
Abstract
Despite limited efficacy of immunotherapy for advanced non-small-cell lung cancer (NSCLC) with driver mutations, whether neoadjuvant immunotherapy could be clinically valuable in those patients warrants further investigation. We utilized 40 oncogene-mutant NSCLC treated with induction immunotherapy from a large consecutive multicenter cohort. Overall response rate was 62.5% while 2 patients had disease progression. Of 39 patients that received surgery, R0 resection rate was 97.4%. The major pathological response (MPR) rate was 37.5% and the pathological complete response (pCR) rate was 12.5%. Pre-treatment PD-L1 expression was not a predictive biomarker in these patients. Median disease-free survival for all oncogenic mutation and EGFR mutation was 28.5 months. Indirect comparison through integrating CTONG1103 cohort showed neoadjuvant immunotherapy plus chemotherapy yielded the most superior efficacy among erlotinib and chemotherapy for resectable EGFR-mutant NSCLC. No MPR patients were identified with neoadjuvant immunotherapy plus chemotherapy for uncommon EGFR insertion or point mutations. Our results indicated the potential clinical feasibility of neoadjuvant immunotherapy for resectable localized oncogene-mutant NSCLC especially for EGFR-mutant NSCLC.
Similar content being viewed by others


Introduction
Anti-PD-1/PD-L1 antibodies, as immune checkpoint blockade therapy (ICB), have been widely applied in advanced lung cancer as a single agent, combined with other checkpoint blockade or platinum-based chemotherapy1,2. Such novel treatment could enhance antitumor immunity by restoring T lymphocytes’ function leading to long-term benefits3. However, for advanced non-small cell lung cancers (NSCLCs) harboring driver mutations such as EGFR or ALK mutations, immunotherapy exhibited impaired response compared to corresponding targeted therapies4,5,6. Even for those rare driver mutations, the efficacy of immunotherapy varied among different mutation types and clinicopathological status such as smoking and tumor mutation burden (TMB), showing significantly inferior response rate to targeted therapies7. The mechanism that caused impaired efficacy in these populations included limited infiltration of CD8+ T lymphocytes, lower PD-L1 expression and strong oncogenic pathway activation8,9. Unlike immunotherapy alone, IMpower 150 is the first trial that has demonstrated comparable efficacy as an immunotherapy-based combination strategy in pretreated EGFR/ALK mutant NSCLCs10. However, the response mechanism of immunotherapy in mutant NSCLC remained controversial.
For resectable NSCLCs, chemotherapy has remained as the standard neoadjuvant treatment for decades11,12,13. In recent years, multiple phase 2 trials of neoadjuvant immunotherapy have shown dramatic pathological response and elevated surgical resection rate14,15,16. However, data on oncogene-mutant NSCLC using novel neoadjuvant modalities are limited. CTONG1103 (EMERGING) (NCT01407822) study compared neoadjuvant targeted therapy and chemotherapy head-to-head and failed to show neither significantly improved major pathological response (MPR) nor overall survival (OS)17. This might be attributed to the highly selective therapeutic mechanism of targeted therapy, intratumor heterogeneity and concomitant mutations which leads to incomplete antitumor response18,19. In a previous study, neoadjuvant atezolizumab and chemotherapy showed dramatic pathological response even in subjects with sensitive EGFR mutations, providing the clinical potentials of immunotherapy-based combination strategy for oncogene-mutant NSCLC16.
Despite the limited efficacy of immunotherapy in advanced mutant NSCLC, little is known about whether neoadjuvant immunotherapy could be clinically valuable for NSCLC positive of driver mutations. Herein, we describe the clinical outcome and safety profile of neoadjuvant immunotherapy followed by surgery in a multi-center cohort of patients with oncogene-mutant localized NSCLC. We also conducted dynamic multi-omics sequencing to provide insight of the potential mechanisms that led to differential response of neoadjuvant immunotherapy.
