
Abstract
Despite advances in treatment regimens that comprise surgery, chemotherapy, and radiation, outcome of many brain tumors remains dismal, more so when they recur. The proximity of brain tumors to delicate neural structures often precludes complete surgical resection. Toxicity and long-term side effects of systemic therapy remain a concern. Novel therapies are warranted. The field of NK cell-based cancer therapy has grown exponentially and currently constitutes a major area of immunotherapy innovation. This provides a new avenue for the treatment of cancerous lesions in the brain. In this review, we explore the mechanisms by which the brain tumor microenvironment suppresses NK cell mediated tumor control, and the methods being used to create NK cell products that subvert immune suppression. We discuss the pre-clinical studies evaluating NK cell-based immunotherapies that target several neuro-malignancies and highlight advances in molecular imaging of NK cells that allow monitoring of NK cell-based therapeutics. We review current and ongoing NK cell based clinical trials in neuro-oncology.
Similar content being viewed by others
Introduction
Brain tumors comprise 1.4% of all cancers, whose treatment options and survival outcomes vary depending on the type of brain tumor1,2. Therapeutic advances in chemotherapy, radiation, and neurosurgical procedures have increased survival of patients with brain tumors3. Yet, malignancies like high-grade gliomas, medulloblastoma, and diffuse intrinsic pontine glioma (DIPG) continue to portend dismal outcomes due to recurrence and/or progression4,5.
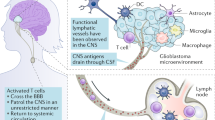
Despite aggressive treatment regimens that include surgery, radiotherapy, and chemotherapy, many central nervous system (CNS) tumors remain incurable. Intra-tumoral heterogeneity and the ability of the brain tumor cells to evade the immune system and suppress anti-tumoral immunity are some of the important limitations1,6,7. Another major limitation to effective therapy is the blood–brain barrier (BBB), which blocks the access of most targeted drugs delivered systemically into tumor sites8, and the significant morbidity and concerning long-term side effects caused by current systemic therapeutic regimens. The lack of lymphatic drainage and low abundance of antigen-presenting cells seem to protect brain tumors from the immune system. However, preclinical, and translational studies in the past decade have changed this perception and showed an important role for immune cells in brain tumors7,9,10,11,12,13.
Natural killer (NK) cells are the predominant innate lymphocyte subset that mediate anti-tumor response. They are known to play important roles in anti-tumor immunity in various cancers14,15, and their efficacy against brain tumors has been shown in preclinical studies16,17,18,19,20,21. NK cell cytotoxicity against cancer cells involves the secretion of perforin, granzyme B22,23,24, and pro-inflammatory cytokines, such as tumor necrosis factor-α and interferon (IFN-γ)25,26,27. As such, NK cell immunotherapy appears as a promising, novel treatment approach for brain tumors28.
This review explores the role of NK cells in the brain tumor microenvironment, emphasizes preclinical research with NK cells, examines effects of NK-cell-based therapies in adult and pediatric patients with brain tumors, and discusses tracking the delivery of infused NK cells through molecular neuroimaging. Obstacles that lie ahead of NK cell therapy advancement and future directions in the fight against brain tumors are further explored.
NK cells in the brain tumor microenvironment
NK cells have been shown to play a fundamental role in brain metastases, meningiomas29, glioblastomas30, craniopharyngiomas31, DIPG32, medulloblastoma, and ependymoma12. The tumoral niche takes a toll on the functioning of NK cells due to the immunosuppressive factors expressed on or released by cancer cells. Single-cell analysis of the immunosuppressive microenvironment of glioblastoma demonstrated those NK cells that infiltrated into the tumor lesions, expressed higher levels of CXC chemokine receptor 3 (CXCR3) and lower levels of IFNγ33.
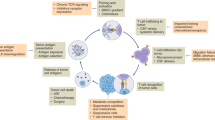
CNS tumor cells can secrete chemokines, cytokines, and growth factors that diminish the anti-tumor cytotoxicity of NK cells in the brain (Fig. 1). This promotes cancer proliferation, spread, and resistance to treatment. In gliomas, anergic NK cells promote glioma differentiation, which results in the resistance to NK cell-mediated cytotoxicity. Sustained release of interleukin (IL)-6 and IL-8, along with decreased IFN-γ secretion post-differentiation further increases immune resistance to NK cell therapy34. Pro-tumoral inflammatory mediators, such as cyclooxygenases (COX) and prostaglandin E2, further weaken NK cell viability and chemokine production35.

Mechanisms of NK cell deactivation by cancer cells include the expression of MHC-I, PD-1, CD155, and CD73 expression on cancer cells that can inhibit NK cells. Cytokine and chemokine release, such as IL-6, IL8, IL-10, VEGF, PDGF, MMP9, IDO, adenosine, PGE2, and TGF-beta can further deactivate NK cells. Proteins expressed on NK cells, such as NKG2E and TIGIT, can lead to brain tumor progression.
The NK cell numbers in the brain tumor microenvironment are often depleted, especially after chemo-radiotherapy because of inflammatory mediators like transforming growth factor-β (TGF-β), COX, and prostaglandin E235,36. In a case series of four patients with DIPG, lymphocyte profiling revealed decreased NK cell levels in all four cases32. In patients with glioblastoma, the number of NK cells in isolated tumor specimens were significantly decreased after being treated with radiation and temozolomide36.
The expression of certain molecules on tumor cells can inhibit NK cell activity. In glioblastoma, major histocompatibility complex class I molecules expressed on tumor cells bind to NK cell inhibitory receptors and suppress their immune function37,38. Gliomas and brain metastases can further downregulate the expression of activating receptors, such as NK Group 2 member D (NKG2D), to evade immune activation of host NK cells39,40. The tumor microenvironment in pediatric solid tumors is rich in immunosuppressive molecules such as transforming growth factor beta (TGFβ)41,42, adenosine43, PGE244, as well as indoleamine 2,3-dioxygenase (IDO)45. All of these factors have been associated with to significant NK cell dysfunction46. As opposed to solid tumors in the adult population, infiltrating and tumor associated macrophages (TAM) are abundantly present in the pediatrics and have been linked to a poor prognosis. Myeloid-derived suppressor cells (MDSC) cells have been also described to commonly infiltrate into the tumor, thereby facilitating the release of various immunosuppressive factors. These include chemokines and cytokines, most of all macrophages-derived IL-10, vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and matrix metalloproteinase-9, all of which contribute to therapy resistance. In addition, TAMs nurture metastatic spread by upregulating the CCL2/CSF-1 signaling axis in childhood gliomas and portending poor outcomes in neuroblastoma47,48. To date, a large body of research has demonstrated that there is a significant number of tumor-infiltrating NK cells that have been associated with good prognosis in many solid tumors46. This is reaffirmed in preclinical models where depletion of NK cell populations prior to tumor transplantation induces a more aggressive phenotype with metastatic tumors. These data have now formed a steppingstone to evaluate NK cell-based therapies in brain tumors, which are rapidly gaining attention as an attractive adoptive immunotherapy in pediatrics49.
Other cytokines and released factors activate NK cells in the brain microenvironment (Fig. 2). IL-15 increases the expression of molecules associated with NK-cell cytotoxicity50. IL-2 and heat shock protein 70 (HSP70) improves NK cell homing within the tumor site in vivo51, which suggests that the administration of IL-15/HSP70/IL-2-treated NK cells may be a promising therapeutic approach to be considered in the treatment of brain tumors. Increasing NK cell-mediated recruitment of dendritic cells through the release of chemokine ligand 5 (CCL5) and X-C Motif Chemokine Ligand 1 (XCL1) can positively impact survival35.

Mechanisms of NK cell activation include the secretion of IFN-γ, TNF-α, perforins, and granzyme B. Cytokines, such as IL-2 and IL-15, can also prime the NK cell immune cytotoxic effect. Protein expression of NKG2D, NKp44, CD226, ABCC3, CD16, CXCR3, and CD96 can help NK cells recognize cancer cell antigens and trigger their cytotoxic effect.
The expression of certain genetic patterns in NK cells or tumor cells can affect brain tumor recurrence and aggressiveness. Silencing of KLRC3 in NK cells, decreases the self-renewal capacity, invasion, proliferation, radio-resistance, and tumorigenicity of the U87-MG glioma cell line52. CRISPR-Cas9-mediated knockout of T cell immunoglobulin mucin family member 3 (TIM3), a marker of dysfunctional NK cells53,54, in human NK cells inhibits glioma cellular growth53. The expression of CD73 or CD155 in glioma suppresses NK cell presence in the tumor microenvironment and contributes to increased tumor migration and aggressiveness55,56. NK cells that express CD16, predominate in glioblastoma patients surviving more than 12 months after surgery without disease progression57. This subtype of NK cells co-expresses increased levels of ATP Binding Cassette Subfamily C Member 3 (ABCC3) and IFN-γ, which are related to a strong, long-term NK cell cytotoxic response57. NK cells can further induce antibody-dependent cell-mediated cytotoxicity (ADCC) to provide anti-tumor cytotoxicity58. This process is mediated through the CD16 receptor on NK cells that binds the Fc portion of IgG antibodies triggering the lysis of tumor cells. The expression of CD1d, an antigen-presenting molecule for NK cells, is common in human glioblastoma but not in glioma stem cells59. Retinoic acid upregulates CD1d expression in glioma stem cells in vitro, sensitizing them to NK-mediated cytotoxicity59.
Hypoxic stress in the tumor microenvironment can lead to immunosuppressed NK cell phenotypes60. Pre-activation of NK cells through exposure to normoxic culture followed by hypoxic conditions allow NK cells to shift metabolically from oxidative phosphorylation into glycolysis61. This shift helps NK cells overcome hypoxia-mediated functional impairment. Hypoxia-inducible factor 1-alpha (HIF-1α) activation can upregulate the expression of the natural cytotoxicity receptor NKp44 receptors to reverse impairment in NK cell survival, proliferation, and tumor cytotoxicity61. The activation of extracellular signal-regulated kinase (ERK)/ signal transducer and activator of transcription 3 (STAT3) pathways can further expand functionally robust NK cells for adoptive cellular therapy by reducing p21/p53 apoptotic pathways, upregulating cell cycle-promoting genes, CCNE1, CDC6, CDC20 and downregulating of cell cycle-arrest genes, CDKN1A, GADD45A, and MDM261.
