. & Curiel, D. T. Synthetic biology approaches for engineering next-generation adenoviral gene therapies. ACS Nano 15, 13970–13979 (2021).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Moyes, K. W. et al. Genetically engineered macrophages: a potential platform for cancer immunotherapy. Hum. Gene Ther. 28, 200–215 (2017).
Boucher, P., Cui, X. & Curiel, D. T. Adenoviral vectors for in vivo delivery of CRISPR-Cas gene editors. J. Control. Release 327, 788–800 (2020).
Zhang, L. et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 13, 153 (2020).
Ackermann, M. et al. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat. Protoc. 17, 513–539 (2022).
Ahmed, N. et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J. Immunother. Cancer 3, O11 (2015).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Yang, M. et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 10, 7622–7634 (2020).
Pan, K. et al. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. CR 41, 119 (2022).
Bielamowicz, K. et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncol. 20, 506–518 (2018).
Henze, A.-T. & Mazzone, M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 126, 3672–3679 (2016).
Morrissey, M. A. et al. Chimeric antigen receptors that trigger phagocytosis. eLife 7, e36688 (2018).
Acknowledgements
This study was funded by Key research and development project of science and technology department of Sichuan Province (2022YFS0321, 2022YFS0320); National Natural Science Foundation of China (82002648); General Program of the National Natural Science Foundation of China (82173175); Knowledge Innovation Program of the Chinese Academy of Sciences (JH2022007) and 1·3·5 project for disciplines of excellence–Clinical Research Incubation Project, West China Hospital, Sichuan University (2020HXFH036). Figures in this review article were created with BioRender.com.
Author information
Authors and Affiliations
Contributions
F.T., Y.W., and Y.Z. searched the relevant literature for analysis and constructed the first draft of this paper. F.T. and Y.W. contributed equally to this work as co-first authors. A.T. and A.X. provided with valuable advice during revising the manuscript. J.X. managed the project and revised the manuscript. All authors approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, F., Wang, Y., Zeng, Y. et al. Tumor-associated macrophage-related strategies for glioma immunotherapy. npj Precis. Onc. 7, 78 (2023). https://doi.org/10.1038/s41698-023-00431-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00431-7
This article is cited by
-
Two-dimensional nanomaterials induced nano-bio interfacial effects and biomedical applications in cancer treatment
Journal of Nanobiotechnology (2024)