Results
Patient characteristics
In total, patients treated with neoadjuvant immunotherapy were preliminarily screened from 26 centers around China, and 40 NSCLC patients that were treated with neoadjuvant immunotherapy and harboring EGFR mutations were collected from 8 thoracic and cancer centers. 60.0% (24/40) were men and 57.5% (23/40) were non-smokers. 32.5% (13/40) patients were pathologically or radiologically confirmed as N2 disease. Only one patient was suspected to have oligometastatic disease of left adrenal gland before neoadjuvant treatment which disappear after neoadjuvant immunotherapy. Among all, there were 47.5% (19/40) patients harboring EGFR mutation including exon 19del, exon 21L858R, exon 17–25 and exon 20 insertion. Other oncogene mutation included KRAS, RET fusion, ALK fusion, ROS1 fusion, BRAF and HER2 insertion. Percentage of residual tumor cells were evaluated in 87.5% (35/40) patients while the other patients did not undergo surgery or comprehensive pathological assessments to determine the specific values of their residual tumors. Clinical information was summarized in Table 1 regarding MPR status. Detailed demographics including therapeutic regimens, genomic profiles, physicians’ consideration for neoadjuvant immunotherapy and adverse events (AE) during neoadjuvant treatment, are shown in Supplementary Table 1.
Clinical efficacy and safety profile of all oncogenic drivers
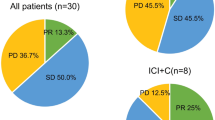
All patients have completed neoadjuvant immunotherapy without treatment discontinuation owing to severe toxicity. No new safety signals were identified regarding previous studies. Only two patients had grade 3 anemia, while others exhibited grade 1-2 adverse events (AEs) such as rash and diarrhea. Detailed AEs profile are shown in Supplementary Table 1. Overall, 62.5% (25/40) patients achieved partial response (PR) while 5.0% (2/40) had disease progression after completion of neoadjuvant immunotherapy. The N2 downstaging rate was 60.0% (9/15) for patients with pathologically or radiologically confirmed N2 disease. Of the two patients with progressive disease, one patient did not receive surgery due to newly-developed metastatic disease, while the other patient’s tumor was still considered to be resectable based on multidisciplinary discussion. Among the patients who underwent surgical resection, only one patient was reported with R2 resection while others achieved R0 resection. The detailed clinicopathological features of enrolled patients upon radiological and pathological response were presented in Fig. 1a. For patients treated with neoadjuvant immunotherapy, 37.5% (15/40) achieved MPR and 12.5% (5/40) achieved pCR.

a Overview of clinicopathological charteristics and clinical outcome of oncogene-mutant NSCLC treated with neoadjuvant immunotherapy. Clinicopathological charateristics including smoking status, therpeutic regimens, PD-L1 expression, treatment duration and preoperative radiologic response (according to Response Evaluation Criteria in Solid Tumors [RECIST]) were annotated for each patients. The horizontal dash line indicates the threshold for a major pathological response (90% regression). Pathological regression was presented in colored bar plot regarding different oncogene mutation. b Correlation between radiological shrinkage and pathological regression. Spearman correlation was used to measure the relevance. c Association between PD-L1 expression and MPR status. Centre line represents the median value of PD-L1 expression. Error bars represent the upper and lower quartiles of PD-L1 expression and whiskers defines the minimal and maximun value. PD Progression disease, SD Stable disease, PR Partial response, MPR Major pathological response, pCR Pathological complete response. d Disease-free survival (DFS) of all patients, EGFR-mutant patients and KRAS-mutant patients treated with neoadjuvant immunotherapy through Kaplan-Meier survival analysis.
We further assessed the correlation between radiological shrinkage and pathological regression of which a strong correlation was found (p-value < 0.001) probably due to most patients (91.2%) receiving neoadjuvant immunotherapy and chemotherapy (Fig. 1b). No significant associations of PD-L1 level were found between MPR and non-MPR patients (Fig. 1c). With a median follow-up time of 15.5 months, median disease-free survival (DFS) for both all oncogenic mutation and EGFR mutation were 28.5 months while median DFS for KRAS mutation was not reached. 1-year DFS for all oncogenic mutation, EGFR mutation and KRAS mutation was 87.3%, 92.9%, 87.3%, and 72.8%, 61.9%, 87.3% for 2-year DFS, respectively (Fig. 1d). Regarding MPR status, patients achieved MPR in both groups showed favorable trending of DFS (Supplementary Fig. 1).