Preclinical advancements in NK cell therapy
Novel immunotherapeutic approaches and cytotoxic agents against brain tumor cells can promote NK cell immune response (Fig. 3). Ex vivo expansion of NK cells and their re-introduction into tumor setting has been tested in preclinical settings. NK cells that have been expanded ex vivo possess the cytolytic ability to target medulloblastoma cells in vitro62,63. Retro-orbital administration of ex vivo expanded and activated NK cells along with IL-2 prolonged the survival of NOG mice bearing subcutaneous U87MG-derived tumors64. The combination of ex vivo expanded NK cells with an IL-15 agonist and dinutuximab, an anti-disialoganglioside GD2 monoclonal antibody, significantly increases cytotoxicity against pediatric glioma cells and improves survival in preclinical models65. Interim analysis of a phase 1 clinical trial using anti-GD2 CAR NK cells, showed that the cells underwent in vivo expansion and localization to the tumor site in patients with relapsed or refractory neuroblastoma (Table 1).

Preclinical approaches involving NK cells for the treatment of brain tumors focus on three modes of action. Tumor-mediated antigen activation includes the activation of NK cell activity by cancer cell antigen recognition. Recognition of stress antigens, expressed as a result of bortezomib treatment, can activate NK cell receptors. Expression of E-cadherin, primed by oHSV-1 G207, enhances NK cell recognition and activation of NK cells through the KLRG1 receptor. The recognition of the RAET1 antigen on cancer cells by the NKG2D receptor on NK cells can also lead to NK cell activation and cytotoxicity. Other receptors on NK cells, such as KLRK1, and KLRC2/3/4 have been reported to play a role in NK cell activation against cancer cells in the brain tumor microenvironment. Growth factors, such as IL2, IL15, and PDGFDD, promote NK cell recruitment and cytotoxicity. Inhibition of TGF-beta, using viral vectors carrying shRNA, has been shown to increase NK cell activation against brain tumor cancer cells in preclinical settings. Checkpoint inhibition and immune checkpoint blockade through antibodies against GD2, PD-L1, PD-1, and B7-H3, has been reported to enhance NK cell activity in brain tumors. Viral vectors carrying siRNA targeting PD-L1 on cancer cells or lipid nanoparticles carrying siRNAs against inhibitory checkpoints, such as Cbl-b, SHP-1, and c-Cbl, on NK cells, have also been reported to activate the NK cell immune response against brain tumor cells.
Anti-tumor immunity in the brain is restrained by multiple tumor-induced immunosuppressive mechanisms. Targeting immunosuppression may unleash NK cell anti-tumor cytotoxicity in the brain. Programmed death 1 (PD-1)/PD-L1 inhibitors can potentiate the NK cell response against brain tumors, such as gliomas66,67. As NK cell inhibition is usually accompanied by the expression of multiple immune checkpoint molecules on T cells68, therapies involving combinations of immunotherapeutic agents and NK cells can be vital in overcoming therapeutic resistance. NK cells that are platelet-derived growth factor D (PDGF-DD) activated and/or highly express killer cell lectin-like receptors (KLR), such as KLRK1 and KLRC2, KLRC3 and/or KLRC4, are associated with better prognosis in low-grade glioma69. Using a lipid nano-carrier encapsulating small interfering RNAs that gene silence the key intrinsic inhibitory NK cell molecules, SHP-1, Cbl-b, and c-Cbl, led to increased NK cell activation, and tumor killing70. Labeling NK cells with a fluorine-19 (19F)-based perfluorocarbon emulsion enhanced the suppression of medulloblastoma growth in mouse orthotopic models while enabling 19F MRI to provide feedback on the delivery of infused NK cells71. TGFβ neutralization sensitize medulloblastoma cells to NK cell cytotoxicity in vitro72. Targeting the αv integrin/TGF-β axis further improves NK cell function against glioblastoma stem cells73. Treatment of glioma stem cell-engrafted mice with allogeneic NK cells in combination with inhibitors of integrin or TGF-β signaling or with TGFBR2 gene-edited allogeneic NK cells prevented glioma-induced NK cell dysfunction and tumor growth73. In glioma, suboptimal doses of N6-isopentenyladenosine upregulate cell surface expression of NKG2D ligands, which facilitates NK cell-mediated cytotoxicity74. The combination of NK cells and temozolomide enhances temozolomide-induced inhibition and apoptosis in U87MG glioma cell lines and overcomes temozolomide resistance74. Resveratrol and IL-2 enhance the cytolytic activity of NK cells by upregulating the expression of c-Myb, a downstream transcription factor of Akt and mTORC275. The administration of resveratrol and IL-2 in humans and mice increased NK cell activity in the blood and effectively inhibited tumor growth and metastasis in mice76. Using hemagglutinating virus of Japan-envelope (HVJ-E) a non-replicating virus-derived vector, it was possible to deliver siRNA that target PD-L1 expression77. HVJ-E provoked a robust anti-tumoral immunity by activating NK cells and CD8 + T lymphocytes77.
Oncolytic viral therapy for brain tumors suffers from poor intra-tumoral viral spread and rapid clearance by NK cells. Oncolytic Herpes Simplex Virus (oHSV) engineered to encode E-cadherin, a ligand for NK cell inhibitory receptor KLRG1, protects virus-infected cells from NK cell killing, thereby enhancing viral spread, facilitating cell-to-cell infection and viral entry, and reducing viral clearance78. Removing the genes that are essential for viral replication in normal brain tissue prevents productive infection of normal cells while permitting conditional replication in tumor cells. oHSV expressing anti(α)-human CD47 IgG1 or IgG4 antibody were capable of blocking CD47, an immune checkpoint in glioblastoma cells, thereby decreasing local tumor size and improving survival in murine models of glioblastoma79. NK cells, along with macrophages, mediated the anti-tumor cytotoxic response and the antibody production by the oHSV79. The engineered oHSV-1 G207 oncolytic virus, which selectively replicates in brain tumor cells80,81, sensitized pediatric high-grade gliomas to virotherapy in preclinical models13. The combination of oncolytic viral therapy with NK cell therapy can also result in a synergistic, therapeutic effect against brain tumors. The combination of bortezomib, a proteasome inhibitor, oHSV, and NK cell therapy, demonstrated synergetic effects and therapeutic efficacy., Treatment of oHSV-infected gliomas with bortezomib induced necroptotic cell death to enhance NK-cell immunotherapy82,83. Bortezomib treatment of cancer cells induces stress antigens that are identified by NK-activating receptors84. This leads to the release of cytolytic molecules, such as IFN-γ, perforin, and granzyme A by NK cells, which disrupts mitochondrial function and leads to glioblastoma cell death84.
In recent years, efforts have been made to provide NK cells with a means of responding to specific cell expressed antigens. This has been achieved through transduction of primary NK or NK cell lines with chimeric antigen receptors (CAR)85,86. CARs consist of an antigen-specific extracellular domain, a transmembrane domain and an intracellular signaling domain that utilizes down-stream signaling adapters such as the CD16 signaling molecules, CD3ζ or FcεR1γ87. One major benefit of using NK cells is that they do not cause graft versus host disease in the allogeneic setting88, making them a safe choice for an off-the-shelf product. Activating cytokines, such as IL-2 or IL-15, may also aid in the persistence, expansion, and trafficking of CAR-NK cells. To date, several CAR-T therapies have been evaluated clinically. These include EGFRvIII, IL13Ra2, and HER2. In the EGFRvIII trial no adverse events were observed up to a dose of 1 × 1010 cells. However, two patients receiving a dose of 3 × 1010 cells resulted in serious adverse events including dyspnea and hypoxia while another patient receiving 6 × 1010 cells died 4 h after administration89. Median progression-free survival in this trial was 1.3 months with an overall median survival of 6.9. Additionally, all patients developed hematologic toxicities from the preparative chemotherapy. The current methodology to generate CAR-T cells requires leukapheresis to acquire the patient’s own T cells to activate and transduce with the desired CAR construct, which then must be expanded and selected for the CAR expressing T cells. This process is lengthy and expensive and leaves the possibility of adverse events including cytokine release syndrome (CRS). Alternatively, NK CAR products can be derived from haploidentical, patient-derived, ex vivo expanded NK90, NK cell lines such as NK9291,92,93 or from iPSC-derived NK cell products94,95 constituting an off-the-shelf product that can be administered regardless of patient MHC background and have demonstrated limited adverse effects compared to CAR-T therapies96. Thus, NK cells represent a robust and flexible platform for a variety of CARs that may target CNS tumor-specific antigens, with less neurotoxicity and cytokine release syndrome seen with CAR-T cell products.
Currently, there are more than 30 ongoing clinical trials using CAR-NK derived from a variety of sources including NK cell-lines, primary peripheral blood NK, umbilical cord blood-derived NK, hematopoietic stem cell (HSC) derived NK97, and iPSC-derived NK targeting a variety of liquid and solid tumors. However, a major challenge to using HSC-derived NK cells is the limited expansion capacity of the initial pool of primary cells while still preserving their stem-cell-like properties. NK cell lines are an attractive NK cell source as, unlike primary cell sources, they require no purification from contaminating cell populations, can be grown in large numbers and are easily transducible98. While there is limited clinical data for CAR-NK products, there are more than 100 pre-clinical studies (www.carnkreview.com) demonstrating good in vitro and in vivo efficacy in animal models87 (Table 2).