Comparison of different treatment modalities for localized EGFR-mutant NSCLC
As the most common driver mutation in lung cancer, we emphatically analyzed the subgroup of EGFR-mutant NSCLC. We integrated the individual data of EGFR-mutant cohort from CTONG1103 (NCT01407822) study to compare the efficacy of different neoadjuvant treatment modalities. Patients were stratified into 3 groups for subsequent analysis: chemotherapy (GC regimen), targeted therapy (erlotinib) and immunotherapy plus chemotherapy (IO + CT). The baseline characteristics were relatively well balanced among groups in terms of smoking status, histology and EGFR mutation subtypes. EGFR exon 21 L858R was the most common subtype among groups (Table 2). In GC group, the response rate was 33.3% (5/15) and 56.3% for erlotinib group while elevated response rate of 63.2% was identified in IO + CT group. Of note, disease control rate (DCR) was 100% for both GC and erlotinib group, and one patient (5.3%) in IO + CT group had disease progress after neoadjuvant immunotherapy plus chemotherapy (Fig. 2). However, this patient was considered for radical resection after multidisplenary discussion and received surgery successfully. No sever postoperative complication was found except for one patient who had blood transfusion in regard to Clavien-Dindo score. Detailed clinicopathological features were summarized in Table 2. Only one patient in IO + CT group did not have specific value of residual viable tumor. The MPR in IO + CT group was significantly higher than erlotinib group (42.1% vs. 12.5%, odds ratio, 5.09; 95%CI, 1.04-26.39; P = 0.071) (Supplementary Fig. 2). Strikingly, while no pCR was found in both GC and erlotinib group, neoadjuvant immunotherapy plus chemotherapy yielded 10.5% pCR (odds ratio, not available; P = 0.489) (Supplementary Fig. 2). Among these patients, 66.7% (4/6) patients with EGFR exon19del achieved MPR and 16.7% (1/6) achieved pCR. On the other hand, 50.0% (4/8) patients of EGFR exon21 L858R had MPR and 12.5% (1/8) achieved pCR. Six patients detected with rare EGFR mutations, including insertions (3 EGFR 20ins, 1 EGFR exon17-25ins and 1 EGFR exon19ins) and rare point mutation (EGFR exon21 L861Q) did not achieve MPR.

Comparison of different treatment modalities for resectable EGFR-mutant NSCLC in regard to radiological response and pathological assessment by integrating chemotherapy and EGFR-TKI cohorts from CTONG1103 study. ADC Adenocarcinoma, SQC Squamous cell carcinoma, SD Stable disease, PR Partial response, PD Progression disease, CR Compelete response, MPR Major pathological response, pCR P athological complete response.
Inter-tumoral heterogeneity of immune contexture between PL and DLNs might reveal diverse response to neoadjuvant immunotherapy
To further illustrate why localized oncogene-mutant NSCLC might be responsive or ineffective to immunotherapy, we investigated the inter-tumoral heterogeneity and immune contexture data of four patients via genomic and transcriptomic analysis of their primary lung cancer (PL) and draining lymph nodes (DLNs). We used ImmuCellAI20 to estimate the abundance of infiltrating immune cells and performed unsupervised-clustering. We found distinct immune microenvironment (IME) between PL and normal lymph nodes while metastatic lymph nodes showed similar IME with PLs. Interestingly, we found a pretreatment metastatic N2 lymph node from patient 2 which exhibited no residual cancer after neoadjuvant treatment showed similar IME with PL instead of other normal lymph nodes. We also applied ImmuCellAI to predict the efficacy of immunotherapy where MPR patient showed infiltrating immune features of both PL and DLNs favored immunotherapy and remarkably less tissues showed response to immunotherapy for non-responders (Fig. 3a). Relatively higher abundance of cytotoxic, Th2 and γδT cells were found in responders (Fig. 3b). For patient 4 with N2 disease, who showed completely no response to neoadjuvant immunotherapy plus chemotherapy (Fig. 3c), we established a phylogenetic tree regarding PL and metastatic lymph nodes. PL retained driver EGFR mutation and other cancer associated genes after neoadjuvant immunotherapy. Although this patient had CD274 amplification which may predict better response to immunotherapy21, we also identified MDM4 amplification in PL which could predict hyperprogression for immunotherapy22 though no MDM4 amplification was found in metastatic DLNs (Fig. 3d). Additionally, we used NeoPredPipe23 to evaluate neoantigen burden, and found notably higher Indel-induced neoantigens burden in PL compared to DLNs while similar for low SNV-induced neoantigens burden (Fig. 3e).

a Non-supervised clustering of infiltrating immune microenviornment among PL and DLNs through ImmuCellAI. Pie chart indicates the proportion of samples which are antipated as responsive to immunotherapy. R Responder, NR Non-responder. b Comparison of specific immune cell subtypes among different patients. Kruskal-Wallis test was used to testify the difference among patients. Centre dots represent the median value of specific immune cell infiltration. Error bars represent the upper and lower quartiles and whiskers defines the range within 1.5IQR. c Radiological evaluation before and after neoadjuvant immunotherapy for PT4. d Genomic evolutionary trajectory between PL and DLNs of PT4. e Indel- and SNV-induced neoantigens burden among PL and DLNs through NeoPredPipe.
To further assess immune microenvironment phenotypes, we applied different functional gene sets upon antigen presenting cells (APC) abundance, T/NK cells abundance, IFN activity and T cell exhaustion among primary lung cancer and draining lymph nodes. Notably high expression of these gene sets was found in both PL and DLNs from responder while relatively lower expression in non-responder. Interestingly, despite low expression of these gene sets was observed in PL of PT2, relatively high expression of APC abundance and IFN activity were enriched in corresponding DLNs, and this patient only had 16% residual tumor left in the PL, which might indicate an underlying role of inflamed phenotype in DLNs for promoting the efficacy of immunotherapy (Fig. 4a). Through the radar plot, we comprehensively presented a multi-dimensional immune index regarding PD-L1 of PLs, T-cell inflamed gene-expression profile (GEP)24 and suppressor cell (SC)25 score in PLs and DLNs, respectively. We identified elevated T-cell inflamed GEP in DLNs along with lower SC score in PL for responders while the opposite in non-responders (Fig. 4b). Gene set enrichment analysis (GSEA) further showed that E2F_TARGETS, G2M_CHECKPOINT enriched in DLNs for non-responders suggesting proliferation arrested of immune cells while IL2_STAT5 and TNFA_SIGNALING for responders which are involved in anti-tumor immune response. For PLs, DNA_REPAIR and FATTY_ACID_METABOLISM were notably enriched in responders while enriched IL6_JAK_STAT3, an immune-suppression pathway, was found in non-responders (Fig. 4c).

a Expression profile of functional genesets including antigen presenting cell abundece, T/NK cell abundence, IFN activity and T cell exhasution were presented through z-score. b Radar plots of comprehensive immune phenotypes including PD-L1 expression in PL, T-cell inflamed GEP and suppressor cell score in PL and DLNs were scaled into 0–100%. T_PD-L1 represents expression of PD-L1 in tumor; T_GEP represents gene-expression profile of infiltrating lymphocytes in tumor; T_SC represents suppressor cell in tumor; LN_GEP represents gene-expression profile of infiltrating lymphocytes in draining lymph nodes; LN_SC represents suppressor cell in draining lymph nodes. c Gene set enrichment analysis (GSEA) of PL and DLNs upon responders and non-responders.
Discussion
Despite being a retrospective study with a heterogeneous population, our study described the clinical efficacy of neoadjuvant immunotherapy in the largest cohort of patients with resectable NSCLC harboring oncogene mutations. We observed both encouraging MPR and pCR rate for oncogene-mutant NSCLC treated with neoadjuvant immunotherapy especially for EGFR-mutant NSCLC as well as well-tolerated safety profile.
Recently released data from Checkmate-81626 showed superior pathological response of neoadjuvant immunotherapy plus chemotherapy compared to chemotherapy alone27. However, most of the neoadjuvant immunotherapy trials precluded EGFR/ALK mutant patients due to the inferior efficacy of immunotherapy in advanced NSCLC harboring EGFR/ALK mutation4,6,8,28. In addition, rare oncogenic mutations were reported to be either insensitive or undetermined efficacy to immunotherapy. Subgroup analysis of NEOSTAR study suggested less efficacy of either neoadjuvant nivolumab or nivolumab plus ipilimumab in canonical oncodriver patients29. However, phase 2 trials evaluating neoadjuvant immunotherapy plus chemotherapy16,30 showed astonishing pathological response in EGFR-mutant NSCLC, although both contained quite a small sample. In our study, we found that the short-term use of neoadjuvant immunotherapy plus chemotherapy in oncogene-mutant NSCLC could still achieve 37.5% MPR generally which were similar to Checkmate-816 suggesting clinical potentials for neoadjuvant immunotherapy used for oncogene mutant NSCLC. Previous study of neoadjuvant EGFR-TKIs in EGFR mutant patients exhibited relatively lower response rate compared to first-line setting in advanced disease, and MPR was unexpectedly lower which might be due to highly selective TKIs and recurrent co-occurring insensitive mutation clones17,31. Interestingly, unlike advanced disease, we found comparable efficacy of neoadjuvant immunotherapy for EGFR-mutant patients with MPR of 42.1%. Despite a lower pCR than in Checkmate-816, 10.5% EGFR-mutant patients could still achieve pCR through neoadjuvant immunotherapy while no pCR was found in the EMERGING study17. To be noticed, patients with EGFR rare point mutation or exon20 insertion did not show satisfactory efficacy rates in response to neoadjuvant immunotherapy plus chemotherapy, suggesting other treatment modalities are required for these patients.