In vitro studies using CAR-NK to target glioblastoma have focused on CAR-targeting of upregulated proteins in brain tumor cells (Fig. 4). CAR KHYG-1 NK cells that can target c-Met also known as hepatocyte growth factor receptor (HGFR) and AXL proteins that are overexpressed in glioblastoma cell lines, led to cytokine secretion and glioblastoma cell lysis99. Epidermal growth factor receptor (EGFR) and it’s variant, EGFRvIII, are well-established therapeutic targets in glioblastoma100 expressed in 40–60% of glioblastoma tumors101 and minimal expression on surrounding tissue102. Similarly, human epidermal growth factor receptor 2 (HER2), a member of the family of EGFR-related receptor tyrosine kinases, is expressed in nearly 80% of glioblastoma tumors103. Preclinical trials with both EGFR/EGFRvIII104 and HER2105 CAR-NK have demonstrated enhanced cytotoxicity against glioblastoma cells in vitro and resulted in increased tumor control and survival in animal models. Use of a HER2 CAR-NK in an orthotopic xenograft glioblastoma mouse model inhibited tumor progression and significantly extended survival105. Furthermore, mice that were re-challenged in the other brain hemisphere rejected the newly implanted tumor more than 120 days after the initial therapy105. In metastatic brain cancer, CAR NK cells that overexpress EGFR in combination with oHSV-1 showed a synergistic lytic effect as compared to monotherapy.106 The combination therapy increased IFN-γ production and significantly improved survival.106 The administration of oHSV that release IL15, a cytokine that activates the immune system, along with EGFR-CAR NK cells improved the survival of NK and CD8 T cells107. This, in turn, inhibited tumor growth and prolonged survival of glioblastoma-bearing mice107. The combination of CAR NK cells designed to target human leukocyte antigen G (HLA-G), an immune checkpoint protein, with low-dose chemotherapy effectively reduced xenograft brain tumor growth and extended median survival in orthotopic mouse models108. This suggested that the reversal of the HLA-G-mediated immunosuppression restores native NK cytolytic functions. CAR NK cells designed to activate the glioblastoma immune-metabolic microenvironment by inhibiting CD73, suppressing adenosine production, and inhibiting autophagy have decreased tumor size in preclinical models using patient-derived xenografts109.

Chimeric antigen receptor (CAR) approaches utilize primary NK cells (isolated from patients) or NK cell lines and their transduction with a CAR gene to produce CARs. CARs consist of an antigen-recognition domain and signaling domain that utilizes down-stream signaling adapters such as the CD16 molecule. Following expansion of the CAR NK cells ex vivo, the cells are injected back into patients with brain tumors. In vitro, CAR KHYG-1 NK cells have been used to target c-Met, FOLR1, and AXL receptors that are overexpressed in glioblastoma cell lines. In preclinical settings, anti-EGFR/EGFRvIII and anti-HER2 CAR-NK cells have also demonstrated enhanced cytotoxicity against glioblastoma cells in vitro and resulted in increased tumor control and survival in animal models. Anti-EGFR CAR NK cells further showed lytic effects in combination with oncolytic virotherapy in metastatic brain cancer.
NK cell therapy for adult brain tumors
Human studies on brain tumors provide evidence to support the use of NK cells for therapy. In patients with glioblastoma, the presence of activated, CD16-positive NK cells were significantly associated with improved survival110. The decrease in levels of activated NK cells correlated to transition from low to high-grade brain tumors, as higher levels of activated NK cells were found in low-grade gliomas than in high-grade gliomas111 (Table 3).
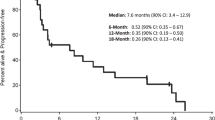
NK cell therapeutics have mostly focused on overcoming the immunosuppressive signals from the tumor microenvironment and amplifying the effect of NK cells with combination therapy. Adoptive immunotherapy using NK cells that have been expanded and activated ex vivo have made it into clinical trials for patients with high-grade gliomas. Administration of autologous NK cells to patients with recurrent malignant gliomas has been shown to be safe and partially effective; four of nine patients recorded tumor regression112. In a phase I/IIa clinical trial, adoptive, ex-vivo-expanded, and activated NK cells and T lymphocytes from peripheral blood mononuclear cells of patients with recurrent glioblastoma was safe, with a median overall survival of 22.5 months and a median progression-free survival of 10 months113. Five of 14 patients (36%) showed a durable response and were alive for over two years113. The combination of NK cell therapy with mTOR inhibitors was safe in adult patients with brain tumors114. At the 6th month interval, disease control rate was 72%114. The combination of monalizumab, a humanized anti-NKG2A antibody, and cetuximab, an EGFR inhibitor, was effective in promoting ADCC in patients with head and neck squamous cell carcinoma115. A phase II trial utilizing this combination therapy showed a 31% objective response rate115. As EGFR is a prime target for gliomas and contribute to temozolomide resistance116,117, the combination therapy can be a viable option for glioma treatment and induction of NK cell anti-tumor activity.
Cancer vaccines designed to prime an anti-tumor, cytotoxic response against using antigen-presenting cells, can employ NK cells as effector cells. In adult patients with newly diagnosed glioblastoma, administering autologous dendritic cells that have been charged with the tumor lysate for an average of seven months was safe but clinically ineffective118. Nevertheless, the vaccine was capable of significantly increasing the levels of IFN-γ, the transcription factor T-bet in the blood, and IL-2 in a dose-dependent manner119. Levels of IFN-γ and CD8 + NK cells in the blood after vaccination correlated significantly with better survival119. The usage of CD4 + T helper cells, activated by DNA-demethylating agents, as antigen-presenting cells can also generate autologous cytotoxic T lymphocytes and NK cells120. Treatment of three of 25 adult patients with recurrent glioblastoma led to tumor regression for up to 27 months, with no treatment-related adverse events120.
Clinical trials with CAR NK-cell-based therapeutics are currently ongoing. The intracranial injection of NK-92/5.28.z CAR NK cells in patients with recurrent HER2-positive glioblastoma is being evaluated for safety and tolerability to determine the maximum tolerated dose for a phase 2 trial (NCT03383978). A trial evaluating CAR-NK cell immunotherapy in relapsed or refractory solid tumors, including malignant gliomas, is enrolling patients to receive a novel specific CAR NK cell targeting the MUC1 antigen (NCT02839954).
NK cell therapy for pediatric brain tumors
Brain tumors in the pediatric population harbor reduced NK cell-mediated immune surveillance and are linked to a less immunosuppressive tumor microenvironment compared to their adult counterparts. Furthermore, the phenotypic characteristics of the pediatric brain tumor microenvironment differ between molecular and histological tumor types121. This emphasizes the importance of a tailored therapeutic approach with immunotherapy against pediatric brain tumors, which should also consider the phenotype of the tumor microenvironment (inhibited immune surveillance) and its complex interaction with the bulk tumor (Table 4).
Medulloblastoma
Extensive genomic, epigenomic, and transcriptomic research efforts have led to the identification of four molecularly distinct subgroups in medulloblastoma. These are not only characterized by distinct biological features but also linked to different clinical outcomes. Various studies have further defined substantial intertumoral heterogeneities within each of the molecular subgroups122. Diverse immune-stromal patterns involving macrophages, T cell-associated mechanisms, and NK cell function, results in distinct mechanisms of immunosuppression in various molecular subgroups of medulloblastoma123,124. This reaffirms the need to better understand the medulloblastoma tumor microenvironment interplay, which could help in improving therapeutic outcomes of NK cell therapy targeting this neoplasm.
Earlier studies have shown some clinical efficacy and safety, using lymphokine-activated killer cells (LAK cells) that were administered intrathecally125. Medulloblastoma express specific ligands that trigger NK activating receptors (commonest being the NKG2D), and thus are susceptible to NK-mediated cytotoxicity. Major histocompatibility complex class I-related chain A (MICA), CD1d and UL16 binding protein 2 (ULBP-2) are cell surface ligands of medulloblastoma, that bind NK cell activating receptor NKG2D. These interactions favor developing NK cell-based therapeutics against these tumors62,126.
TGF-β is a potent mechanism for immune invasion and its pro-migratory potential induces metastasis in medulloblastoma127. Group 3 with c-MYC amplification is the most virulent and formidable subtype of medulloblastoma. TGFβ signaling is upregulated by the exopolyphosphatase PRUNE 1 inducing metastatic medulloblastoma128. Engineered cord blood-derived NK cells expressing a TGFβ dominant-negative receptor-2 (TGF-β DNRII), significantly undermined the inhibitory effects of the tumor microenvironment induced by TGFβ. This efficacy could now be pursued as a mode of targeted therapy in these tumors72. The recently completed first-in-human trial with intraventricular infusions of NK cells in pediatric malignant brain tumors, including medulloblastoma, has demonstrated safety and some transient responses12.
The immune checkpoint B7-H3 is expressed in nearly 95% of medulloblastoma129,130. This has motivated the development of novel B7-H3 directed immunotherapeutic methods for refractory or recurrent medulloblastoma. Recent preclinical data showed successful development of a tri-specific killer engager platform (TriKETM), assembled by DNA shuffling and ligation using DNA encoding a camelid anti-CD16 antibody fragment, a wild-type IL-15 moiety and an anti-B7-H3 scFV. These TriKE activated and targeted NK cells to inhibit the growth of various B7-H3-positive human cancer cells. This promising data provides an opportunity for this molecule to target B7-H3 in medulloblastoma131. A radioimmunotherapy Phase I/II trial of intracerebroventricular infusion of 177Lu-DTPA-omburtamab (antibody to B7-H3) was initiated in recurrent medulloblastoma. Preclinical data showed NK cell migration continued into tumors, as these infusions were given, suggesting an immune benefit (NCT04167618).
Pediatric high-grade glioma
Pediatric high-grade glioma (pHGG) comprise approximately 11% of all pediatric brain tumors132. Despite multimodal therapy; including surgery, systemic chemotherapy and irradiation; the 5-year overall survival for recurrent pHGG is dismal133. Lack of sustainable efficacious modalities has led to innovative research to improve these outcomes. The tumor microenvironment, particularly the immune microenvironment, is being examined for possible therapeutic implications.