Although LCMC4, an umbrella trial evaluating corresponding TKIs for different driver mutations as neoadjuvant treatment, was about to initiated, there remains an open question as to whether neoadjuvant immunotherapy might be feasible for these rare oncogene-mutant patients in consideration of one third of these patients achieved MPR in our cohort and no available neoadjuvant targeted therapy data for these patients. Indeed, limited sample size of these patients might be underpowered to draw solid conclusions.
A number of prior studies had evaluated potential biomarkers for MPR, but no agreement was made upon predictive biomarkers currently. PD-L1 status, the most well-known predictive biomarkers of CPIs in advanced NSCLC32,33,34, did not show correlation with MPR/pCR in some trials27,35. In the present study, we did not observe a significant correlation between pretreatment PD-L1 status and MPR either. For CA209-159 trial, although TMB was found to be relevant to the efficacy of neoadjuvant CPI15, TMB measures were not available for present study since few patients were performed WES or large panel NGS.
Owing to its retrospective nature, this study had two primary limitations of this study. First of all, the sample size is relatively small with diverse oncogene mutations and PD-1 blockades involved. Nevertheless, this is so far the largest cohort of localized oncogene-mutant NSCLC treated with neoadjuvant PD-1 blockades with median follow-up over 1 year. Besides, 19 patients harboring EGFR mutations, the most common driver mutation of lung cancer, were involved which could shed light on neoadjuvant treatment for EGFR-mutant NSCLC in a way. Last but not least, the lack of central evaluation of pathological response might lead to disparity of MPR which would potentially cause overestimation of MPR. However, evaluation of pCR would be barely affected due to its definition of no residual tumor cell at all. Moreover, pCR of EGFR-mutant NSCLC treated with neoadjuvant immunotherapy plus chemotherapy was notably higher than first-generation EGFR-TKI (CTONG1103) (MPR 9.7%; pCR 0.0%) and third-generation EGFR-TKI (NEOS, 2022 ELCC) (MPR 10.7%; pCR 3.6%).
In conclusion, we found that neoadjuvant immunotherapy plus chemotherapy could be clinically valuable for EGFR-mutant patients and potentially be extended to other rare driver mutations. Considering the underlying influence of tumor heterogeneity to targeted therapy, relatively low pathological response of targeted therapy in neoadjuvant setting and long-term benefit of immunotherapy, we assume front-line use of neoadjuvant immunotherapy plus chemotherapy may provide higher cured potentials and long-term survival benefit. Therefore, we proposed an underlying mode of neoadjuvant immunotherapy in optimizing whole-course treatment for localized oncogene-mutant NSCLC (Supplementary Fig. 3). Indeed, for localized NSCLC, multiplex genotyping is getting increasingly pivotal to guide personalized perioperative treatment. Ongoing trials such as LCMC4 (NCT04712877), NeoADAURA (NCT04351555) and NAUTIKA1 (NCT04302025) trials are evaluating matched neoadjuvant targeted therapies regarding molecular genotyping. However, neither MPR nor pCR which may indicate curative conditions from published prospective trials of neoadjuvant targeted therapy were satisfactory till now. An ongoing phase 2 trial called Neo-DIANA (NCT04512430) would evaluate neoadjuvant atezolizumab combined with chemotherapy and bevacizumab for EGFR-mutated NSCLC. Matched umbrella trials evaluating neoadjuvant targeted therapies should be expected and future trials of combination immunotherapy are warranted to further clarify its clinical significance, identify potential beneficiaries and tailor more optimal treatment modalities for oncogene-mutant localized NSCLC.
Methods
Patients and evaluation
We initiated a national questionnaire of real-world neoadjuvant immunotherapy in resectable NSCLC across 26 cancer research centers and 8 of them reported patients with oncogenic mutations who were treated with neoadjuvant immunotherapy under the real-world circumstances including inadequate biopsy specimen for genetic testing, negative findings of driver mutation from ctDNA, or no available TKI treatment. In case of any potential selection bias, all patients were screened and collected consecutively. Oncogenic mutations included EGFR mutations, KRAS mutations, ALK fusions, RET fusions, ROS1 fusions, the BRAF V600E mutation and HER2 mutations. Additionally, we applied data of Guangdong Lung Cancer Institute cohort from CTONG-1103 trial (NCT01407822) which evaluated either neoadjuvant gemcitabine plus cisplatin or erlotinib in EGFR-mutant stage III NSCLC. Informed consent was written and received from each patient before treatment as well as informed consents for experimentation with human subjects. This study was approved by the ethical committee of Guangdong Provincial People’s Hospital (KY-Z-2021-567-03).