Historically, NK cell function within pHGG has been limited by the immunosuppressive landscape of these tumors7. Factors such as PD-L1, B7-H3, IL-8, and TGFβ restrict the brain’s anti-tumor response, furthering the tumor’s aggressive nature134,135,136,137. TGFβ and IL-8 are readily expressed in pHGG, and directly reduce NK cell activating receptors; as well as antagonize IL-15, which increases NK cell proliferation, activation and decreases apoptosis138,139. In glioblastoma there is evidence suggesting that these tumors have increased expression of CD99, which is induced by TGFβ, inferring higher TGF-β in the tumor microenvironment68. Furthermore, pre-clinical data demonstrates that NK cells retrovirally transduced to express TGF-β-dominant-negative receptor II (DNRII) are efficacious in evading TGF-β mediated mechanisms of NK cell suppression, making them an attractive therapeutic modality140,141,142.
For NK cell therapy to be efficacious in these highly aggressive tumors, it is imperative to mitigate the cytotoxic activity TGF-β exerts on adoptive immunity143,144,145. A preclinical study showed that treatment of glioblastoma stem cell-engrafted mice, with allogeneic NK cells in combination with inhibitors of integrin or TGF-β signaling or with TGFBR2 gene-edited allogeneic NK cells, prevented GSC-induced NK cell dysfunction and tumor growth. This phenomenon should now be explored in clinical trials73.
Additional novel immune therapies, such as oncolytic viral therapy and CAR-NK therapy, are also being explored in these tumors. Oncolytic viral therapy induces tumor killing, both through direct cytotoxic selective viral replication in cancer cells as well as through immune recruitment. In fact, studies have demonstrated that immunosuppressive cytokine expression decreases oncolytic viral efficacy and overall survival, while immunostimulatory expression improves survival in pre-clinical glioma models146,147. Recently, Jennings et al examined the use of histone deacetylase (HDAC) inhibitor priming to overcome the immunoregulatory impact of these factors on oncolytic viral therapy. In this study the HDAC inhibitor, valproic acid, augmented the expression of activating NK ligands on human melanoma cells, and when co-cultured with a herpes simplex oncolytic virus, NK-mediated tumor lysis was increased148.
Based on its utility and efficacy in melanoma, oncolytic viral therapy is now being translated to early phase clinical trials in adults with high-grade glioma149, though it remains in infancy in the pediatric neuro-oncology world. A recently completed oncolytic HSV-1G207 immunotherapy for pediatric HGG has been completed in 12 patients. Radiographic or clinical responses was seen in 11 patient’s and there was no dose-limiting toxicity. G207 infusions converted this immunological cold formidable tumor to a hot phenotype. Furthermore, significant infiltration of lymphocytes including NK cells into the tumor were observed13.
Elimination of glioblastoma by NK cells, was markedly enhanced through expression of chimeric antigen receptor (CARs), targeting relevant antigens such as ErbB2 (HER2). These genetically engineered cells enhance efficacy, not only by directly killing tumor cells, but also by augmenting tumor killing through crosstalk with dendritic cells150. Encouraging pre-clinical data of CAR-NK cells in glioblastoma (as well as solid tumors) have led to two adult clinical trials (NCT04489420, NCT03383978)105,150,151. These trials however are limited to adult subjects, once again demonstrating the need for pediatric-specific clinical trials.
Challenges facing NK cell therapeutics in brain tumors
NK cell therapy in brain tumors is emerging as a promising tool, due to their superior safety profile, absence of graft-versus-host disease, cytokine storm and neurotoxicity compared to T cell-based therapies152. Nevertheless, NK cell therapies are hindered by some limitations. NK cells comprise 5–15% of circulating lymphocytes in adult humans153. The cost of treatment and the difficulties to meet optimal clinical-grade ex vivo expansion have posed challenges to the advancement of NK cell therapies7,154. CAR NK therapies use NK cells derived from peripheral or umbilical cord blood155; however, transfecting these NK cells with the CAR constructs is more difficult than T cells156,157. Gene editing NK cells using CRISPR Cas9 techniques can help overcome this problem158. The life span of NK cells with the CAR constructs may be shorter than that of CAR T cells159. Designing CAR NK cells that overexpress IL-15 and directed towards CD19 prolonged NK cell survival to at least 4 weeks in preclinical mouse models86. In human clinical trials, higher or multiple doses of NK cells could be needed to induce a meaningful therapeutic effect160. Tumor heterogeneity poses another limitation, as it becomes necessary to identify a universal tumor antigen/target for CAR NK cell constructs. Brain tumors, such as gliomas, can have decreased expression levels of proteins that inhibit NK cell activity16,37. The addition of CAR NK cells to the natural NK cells in circulation can lead to increased cytotoxicity140,161, and immune side effects. The BBB restricts the passage and trafficking of immune cells, such as NK cells, into the brain8. Intrinsic factors, such as transcription factors, and extrinsic factors, such as integrins, selectins, cytokines, and chemokines, in the cell control trafficking and homing of NK cells to the tumor microenvironment162. Immune cell infiltration varies in patients with brain tumors163,164,165; higher infiltration is associated with better survival in glioma37. Local delivery of NK cell therapeutics via intraventricular and/or intrathecal routes can aide with tumor coverage and NK cell distribution.
Imaging tools to understand homing of therapeutic NK cells
In parallel to the surge of NK cell therapeutics, recent years have seen a constant improvement of molecular imaging techniques, to evaluate the tracking of infused NK cells. These include quantitative dynamic footprinting (qDF) and total internal reflection fluorescence (TIRF) microscopy166,167,168, Förster resonance energy transfer (FRET) imaging169,170,171, optical live-cell imaging (including multiphoton and confocal imaging)172,173,174, light-sheet microscopy175,176, and super-resolution microscopy177,178,179. These techniques facilitate the visualization of immune responses at subcellular and molecular level in real time. This will facilitate profiling optimal and effective NK cell therapies180. Conventional cytological and histopathological methods usually rely on chemical fixation and are limited in the resolution of dynamic molecular and cellular processes. However, NK cell delivery, homing, and trafficking to and within the tumor are dynamic, time-dependent contributory factors. Monitoring of the ongoing differentiation of transferred NK cells as well as their persistence and activity in the tumor are of particular interest to facilitate therapy assessment and profile clinical optimization (Table 5).
Intense interrogation of the molecular interactions between natural killer cells and the target cells, have unfolded the functional heterogeneity and subclasses of NK cell behavior, as they migrate into the tumor cells. This dynamic data has allowed categorization of NK cells into 5 distinct classes as they migrate towards the target cells, following their infusion181,182. Thus, imaging technologies of NK cell-based brain tumor therapies are being investigated to understand the real-time spatiotemporal distribution and concentration of transferred NK cells to and within tumor sites.
Various methods have been developed in recent years for the noninvasive in vivo tracking of NK cells by molecular imaging183. These technologies can provide visualization and allow for characterization as well as quantification of biological, pathophysiological processes involved in NK cell-based treatments184. Future efforts should continue to improve NK cell molecular imaging strategies, to improve understanding of in the in vivo kinetics (trafficking, persistence) of NK cells to the tumor, potential therapeutic efficacy, off-target effects, and dynamics of the immunosuppressive microenvironment184. To date, these methods are restricted to preclinical models of brain tumors. The stage is now being set for the gradual translation of in vitro to in vivo settings in animals and humans to facilitate an in-depth exploration of NK cell behavior in the context of brain tumor biology and specific microenvironments185, with the most promising strategies being summarized in the following section of this review.
A growing body of noninvasive imaging modalities are currently being investigated to track immune cells, including optical imaging using fluorescence lifetime imaging (FLI), bioluminescent imaging (BLI), radionuclide imaging using single photon emission computed tomography (SPECT), positron emission tomography (PET) and magnetic resonance imaging (MRI)186. Many of these techniques rely on directly labeling the NK cell surface with fluorophores or loading of NK cells with contrast agents, radioisotopes, or cell-permeable fluorophores to allow for real-time visualization of NK cell-based immunotherapies187,188.
Direct or indirect labeling of cells are potential ways to track therapeutic NK cells via optical imaging. For example, NK cells can be directly labeled with exogenous fluorescent tracers for FLI. Fluorescent dyes commonly used in FLI include endogenous fluorophores, such as phenylalanine, NAD(P)H and flavin mononucleotide, organic dyes, fluorescent proteins, fullerenes, and quantum dots, all of which are applicable to permanently label NK cells through covalent and/or noncovalent binding to DNA, RNA, and proteins, reactions in the cytosol, and cell membrane insertion189,190. Indirect NK cell labeling is performed by transfection of reporter genes that induce formation of a protein product detectable via BLI (e.g., luciferase) or via FLI (e.g., green fluorescent protein)191,192. These NK cell labeling methods exhibit high sensitivity and specificity in small animal models, including mice and rats. Optical imaging has demonstrated a very high signal-to-noise ratio, the potential of providing multiplex imaging using various probes with diverse spectral characteristics and is accompanied with low instrumentation expenses193. Although appealing and widely used in the preclinical field, optical imaging is challenged by the small depth of tissue penetration (1 mm in FLI, 3 mm in BLI), restricted spatial resolution (2–3 mm) and poor anatomical discrimination, which limits its use to small animal studies and precludes its evaluation in larger animals and humans188.
The two nuclear imaging modalities used to track immune cells (PET and SPECT) are well integrated in the clinic and can be applied to visualize NK cell dynamics in animals and humans due to their capability of visualizing in vivo cell migration anywhere in living organisms and providing three-dimensional imaging data. PET and SPECT have the advantages of high sensitivity and unlimited depth penetration, excellent signal-to-background ratios, and a broad range of clinically applicable probes, many of which are FDA-approved194,195. From a technological perspective, these modalities are limited by its poor spatial resolution of approximately 1 to 5 mm, restricted anatomical information, and rapid decay of radionuclide tracers (e.g., 2 to 4 h for 18FDG), which limits the number of scanning sessions. Furthermore, these imaging modalities are associated with high cost and patients are exposed to ionizing radiation, which presents a concern196,197. Computed tomography can be used to improve PET and SPECT resolution, through attenuation correction and allows for the correlation of tracer signal with anatomical structures198.