Patients’ demographic and clinical data were retrospectively reviewed, including clinical information, tumor histology, molecular profile, treatment modality, radiological and pathological response assessment. Genotyping assessments were performed through various approaches, including PCR, immunohistochemistry (IHC) (for ALK fusion) and large panel next-generation sequencing (NGS). Radiological response was assessed following formal Response Evaluation Criteria in Solid Tumors (RECIST) measurements. Due to the retrospective nature, pathological assessment was performed independently in the Department of Pathology of each center following recommended assessment process36 to evaluate residual viable tumor (RVT) except for multiple slides. MPR and pathological complete response (pCR) was defined as less than 10% viable tumor cells in primary lesions and no viable tumor cells in both primary and lympho nodes, respectively.
Expression of PD-L1 was independently scored through Dako PD-L1 22C3 (pharmDx) assay. PD-L1 expression was quantified as the proportion of PD-L1-positive tumor cells. Positive PD-L1 expression in a given specimen was defined as ≥1% for tumor cell and ≥50% for high expression. Cases with <100 total tumor cells for scoring were defined as not applicable (NA).
Comprehensive genomic and immune profiling of blood and tumor samples
We collected serial blood samples, post-treatment primary tumor samples, and draining lymph nodes specimens from four patients. Genomic DNA was extracted from frozen tumor and normal tissue sections using the QIAamp DNA FFPE Tissue Kit (Qiagen) following the manufacturer’s instructions. A minimum of 1ug of DNA was used for WES profiling experiment. The DNA quality was assessed by Nanodrop2000 (Thermo Fisher Scientific), and the quantity was measured by the dsDNA HS Assay Kit using Qubit 2.0 (Life Technologies). WES was only performed when the tumor proportion was above 20%. WES library was sequenced using an Illumina Novaseq 4000 platform according to the manufacturer’s instructions.
Total RNA from frozen section samples was extracted using RNeasy FFPE kit (QIAGEN). Ribosomal RNA was depleted using RNase H followed by library preparation using KAPA Stranded RNA -seq Kit with RiboErase (HMR) (KAPA Biosystems), and library quality was accessed by Agilent High Sensitivity DNA kit on Bioanalyzer 2100 (Agilent Technologies), which was then sequenced on Illumina Novaseq NGS platforms.
Gene expression analysis and immune construction
Base calling was performed on bcl2fastq v2.16.0.10 (Illumina, Inc.) to generate sequence reads in FASTQ format (Illumina 1.8+ encoding). Quality control (QC) was performed with Trimmomatic (version 0.33). STAR (version 2.5.3a) is used for transcriptome mapping followed by isoform and gene level quantification performed by RSEM (version 1.3.0). Corresponding heatmaps were generated by in-house R scripts. To evaluate the immune construction of primary lung cancer and draining lymph nodes, we applied ImmuCellAI20 to comprehensively decode distribution of different infiltrating immune cells.
Reconstruction of phylogenetic trees
SAMtools v1.3.137 mpileup with parameter “-p”:20; “-P”: 20 was used to get mutation supporting reads counts across all tumor regions where a variant (SSNV or InDel) was detected in one or more regions in one patient. For those somatic variants that were not called ubiquitously across tumor regions, the missing variants were picked back up if the mutant read count was ≥3, as well as the read depth, was >10. Sites with sequencing depth ≤10 were marked as “NA”. The neighbor joining phylogenetic trees were constructed using R package named “MesKit”38. The recalled mutation file (.maf format) was set as input and method = “NJ” with bootstrap 100 times were set in the function “getPhyloTree”.
Statistical analysis
Clinical and genomic data were analyzed using R Package (Version 3.3.0) or Prism 5.0 (Graph Pad Software Inc., La Jolla, CA, USA) software. Kruskal-Wallis test was applied to compare the significance among groups and Student’s t-test was used for comparison between groups. Correlations among different variables were examined by Pearson and presented through Pearson r and p-values. Odds ratios were used to compare pathological response between different treatment modalities and Fisher exact test was used to calculate the significance. A two-sided p < 0.05 was determined to be statistically significance.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Partial clinicopathological data is presented in the manuscript. Sequential sequencing data including WES and RNA are available in the GSA-Human repository (Accession code: HRA002653). Other detailed informations are available from corresponding author (W-Z.Z., syzhongwenzhao@scut.edu.cn) of this study upon reasonable request.
Code availability
No custom code was used in this study. WES sequencing data was analyzed using Trimmomatic, Burrows-Wheller Aligner(v0.7.12), Genome Analysis Toolkit (GATK 3.4.0) and VarScan 2. RNAseq data was analyzed using RSEM(v1.3.0), STAR(v2.5.3a), and ClusterProfiler(v3.4.4). TCR sequencing data was analyzed using Cutadapt (v1.18), PEAR (v0.9.10), and MiXCR(v2.1.11). All statistical analyses were performed in R version 3.5.0.
References
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science (N.Y., NY) 359, 1350–1355 (2018).
Hargadon, K. M., Johnson, C. E. & Williams, C. J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 62, 29–39 (2018).
Postow, M. A., Callahan, M. K. & Wolchok, J. D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol.: Off. J. Am. Soc. Clin. Oncol. 33, 1974–1982 (2015).
Garassino, M. C. et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 19, 521–536 (2018).
Mazieres, J. et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann. Oncol.: Off. J. Eur. Soc. Med. Oncol. 30, 1321–1328 (2019).
Lee, C. K. et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J. Thorac. Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 12, 403–407 (2017).
Ng, T. L. et al. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non-small cell lung cancer. Cancer 125, 1038–1049 (2019).
Gainor, J. F. et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 22, 4585–4593 (2016).
Lin, A. et al. Role of the dynamic tumor microenvironment in controversies regarding immune checkpoint inhibitors for the treatment of non-small cell lung cancer (NSCLC) with EGFR mutations. Mol. Cancer 18, 139 (2019).
Socinski, M. A. et al. Abstract CT216: IMpower150 final analysis: Efficacy of atezolizumab (atezo) + bevacizumab (bev) and chemotherapy in first-line (1L) metastatic nonsquamous (nsq) non-small cell lung cancer (NSCLC) across key subgroups. 80, CT216-CT216 (2020).
Nagai, K. et al. A randomized trial comparing induction chemotherapy followed by surgery with surgery alone for patients with stage IIIA N2 non-small cell lung cancer (JCOG 9209). J. Thorac. Cardiovascular Surg. 125, 254–260 (2003).
Blumenthal, G. M. et al. Current Status and Future Perspectives on Neoadjuvant Therapy in Lung Cancer. J. Thorac. Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 13, 1818–1831 (2018).
Detterbeck, F. C. et al. Neoadjuvant chemotherapy with gemcitabine-containing regimens in patients with early-stage non-small cell lung cancer. J. Thorac. Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 3, 37–45 (2008).
Gao, S. et al. Neoadjuvant PD-1 inhibitor (Sintilimab) in NSCLC. J. Thorac. Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 15, 816–826 (2020).
Forde, P. M. et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 378, 1976–1986 (2018).
Shu, C. A. et al. Neoadjuvant atezolizumab and chemotherapy in patients with resectable non-small-cell lung cancer: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 21, 786–795 (2020).
Zhong, W. Z. et al. Erlotinib Versus Gemcitabine Plus Cisplatin as Neoadjuvant Treatment of Stage IIIA-N2 EGFR-Mutant Non-Small-Cell Lung Cancer (EMERGING-CTONG 1103): A Randomized Phase II Study. J. Clinical Oncol.: Off. J. American Soc. Clinical Oncol. 2019:Jco1900075.
Jiang, S. X. et al. EGFR genetic heterogeneity of nonsmall cell lung cancers contributing to acquired gefitinib resistance. Int. J. Cancer 123, 2480–2486 (2008).
Dong, Z. Y. et al. Mixed responses to systemic therapy revealed potential genetic heterogeneity and poor survival in patients with non-small cell lung cancer. Oncologist 22, 61–69 (2017).
Miao, Y. R. et al. ImmuCellAI: A unique method for comprehensive T-Cell subsets abundance prediction and its application in cancer immunotherapy. Adv. Sci. (Weinh., Baden.-Wurtt., Ger.) 7, 1902880 (2020).
Inoue, Y. et al. Evaluation of Programmed Death Ligand 1 (PD-L1) gene amplification and response to nivolumab monotherapy in non-small cell lung cancer. JAMA Netw. open 3, e2011818 (2020).
Kato, S. et al. Hyperprogressors after Immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 23, 4242–4250 (2017).
Schenck, R. O. et al. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinforma. 20, 264 (2019).
Ayers, M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 127, 2930–2940 (2017).