MRI is well-established in the clinical practice and readily translatable from preclinical to clinical applications. This imaging modality offers high-resolution anatomical information good depth of penetration in tissue, well-defined soft tissue contrast and specificity, and lacks exposure to ionizing radiation. Constraints of MRI compared to optical imaging and radiotracer-based imaging modalities are the low sensitivity for molecular detection, including small NK cell populations, long scan times, and relatively high costs199. In vivo quantitation of NK cells is feasible using relaxation rate maps. However, this is an indirect quantification approach, as the detected MRI signal has to be computationally related to perturbations in tissue proton magnetization rather than directly to cell concentration188. 19F MRI is another MRI-based cell tracking technique that has received increasing attention in recent years. This technology builds upon the natural abundance of the tracer and offers high MRI sensitivity and background free images. 19F MRI, which is a promising quantitative molecular imaging approach due to the high sensitivity and low endogenous background signal of the 19F atom in vivo, has recently been approved by the FDA after several studies showed its potential for tracking NK cell-based immunotherapies200,201. 19F perfluorocarbon has been successfully used in preclinical models of medulloblastoma to demonstrate intracranial delivery of therapeutic NK cells. 19F MRI provided adequate imaging feedback of 19F-labeled NK cells, which distributed throughout the target area. Importantly, 19F-labeling did not undermine NK cell-cytotoxicity71 and suppressed medulloblastoma growth. A separate study supported these findings, demonstrating that NK cell labeling with the 19F isotope caused no detrimental effect on cellular function as indicated by maintained cytotoxicity of leukemia cells202.
Nanoparticles, such as superparamagnetic iron oxide and ultra-small superparamagnetic iron oxide nanoparticles, have also been used to label NK cells for MRI203,204. Loading NK cells with iron oxide nanoparticle is relatively easy and involves simple incubation, electroporation, or use of a transfection agent205. Iron oxide-labeled NK cells produce strong hypointense signals in T2 and T2* weighted images, and depending on how long the adoptively transferred NK cells survive, the iron oxide label can be retained for up to 4 days206. Several iron oxide nanoparticles are FDA-approved and in clinical use (ferumoxide, ferumoxytol, and ferucarbotran), underscoring the promising translation potential of paramagnetic particles-labeled NK cells. Nevertheless, the accurate interpretation of iron oxide nanoparticle-induced loss in MRI signal intensity remains a challenging task since other factors also contribute to hypointensity on T2 and T2* images, such as susceptibility artifacts due to air–tissue interfaces, magnetic field inhomogeneities due to poor shimming, presence of blood, and others.
Although each of the discussed imaging modalities individually is saddled with limitations, which impede their development into a gold standard for quantitatively monitoring in vivo distribution and timing of NK cell trafficking, multimodal hybrid imaging of two or more imaging technologies holds promise for future clinical applications. Specifically, MRI and PET, or optical imaging and MRI, offers the most desirable characteristics for tracking NK cells qualitatively and quantitatively, measuring cellular viability through visualization of signal colocalization, and assessing therapeutic response to NK cell adoptive therapy. The last few years have yielded promising results in the expansion, labeling, and in vivo tracking of NK cells199. Future research efforts should focus on refined, optimal, and robust imaging modalities to precisely quantify the homing, bio-distribution, and persistence of adoptively transferred NK cells in humans with an intent to improve clinical outcome188.
Conclusion and future perspectives
Despite technological advances in the multimodal treatments for patients with brain tumors, significant relapses and treatment-led toxicities continue to undermine therapeutic efficacy. The last decade has witnessed a surge in cellular therapy for these formidable neoplasms. As an alternative to T cell therapy, use of NK and CAR-NK cell therapeutics are gaining momentum in solid tumors, due to the absence of graft-versus-host disease, potential to generate “off-the-shelf” products with NK cell lines, iPSC-NK or expanded NK cells from blood, umbilical cord blood or other hematopoietic progenitors. The immediate availability of product is critically important in aggressive CNS tumors where therapy cannot wait for production delays. Continuing efforts should also focus on strategies using loco-regional infusions of NK cells (intra-tumoral and intraventricular), enhancing its tumor delivery by overcoming blood–brain barrier, minimizing risk of rejection, and undermining the suboptimal systemic delivery of cellular therapy into these CNS tumors. The future should also see significant advances in labeling and in vivo tracking of NK cells to tumor sites in real time, to optimize clinical efficacy of NK cell-based therapeutics in neuro-oncology.
References
Tallman, M. M., Zalenski, A. A. & Venere, M. In Gliomas (ed. Debinski, W.) (2021).
Dolecek, T. A., Propp, J. M., Stroup, N. E. & Kruchko, C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro Oncol. 14(Suppl 5), v1–v49 (2012).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Mathew, R. K. & Rutka, J. T. Diffuse intrinsic pontine glioma: clinical features, molecular genetics, and novel targeted therapeutics. J. Korean Neurosurg. Soc. 61, 343–351 (2018).
Kmiecik, J., Zimmer, J. & Chekenya, M. Natural killer cells in intracranial neoplasms: presence and therapeutic efficacy against brain tumours. J. Neurooncol. 116, 1–9 (2014).
Fares, J., Fares, M. Y. & Fares, Y. Immune checkpoint inhibitors: Advances and impact in neuro-oncology. Surg. Neurol. Int. 10, 9 (2019).
Fares, J., Fares, M. Y. & Fares, Y. Natural killer cells in the brain tumor microenvironment: defining a new era in neuro-oncology. Surgical Neurol. Int. 10, 43 (2019).
Fares, J., Kanojia, D., Rashidi, A., Ulasov, I. & Lesniak, M. S. Genes that mediate metastasis across the blood-brain barrier. Trends cancer 6, 660–676 (2020).
Sonabend, A. M., Rolle, C. E. & Lesniak, M. S. The role of regulatory T cells in malignant glioma. Anticancer Res. 28, 1143–1150 (2008).
Yang, I., Han, S. J., Kaur, G., Crane, C. & Parsa, A. T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 17, 6–10 (2010).
Dunn, G. P., Dunn, I. F. & Curry, W. T. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human glioma. Cancer Immun. 7, 12 (2007).
Khatua, S. et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro Oncol. 22, 1214–1225 (2020).
Friedman, G. K. et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 384, 1613–1622 (2021).
Smyth, M. J., Hayakawa, Y., Takeda, K. & Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2, 850–861 (2002).
Hsia, J. Y. et al. Prognostic significance of intratumoral natural killer cells in primary resected esophageal squamous cell carcinoma. Chang Gung Med. J. 28, 335–340 (2005).
Castriconi, R. et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 182, 3530–3539 (2009).
Avril, T. et al. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain Pathol. 22, 159–174 (2012).
Poli, A. et al. Targeting glioblastoma with NK cells and mAb against NG2/CSPG4 prolongs animal survival. Oncotarget 4, 1527–1546 (2013).
Blaylock, R. L. Cancer microenvironment, inflammation and cancer stem cells: A hypothesis for a paradigm change and new targets in cancer control. Surg. Neurol. Int. 6, 92 (2015).
Alizadeh, D. et al. Induction of anti-glioma natural killer cell response following multiple low-dose intracerebral CpG therapy. Clin. Cancer Res. 16, 3399–3408 (2010).
Friese, M. A. et al. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 64, 7596–7603 (2004).
Campbell, K. S. & Hasegawa, J. Natural killer cell biology: an update and future directions. J. Allergy Clin. Immunol. 132, 536–544 (2013).
Cho, D. & Campana, D. Expansion and activation of natural killer cells for cancer immunotherapy. Korean J. Lab Med. 29, 89–96 (2009).
Luetke-Eversloh, M., Killig, M. & Romagnani, C. Signatures of human NK cell development and terminal differentiation. Front. Immunol. 4, 499 (2013).
Caligiuri, M. A. Human natural killer cells. Blood 112, 461–469 (2008).
Moretta, A., Locatelli, F. & Moretta, L. Human NK cells: from HLA class I-specific killer Ig-like receptors to the therapy of acute leukemias. Immunol. Rev. 224, 58–69 (2008).
Vivier, E. & Ugolini, S. Natural killer cells: from basic research to treatments. Front. Immunol. 2, 18 (2011).
Golan, I., Rodriguez de la Fuente, L. & Costoya, J. A. NK Cell-Based Glioblastoma Immunotherapy. Cancers 10, 522 (2018).
Domingues, P. H. et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am. J. Pathol. 181, 1749–1761 (2012).
Yang, I., Han, S. J., Sughrue, M. E., Tihan, T. & Parsa, A. T. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J. Neurosurg. 115, 505–511 (2011).
Stevens, A., Kloter, I. & Roggendorf, W. Inflammatory infiltrates and natural killer cell presence in human brain tumors. Cancer 61, 738–743 (1988).
Zhang, L., Yu, H., Xue, Y. & Liu, Y. Decreased natural killer cells in diffuse intrinsic pontine glioma patients. Childs Nerv. Syst. 36, 1345–1346 (2020).
Fu, W. et al. Single-cell atlas reveals complexity of the immunosuppressive microenvironment of initial and recurrent glioblastoma. Front. Immunol. 11, 835 (2020).
Kozlowska, A. K. et al. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-gamma after differentiation of glioblastoma by human natural killer cells. Cancer Immunol. Immunother. 65, 1085–1097 (2016).
Bottcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037.e1014 (2018).
Fadul, C. E. et al. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 13, 393–400 (2011).
Kmiecik, J. et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 264, 71–83 (2013).
Mittelbronn, M. et al. Elevated HLA-E levels in human glioblastomas but not in grade I to III astrocytomas correlate with infiltrating CD8+ cells. J. Neuroimmunol. 189, 50–58 (2007).