Charoentong, P. et al. Pan-cancer Immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017).
Forde, P. M. et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. New England J. Med. 386,1973–1985 (2022).
Spicer, J. Surgical outcomes from the phase 3 CheckMate 816 trial: Nivolumab (NIVO) + platinum-doublet chemotherapy (chemo) vs chemo alone as neoadjuvant treatment for patients with resectable non-small cell lung cancer (NSCLC). In: Jonathan Spicer CWFTGBSK-NCMLEEVNGSLMPTMMMAEF, ed.ASCO Annual Meeting: American Soc. Clinical Oncol.; 2021 Available at https://meetinglibrary.asco.org/record/195951/abstract.
Uprety, D. et al. Neoadjuvant Immunotherapy for NSCLC: Current Concepts and Future Approaches. J. Thorac. Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 15, 1281–1297 (2020).
Zhou, N. et al. Impact of genomic aberrations and additional therapies on survival outcomes of patients with operable non-small cell lung cancer (NSCLC) from the NEOSTAR study. J. Clinical Oncol. 39, 8542–8542 (2021).
Provencio, M. et al. Neoadjuvant chemotherapy and nivolumab in resectable non-small-cell lung cancer (NADIM): An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 21, 1413–1422 (2020).
Skoulidis, F. & Heymach, J. V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19, 495–509 (2019).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (Lond., Engl.) 387, 1540–1550 (2016).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Brahmer, J. et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015).
Rothschild, S. I. et al. SAKK 16/14: Durvalumab in Addition to Neoadjuvant Chemotherapy in Patients With Stage IIIA(N2) Non-Small-Cell Lung Cancer-A Multicenter Single-Arm Phase II Trial. J. Clinical Oncol.: Off. J. American Soc. Clinical Oncol. 2021:Jco2100276.
Travis, W. D. et al. IASLC multidisciplinary recommendations for pathologic assessment of lung cancer resection specimens after neoadjuvant therapy. J. Thorac. Oncol.: Off. Publ. Int. Assoc. Study Lung Cancer 15, 709–740 (2020).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinforma. (Oxf., Engl.) 25, 2078–2079 (2009).
Liu, M. et al. MesKit: A tool kit for dissecting cancer evolution of multi-region tumor biopsies through somatic alterations. GigaScience 10, giab036 (2021).
Acknowledgements
We thank all the contributing authors for their great effort on this manuscript and all enrolled patients for their support. This study was supported by National Natural Science Foundation (grant 81872510 to W-ZZ) and High-level Hospital Construction Project (grant DFJH201801 to W-ZZ).
Author information
Authors and Affiliations
Contributions
Conception and design: C.Z., W.-Z.Z., Y.-L.W.; Provision of patients: C.Z., H.-F.C., S.Y., L.W., X.-L.Y., D.-S.Y., M.Z., J.-S.L., B.-Y.J., W.-Z.Z.; Data analysis and interpretation: C.Z., L.-X.Y., L.-L.Y.; Manuscript writing: C.Z., Z.-B.Q., Q.-X.O.; Final approval of manuscript: All authors.
Corresponding author
Ethics declarations
Competing interests
W.-Z.Z. received speech honoraria from AstraZeneca, Roche, Eli Lilly, and Pfizer. Y.-L.W. received research funding from Roche and speech honoraria from AstraZeneca, Roche, Eli Lilly, Pfizer and Sanofi, and he was a research consultant for AstraZeneca. L.W. and D.S.Y. received speech honoraria from AstraZeneca and Roche. L.-L.Y., Q.-X.O., and Y.S. are employees of Nanjing Geneseeq Technology Inc. The remaining authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, C., Chen, HF., Yan, S. et al. Induction immune-checkpoint inhibitors for resectable oncogene-mutant NSCLC: A multicenter pooled analysis. npj Precis. Onc. 6, 66 (2022). https://doi.org/10.1038/s41698-022-00301-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-022-00301-8
This article is cited by
-
Impact of chronic obstructive pulmonary disease on the efficacy and safety of neoadjuvant immune checkpoint inhibitors combined with chemotherapy for resectable non-small cell lung cancer: a retrospective cohort study
BMC Cancer (2024)
-
Emerging precision neoadjuvant systemic therapy for patients with resectable non-small cell lung cancer: current status and perspectives
Biomarker Research (2023)
-
The impact of oncogenic driver mutations on neoadjuvant immunotherapy outcomes in patients with resectable non-small cell lung cancer
Cancer Immunology, Immunotherapy (2023)