Jachimowicz, R. D. et al. Induction of in vitro and in vivo NK cell cytotoxicity using high-avidity immunoligands targeting prostate-specific membrane antigen in prostate carcinoma. Mol. Cancer Ther. 10, 1036–1045 (2011).
Fluh, C., Mafael, V., Adamski, V., Synowitz, M. & Held-Feindt, J. Dormancy and NKG2D system in brain metastases: Analysis of immunogenicity. Int. J. Mol. Med. 45, 298–314 (2020).
Ghiringhelli, F. et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 202, 1075–1085 (2005).
Wahl, S. M., Wen, J. & Moutsopoulos, N. M. The kiss of death: interrupted by NK-cell close encounters of another kind. Trends Immunol. 27, 161–164 (2006).
Vigano, S. et al. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol. 10, 925 (2019).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Frumento, G. et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 196, 459–468 (2002).
Melaiu, O., Lucarini, V., Cifaldi, L. & Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front Immunol. 10, 3038 (2019).
Roman, F., Pascaud, X., Vauche, D. & Junien, J. L. Evidence for a non-opioid sigma binding site in the guinea-pig myenteric plexus. Life Sci. 42, 2217–2222 (1988).
Dysthe, M. & Parihar, R. Myeloid-derived suppressor cells in the tumor microenvironment. Adv. Exp. Med. Biol. 1224, 117–140 (2020).
Kimpo, M. S., Oh, B. & Lee, S. The role of natural killer cells as a platform for immunotherapy in pediatric cancers. Curr. Oncol. Rep. 21, https://doi.org/10.1007/s11912-019-0837-8 (2019).
Zhu, L. et al. Enhancement of antitumor potency of extracellular vesicles derived from natural killer cells by IL-15 priming. Biomaterials 190-191, 38–50 (2019).
Sharifzad, F. et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int. J. Mol. Sci. 21, 2263 (2020).
Cheray, M. et al. KLRC3, a Natural Killer receptor gene, is a key factor involved in glioblastoma tumourigenesis and aggressiveness. J. Cell Mol. Med. 21, 244–253 (2017).
Morimoto, T. et al. CRISPR-Cas9-mediated TIM3 knockout in human natural killer cells enhances growth inhibitory effects on human glioma cells. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22073489 (2021).
Wolf, Y., Anderson, A. C. & Kuchroo, V. K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 20, 173–185 (2020).
Wang, J. & Matosevic, S. NT5E/CD73 as correlative factor of patient survival and natural killer cell infiltration in glioblastoma. J. Clin. Med. 8, 1526 (2019).
Lupo, K. B. & Matosevic, S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J. Hematol. Oncol. 13, 76 (2020).
Pellegatta, S. et al. ABCC3 expressed by CD56dim CD16+ NK cells predicts response in glioblastoma patients treated with combined chemotherapy and dendritic cell immunotherapy. Int. J. Mol. Sci. 20, 5886 (2019).
Lo Nigro, C. et al. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann. Transl. Med. 7, 105 (2019).
Hara, A. et al. CD1d expression in glioblastoma is a promising target for NKT cell-based cancer immunotherapy. Cancer Immunol. Immunother. 70, 1239–1254 (2021).
Parodi, M. et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front Immunol. 9, 2358 (2018).
Lim, S. C., Geleta, B., Maleki, S., Richardson, D. R. & Kovacevic, Z. The metastasis suppressor NDRG1 directly regulates androgen receptor signaling in prostate cancer. J. Biol. Chem. 297, 101414 (2021).
Perez-Martinez, A., Fernandez, L. & Diaz, M. A. The therapeutic potential of natural killer cells to target medulloblastoma. Expert Rev. Anticancer Ther. 16, 573–576 (2016).
Fernandez, L. et al. In vitro Natural Killer Cell Immunotherapy for Medulloblastoma. Front. Oncol. 3, 94 (2013).
Shida, Y. et al. Ex vivo expanded and activated natural killer cells prolong the overall survival of mice with glioblastoma-like cell-derived tumors. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22189975 (2021).
Chu, Y. et al. Combinatorial immunotherapy of N-803 (IL-15 superagonist) and dinutuximab with ex vivo expanded natural killer cells significantly enhances in vitro cytotoxicity against GD2(+) pediatric solid tumors and in vivo survival of xenografted immunodeficient NSG mice. J. Immunother. Cancer 9, https://doi.org/10.1136/jitc-2020-002267 (2021).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 128, 4654–4668 (2018).
Huang, B. Y. et al. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS ONE 10, e0134715 (2015).
Close, H. J. et al. Expression profiling of single cells and patient cohorts identifies multiple immunosuppressive pathways and an altered NK cell phenotype in glioblastoma. Clin. Exp. Immunol. 200, 33–44 (2020).
Sun, Y., Sedgwick, A. J., Palarasah, Y., Mangiola, S. & Barrow, A. D. A transcriptional signature of PDGF-DD activated natural killer cells predicts more favorable prognosis in low-grade glioma. Front Immunol. 12, 668391 (2021).
Biber, G. et al. Modulation of intrinsic inhibitory checkpoints using nano-carriers to unleash NK cell activity. EMBO Mol. Med. 14, e14073 (2022).
Kennis, B. A. et al. Monitoring of intracerebellarly-administered natural killer cells with fluorine-19 MRI. J. Neurooncol. 142, 395–407 (2019).
Powell, A. B. et al. Medulloblastoma rendered susceptible to NK-cell attack by TGFbeta neutralization. J. Transl. Med. 17, 321 (2019).
Shaim, H. et al. Targeting the alpha v integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Invest. 131, https://doi.org/10.1172/JCI142116 (2021).
Tanaka, Y. et al. Ex vivo-expanded highly purified natural killer cells in combination with temozolomide induce antitumor effects in human glioblastoma cells in vitro. PLoS ONE 14, e0212455 (2019).
Lee, Y. J. & Kim, J. Resveratrol activates natural killer cells through Akt- and mTORC2-mediated c-Myb upregulation. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21249575 (2020).
Lee, Y., Shin, H. & Kim, J. In vivo anti-cancer effects of resveratrol mediated by NK cell activation. J. Innate Immun. 13, 94–106 (2021).
Sugii, N. et al. Hemagglutinating virus of Japan-envelope containing programmed cell death-ligand 1 siRNA inhibits immunosuppressive activities and elicits antitumor immune responses in glioma. Cancer Sci. 112, 81–90 (2021).
Xu, B. et al. An oncolytic herpesvirus expressing E-cadherin improves survival in mouse models of glioblastoma. Nat. Biotechnol. https://doi.org/10.1038/nbt.4302 (2018).
Xu, B. et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat. Commun. 12, 5908 (2021).
He, B., Gross, M. & Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl Acad. Sci. USA 94, 843–848 (1997).
Goldstein, D. J. & Weller, S. K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 62, 196–205 (1988).
Suryadevara, C. M., Riccione, K. A. & Sampson, J. H. Immunotherapy gone viral: bortezomib and oHSV enhance antitumor NK-cell activity. Clin. Cancer Res. 22, 5164–5166 (2016).
Yoo, J. Y. et al. Bortezomib treatment sensitizes oncolytic HSV-1-treated tumors to NK cell immunotherapy. Clin. Cancer Res. 22, 5265–5276 (2016).
Gras Navarro, A. et al. Pretreatment of glioblastoma with bortezomib potentiates natural killer cell cytotoxicity through TRAIL/DR5 mediated apoptosis and prolongs animal survival. Cancers 11, 996 (2019).
Han, J. et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 5, 11483 (2015).
Liu, E. et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 32, 520–531 (2018).
Gong, Y., Klein Wolterink, R. G. J., Wang, J., Bos, G. M. J. & Germeraad, W. T. V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 14, 73 (2021).
Simonetta, F., Alvarez, M. & Negrin, R. S. Natural killer cells in graft-versus-host-disease after allogeneic hematopoietic cell transplantation. Front Immunol. 8, 465 (2017).
Goff, S. L. et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 42, 126–135 (2019).
Ciurea, S. O. et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 130, 1857–1868 (2017).
Mitwasi, N. et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 10, 2141 (2020).
Romanski, A. et al. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell Mol. Med. 20, 1287–1294 (2016).
Ao, X. et al. Anti-alphaFR CAR-engineered NK-92 cells display potent cytotoxicity against alphafr-positive ovarian cancer. J. Immunother. 42, 284–296 (2019).
Li, Y., Hermanson, D. L., Moriarity, B. S. & Kaufman, D. S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 23, 181–192.e185 (2018).
Tang, S. Y. et al. Targeted integration of EpCAM-specific CAR in human induced pluripotent stem cells and their differentiation into NK cells. Stem Cell Res. Ther. 12, 580 (2021).
Tang, X. et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 8, 1083–1089 (2018).
Arias, J., Yu, J., Varshney, M., Inzunza, J. & Nalvarte, I. Hematopoietic stem cell- and induced pluripotent stem cell-derived CAR-NK cells as reliable cell-based therapy solutions. Stem Cells Transl. Med. 10, 987–995 (2021).
Davis, Z. B., Felices, M., Verneris, M. R. & Miller, J. S. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J. 21, 486–491 (2015).
Kang, C. H., Kim, Y., Lee, S. M., Choi, S. U. & Park, C. H. Development of antigen-specific chimeric antigen receptor KHYG-1 cells for glioblastoma. Anticancer Res. 41, 1811–1819 (2021).
Ronellenfitsch, M. W., Luger, A. L. & Steinbach, J. P. EGFR and mTOR as therapeutic targets in glioblastoma. Oncotarget 10, 4721–4723 (2019).
Bigner, S. H. et al. Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 50, 8017–8022 (1990).
Salomon, D. S., Brandt, R., Ciardiello, F. & Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 19, 183–232 (1995).
Schlegel, J. et al. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J. Neurooncol. 22, 201–207 (1994).
Genssler, S. et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 5, e1119354 (2016).
Zhang, C. et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl Cancer Inst. 108, https://doi.org/10.1093/jnci/djv375 (2016).
Chen, X. et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 7, 27764–27777 (2016).
Ma, R. et al. An oncolytic virus expressing IL15/IL15Ralpha combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 81, 3635–3648 (2021).
Jan, C. I. et al. Targeting human leukocyte antigen G with chimeric antigen receptors of natural killer cells convert immunosuppression to ablate solid tumors. J. Immunother. Cancer 9, https://doi.org/10.1136/jitc-2021-003050 (2021).
Wang, J. et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc. Natl Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2107507118 (2021).
Mostafa, H. et al. Immune phenotypes predict survival in patients with glioblastoma multiforme. J. Hematol. Oncol. 9, 77 (2016).
Lu, J. et al. Identification of 3 subpopulations of tumor-infiltrating immune cells for malignant transformation of low-grade glioma. Cancer Cell Int. 19, 265 (2019).
Ishikawa, E. et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 24, 1861–1871 (2004).
Lim, J. et al. Autologous adoptive immune-cell therapy elicited a durable response with enhanced immune reaction signatures in patients with recurrent glioblastoma: An open label, phase I/IIa trial. PLoS ONE 16, e0247293 (2021).
Thakar, M. S. et al. Phase II trial using haploidentical hematopoietic cell transplantation (HCT) followed by donor natural killer (NK) cell infusion and sirolimus maintenance for patients with high-risk solid tumors. J. Clin. Oncol. 38, e23551–e23551 (2020).
Andre, P. et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743 (2018).
Yeom, S. Y., Nam, D. H. & Park, C. RRAD promotes EGFR-mediated STAT3 activation and induces temozolomide resistance of malignant glioblastoma. Mol. Cancer Ther. 13, 3049–3061 (2014).
Zadeh, G., Bhat, K. P. & Aldape, K. EGFR and EGFRvIII in glioblastoma: partners in crime. Cancer Cell 24, 403–404 (2013).
Buchroithner, J. et al. Audencel immunotherapy based on dendritic cells has no effect on overall and progression-free survival in newly diagnosed glioblastoma: a phase II randomized trial. Cancers 10, 372 (2018).
Erhart, F. et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropathol. Commun. 6, 135 (2018).
Kirkin, A. F. et al. Adoptive cancer immunotherapy using DNA-demethylated T helper cells as antigen-presenting cells. Nat. Commun. 9, 785 (2018).
Griesinger, A. M. et al. Characterization of distinct immunophenotypes across pediatric brain tumor types. J. Immunol. 191, 4880–4888 (2013).
Cavalli, F. M. G. et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31, 737–754 (2017).
Bockmayr, M. et al. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 7, https://doi.org/10.1080/2162402X.2018.1462430 (2018).
Margol, A. S. et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin. Cancer Res. 21, 1457–1465 (2015).
Shimizu, K. et al. Adoptive immunotherapy in patients with medulloblastoma by LAK cells. No Shinkei 41, 991–995 (1989).
Kabir, T. F., Kunos, C. A., Villano, J. L. & Chauhan, A. Immunotherapy for medulloblastoma: current perspectives. Immunotargets Ther. 9, 57–77 (2020).
Gate, D. et al. T-cell TGF-beta signaling abrogation restricts medulloblastoma progression. Proc. Natl Acad. Sci. USA 111, E3458–E3466 (2014).
Ferrucci, V. et al. Metastatic group 3 medulloblastoma is driven by PRUNE1 targeting NME1-TGF-beta-OTX2-SNAIL via PTEN inhibition. Brain 141, 1300–1319 (2018).
Li, S. et al. Pediatric medulloblastoma express immune checkpoint B7-H3. Clin. Transl. Oncol. https://doi.org/10.1007/s12094-021-02762-y (2022).
Castriconi, R. et al. Both CD133(+) and CD133(-) medulloblastoma cell lines express ligands for triggering NK receptors and are susceptible to NK-mediated cytotoxicity. Eur. J. Immunol. 37, 3190–3196 (2007).
Vallera, D. A. et al. NK-cell-mediated targeting of various solid tumors using a B7-H3 tri-specific killer engager in vitro and in vivo. Cancers 12, https://doi.org/10.3390/cancers12092659 (2020).
Ostrom, Q. T. et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncol. 16, x1–x36 (2015).
El-Ayadi, M. et al. High-grade glioma in very young children: a rare and particular patient population. Oncotarget 8, 64564–64578 (2017).
Price, G., Bouras, A., Hambardzumyan, D. & Hadjipanayis, C. G. Current knowledge on the immune microenvironment and emerging immunotherapies in diffuse midline glioma. EBioMedicine 69, 103453 (2021).
Griesinger, A. M., Donson, A. M. & Foreman, N. K. Immunotherapeutic implications of the immunophenotype of pediatric brain tumors. Oncoimmunology 3, e27256 (2014).
Johnson, A. et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist 22, 1478–1490 (2017).
Taurone, S. et al. Brain gliomas and growth factors: immunohistochemical, immunofluorescence, flow cytometry and RT-PCR profile in pediatric age. J. Biol. Regul. Homeost. Agents 33, 1451–1463 (2019).
Ross, J. L. et al. Platelet-derived growth factor beta is a potent inflammatory driver in paediatric high-grade glioma. Brain 144, 53–69 (2021).
Mao, Y. et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 128, 1475–1489 (2016).
Gras Navarro, A. et al. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J. Immunol. 193, 6192–6206 (2014).
Giotta Lucifero, A. & Luzzi, S. Against the resilience of high-grade gliomas: the immunotherapeutic approach (Part I). Brain Sci. 11, https://doi.org/10.3390/brainsci11030386 (2021).
Yvon, E. S. et al. Cord blood natural killer cells expressing a dominant negative TGF-β receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 19, 408–418 (2017).
Wilson, E. B. et al. Human tumour immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS ONE 6, e22842 (2011).
Haberthur, K. et al. NKG2D ligand expression in pediatric brain tumors. Cancer Biol. Ther. 17, 1253–1265 (2016).
Viel, S. et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 9, ra19 (2016).
Pearl, T. M., Markert, J. M., Cassady, K. A. & Ghonime, M. G. Oncolytic virus-based cytokine expression to improve immune activity in brain and solid tumors. Mol. Ther. Oncolytics 13, 14–21 (2019).
Andreansky, S. et al. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 5, 121–130 (1998).
Jennings, V. A. et al. Potentiating oncolytic virus-induced immune-mediated tumor cell killing using histone deacetylase inhibition. Mol. Ther. 27, 1139–1152 (2019).
Bernstock, J. D. et al. The current landscape of oncolytic herpes simplex viruses as novel therapies for brain malignancies. Viruses 13, https://doi.org/10.3390/v13061158 (2021).
Burger, M. C. et al. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front. Immunol. 10, 2683 (2019).
Schönfeld, K. et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 23, 330–338 (2015).
Balatsoukas, A., Rossignoli, F. & Shah, K. NK cells in the brain: implications for brain tumor development and therapy. Trends Mol. Med. 28, 194–209 (2022).
Angelo, L. S. et al. Practical NK cell phenotyping and variability in healthy adults. Immunol. Res. 62, 341–356 (2015).
Baggio, L., Laureano, A. M., Silla, L. M. D. R. & Lee, D. A. Natural killer cell adoptive immunotherapy: Coming of age. Clin. Immunol. 177, 3–11 (2017).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Lin, C. Y., Gobius, I. & Souza-Fonseca-Guimaraes, F. Natural killer cell engineering—a new hope for cancer immunotherapy. Semin. Hematol. 57, 194–200 (2020).
Rezvani, K., Rouce, R., Liu, E. & Shpall, E. Engineering natural killer cells for cancer immunotherapy. Mol. Ther. 25, 1769–1781 (2017).
Rautela, J., Surgenor, E. & Huntington, N. D. Drug target validation in primary human natural killer cells using CRISPR RNP. J. Leukoc. Biol. 108, 1397–1408 (2020).
Zhang, Y. et al. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology 121, 258–265 (2007).
Arnesen, V. S., Navarro, A. G. & Chekenya, M. Challenges and prospects for designer T and NK cells in glioblastoma immunotherapy. Cancers 13, https://doi.org/10.3390/cancers13194986 (2021).
Haspels, H. N., Rahman, M. A., Joseph, J. V., Navarro, A. G. & Chekenya, M. Glioblastoma stem-like cells are more susceptible than differentiated cells to natural killer cell lysis mediated through killer immunoglobulin-like receptors-human leukocyte antigen ligand mismatch and activation receptor-ligand interactions. Front. Immunol. 9, https://doi.org/10.3389/fimmu.2018.01345 (2018).
Ran, G. H. et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct. Target Ther. 7, 205 (2022).
Waziri, A. et al. Preferential in situ CD4(+)CD56(+) T cell activation and expansion within human glioblastoma. J. Immunol. 180, 7673–7680 (2008).
Rossi, M. L., Hughes, J. T., Esiri, M. M., Coakham, H. B. & Brownell, D. B. Immunohistological study of mononuclear cell infiltrate in malignant gliomas. Acta Neuropathol. 74, 269–277 (1987).
Kim, Y. H. et al. Tumour-infiltrating T-cell subpopulations in glioblastomas. Br. J. Neurosurg. 26, 21–27 (2012).
Fan, Z. et al. Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nat. Commun. 7, 12658 (2016).
Jung, Y. et al. Three-dimensional localization of T-cell receptors in relation to microvilli using a combination of superresolution microscopies. Proc. Natl Acad. Sci. USA 113, E5916–e5924 (2016).
Sundd, P. et al. Quantitative dynamic footprinting microscopy reveals mechanisms of neutrophil rolling. Nat. Methods 7, 821–824 (2010).
Ma, Y. et al. An intermolecular FRET sensor detects the dynamics of T cell receptor clustering. Nat. Commun. 8, 15100 (2017).
Radbruch, H. et al. Intravital FRET: probing cellular and tissue function in vivo. Int. J. Mol. Sci. 16, 11713–11727 (2015).
Zhang, J., Campbell, R. E., Ting, A. Y. & Tsien, R. Y. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 3, 906–918 (2002).
Moseman, E. A., Wu, T., de la Torre, J. C., Schwartzberg, P. L. & McGavern, D. B. Type I interferon suppresses virus-specific B cell responses by modulating CD8(+) T cell differentiation. Sci. Immunol. 1, https://doi.org/10.1126/sciimmunol.aah3565 (2016).
Thestrup, T. et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods 11, 175–182 (2014).
Pittet, M. J., Garris, C. S., Arlauckas, S. P. & Weissleder, R. Recording the wild lives of immune cells. Sci. Immunol. 3 https://doi.org/10.1126/sciimmunol.aaq0491 (2018).
Liu, T. L. et al. Observing the cell in its native state: imaging subcellular dynamics in multicellular organisms. Science 360 https://doi.org/10.1126/science.aaq1392 (2018).
O’Shaughnessy, E. C. et al. Software for lattice light-sheet imaging of FRET biosensors, illustrated with a new Rap1 biosensor. J. Cell Biol. 218, 3153–3160 (2019).
Chen, Y. et al. Multi-color live-cell super-resolution volume imaging with multi-angle interference microscopy. Nat. Commun. 9, 4818 (2018).
Fan, Z. et al. High-affinity bent β(2)-integrin molecules in arresting neutrophils face each other through binding to ICAMs in cis. Cell Rep. 26, 119–130.e115 (2019).
Margraf, A., Ley, K. & Zarbock, A. Neutrophil recruitment: from model systems to tissue-specific patterns. Trends Immunol. 40, 613–634 (2019).
Oh, S., Lee, J. H., Kwack, K. & Choi, S. W. Natural killer cell therapy: a new treatment paradigm for solid tumors. Cancers 11 https://doi.org/10.3390/cancers11101534 (2019).
Vitale, M., Cantoni, C., Pietra, G., Mingari, M. C. & Moretta, L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 44, 1582–1592 (2014).
Vanherberghen, B. et al. Classification of human natural killer cells based on migration behavior and cytotoxic response. Blood 121, 1326–1334 (2013).
Shapovalova, M., Pyper, S. R., Moriarity, B. S. & LeBeau, A. M. The molecular imaging of natural killer cells. Mol. Imaging 17, 1536012118794816 (2018).
Youn, H. & Hong, K. J. In vivo noninvasive small animal molecular imaging. Osong Public Health Res. Perspect. 3, 48–59 (2012).
Lee, H. W., Gangadaran, P., Kalimuthu, S. & Ahn, B. C. Advances in molecular imaging strategies for in vivo tracking of immune cells. Biomed. Res. Int. 2016, 1946585 (2016).
Galli, F. et al. In vivo imaging of natural killer cell trafficking in tumors. J. Nucl. Med. 56, 1575–1580 (2015).
Gangadaran, P. & Ahn, B. C. Molecular imaging: a useful tool for the development of natural killer cell-based immunotherapies. Front. Immunol. 8, 1090 (2017).
Sta Maria, N. S., Barnes, S. R. & Jacobs, R. E. In vivo monitoring of natural killer cell trafficking during tumor immunotherapy. Magn. Reson Insights 7, 15–21 (2014).
Parish, C. R. Fluorescent dyes for lymphocyte migration and proliferation studies. Immunol. Cell Biol. 77, 499–508 (1999).
Berezin, M. Y. & Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 110, 2641–2684 (2010).
Tavri, S. et al. Optical imaging of cellular immunotherapy against prostate cancer. Mol. Imaging 8, 15–26 (2009).
Edinger, M. et al. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood 101, 640–648 (2003).
Massoud, T. F. & Gambhir, S. S. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 17, 545–580 (2003).
Jha, P. et al. Monitoring of natural killer cell immunotherapy using noninvasive imaging modalities. Cancer Res. 70, 6109–6113 (2010).
Neves, A. A. & Brindle, K. M. Assessing responses to cancer therapy using molecular imaging. Biochim. Biophys. Acta 1766, 242–261 (2006).
Meier, R. et al. Tracking of [18F]FDG-labeled natural killer cells to HER2/neu-positive tumors. Nucl. Med. Biol. 35, 579–588 (2008).
Meller, B. et al. Monitoring of a new approach of immunotherapy with allogenic (111)In-labelled NK cells in patients with renal cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging 31, 403–407 (2004).
Rahmim, A. & Zaidi, H. PET versus SPECT: strengths, limitations and challenges. Nucl. Med. Commun. 29, 193–207 (2008).
Galli, F., Histed, S. & Aras, O. NK cell imaging by in vitro and in vivo labelling approaches. Q J. Nucl. Med. Mol. Imaging 58, 276–283 (2014).
Ahrens, E. T. & Bulte, J. W. Tracking immune cells in vivo using magnetic resonance imaging. Nat. Rev. Immunol. 13, 755–763 (2013).
Somanchi, S. S., Kennis, B. A., Gopalakrishnan, V., Lee, D. A. & Bankson, J. A. In vivo (19)F-magnetic resonance imaging of adoptively transferred NK cells. Methods Mol. Biol. 1441, 317–332 (2016).
Bouchlaka, M. N. et al. (19)F-MRI for monitoring human NK cells in vivo. Oncoimmunology 5, e1143996 (2016).
Meier, R. et al. Depicting adoptive immunotherapy for prostate cancer in an animal model with magnetic resonance imaging. Magn. Reson Med. 65, 756–763 (2011).
Sheu, A. Y., Zhang, Z., Omary, R. A. & Larson, A. C. MRI-monitored transcatheter intra-arterial delivery of SPIO-labeled natural killer cells to hepatocellular carcinoma: preclinical studies in a rodent model. Invest Radio. 48, 492–499 (2013).
Daldrup-Link, H. E. et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur. Radio. 15, 4–13 (2005).
Mallett, C. L., McFadden, C., Chen, Y. & Foster, P. J. Migration of iron-labeled KHYG-1 natural killer cells to subcutaneous tumors in nude mice, as detected by magnetic resonance imaging. Cytotherapy 14, 743–751 (2012).
Sharifzad, F. et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21072263 (2020).
Ciaglia, E. et al. Recognition by natural killer cells of N6-isopentenyladenosine-treated human glioma cell lines. Int. J. Cancer 142, 176–190 (2018).
Yoo, J. Y. et al. Bortezomib treatment sensitizes oncolytic HSV-1-treated tumors to NK cell immunotherapy. Clin. Cancer Res. 22, 5265–5276 (2016).
Alkins, R., Burgess, A., Kerbel, R., Wels, W. S. & Hynynen, K. Early treatment of HER2-amplified brain tumors with targeted NK-92 cells and focused ultrasound improves survival. Neuro. Oncol. 18, 974–981 (2016).
Esser, R. et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell Mol. Med. 16, 569–581 (2012).
Muller, N. et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1alpha-secreting glioblastoma. J. Immunother. 38, 197–210 (2015).
Murakami, T. et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 38, 5049–5056 (2018).
Nakazawa, T. et al. KHYG-1 cells with EGFRvIII-specific CAR induced a pseudoprogression-like feature in subcutaneous tumours derived from glioblastoma-like cells. Anticancer Res. 40, 3231–3237 (2020).
Seidel, D. et al. Disialoganglioside-specific human natural killer cells are effective against drug-resistant neuroblastoma. Cancer Immunol. Immunother. 64, 621–634 (2015).
Altvater, B. et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 15, 4857–4866 (2009).
Trapani, J. A. & Smyth, M. J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2, 735–747 (2002).
Kim, H. W. et al. Enhancement of natural killer cell cytotoxicity by sodium/iodide symporter gene-mediated radioiodine pretreatment in breast cancer cells. PLoS ONE 8, e70194 (2013).
Chin, B. B. et al. 111In oxine labelled mesenchymal stem cell SPECT after intravenous administration in myocardial infarction. Nucl. Med. Commun. 24, 1149–1154 (2003).
Lichtenfels, R., Biddison, W. E., Schulz, H., Vogt, A. B. & Martin, R. CARE-LASS (calcein-release-assay), an improved fluorescence-based test system to measure cytotoxic T lymphocyte activity. J. Immunol. Methods 172, 227–239 (1994).
Somanchi, S. S., McCulley, K. J., Somanchi, A., Chan, L. L. & Lee, D. A. A novel method for assessment of natural killer cell cytotoxicity using image cytometry. PLoS ONE 10, e0141074 (2015).
Ferreira-Teixeira, M. et al. Natural killer cell-based adoptive immunotherapy eradicates and drives differentiation of chemoresistant bladder cancer stem-like cells. BMC Med. 14, 163 (2016).
Chandrasekaran, S., Chan, M. F., Li, J. & King, M. R. Super natural killer cells that target metastases in the tumor draining lymph nodes. Biomaterials 77, 66–76 (2016).
Acknowledgements
No individuals other than the listed co-authors contributed to this publication. This research received no funding support. Figures are original and were created with Biorender.com.
Author information
Authors and Affiliations
Contributions
J.F., Z.B.D., J.S.R., J.S.M., and S.K. contributed to the conception, drafting, and design of the paper. S.A.T., J.D.S., and D.J.D. contributed to the drafting and design of the paper. All authors participated in interactive teleconferences, writing and editing. Final paper assembly and editing as well as approval of the final submitted version of manuscript was done by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fares, J., Davis, Z.B., Rechberger, J.S. et al. Advances in NK cell therapy for brain tumors. npj Precis. Onc. 7, 17 (2023). https://doi.org/10.1038/s41698-023-00356-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00356-1
This article is cited by
-
Immune cell infiltration and inflammatory landscape in primary brain tumours
Journal of Translational Medicine (2024)
