
Abstract
Information regarding the molecular features of pulmonary pleomorphic carcinoma (PPC) is insufficient. Here, we performed next-generation sequencing to determine the genomic and transcriptomic profiles of PPC. We sequenced the DNAs and RNAs of 78 specimens from 52 patients with PPC. We analyzed 15 PPC cases to identify intratumoral differences in gene alterations, tumor mutation burden (TMB), RNA expression, and PD-L1 expression between epithelial and sarcomatoid components. The genomic alterations of six cases of primary tumors and corresponding metastatic tumors were analyzed. KRAS mutations (27%) were the most common driver mutations, followed by EGFR (8%), and MET (8%) mutations. Epithelial and sarcomatoid components shared activating driver mutations, and there were no significant differences in CD274 expression or TMB between the two components. However, PD-L1 was highly expressed in the sarcomatoid component of several cases compared with the epithelial component. Primary and metastatic tumors shared oncogenic mutations among genes such as KRAS and TP53, and additional alterations including NOTCH4 mutations were specifically identified in the metastatic regions. Our data suggest that therapies targeting activating driver mutations may be effective for patients with PPC and that immune checkpoint inhibitors of PPC may be recommended after careful assessment of PD-L1 expression in each epithelial and sarcomatoid component.
Similar content being viewed by others

Introduction
Pulmonary pleomorphic carcinoma (PPC) is a rare subtype of non-small cell lung cancer (NSCLC) that accounts for 0.4–1.6% of malignant lung tumors1,2. According to the 4th edition of the World Health Organization Classification of Lung Tumors3, PPC is defined as a poorly differentiated NSCLC comprising ≥10% spindle or giant cells. These tumors, which predominantly arise in men who heavily smoke, are characterized by a poor response to cytotoxic chemotherapy and a worse outcome than other types of NSCLC4,5. Two studies identified EGFR-activating mutations in approximately 20% of PPCs6,7, some of which exhibit a partial response to gefitinib8. However, insufficient information is available regarding the molecular features of PPC and effective therapeutic targets.
Many PPCs comprise an admixture of sarcomatoid (spindle or giant cell elements or both) and epithelial components (adenocarcinoma, squamous cell carcinoma, or undifferentiated NSCLC). Previous studies indicated that genomic intratumoral heterogeneity in cancers is one of the leading determinants of treatment failure and drug resistance9,10. Intratumoral heterogeneity in large-cell neuroendocrine carcinoma (LCNEC) combined with NSCLC is characterized by a relatively high (71%) median concordance rate of genomic mutations between these components11. To our knowledge, however, published studies do not comprehensively define the intratumor heterogeneity of PPC.
Immunotherapies targeting the programmed death-1 (PD-1)/PD ligand 1 (PD-L1) axis yielded promising results for patients with NSCLC, and several studies suggest that PD-L1 expression may predict the response to this type of immunotherapy12,13. Interestingly, >90% of patients with PPC have PD-L1-positive disease, supporting the conclusion that immunotherapy may serve as a potential option for this patient population14. However, this study14 demonstrates higher levels of PD-L1 in sarcomatoid vs epithelial components, and the potential effect of this intratumoral difference in PD-L1 expression on treatment efficacy is unknown. Further, a high tumor mutation burden (TMB) serves as a biomarker of the tumor response to PD-1/PD-L1 targeted-immunotherapy15,16. However, little is known about the effect of the TMB in PPC.
Here we performed next-generation sequencing to analyze the molecular profiles of PPCs. For certain PPC samples, we extracted genomic DNA and RNA from the sarcomatoid and epithelial components and compared them to detect intratumoral differences in gene mutations, RNA expression, and PD-L1 expression. We further compared the gene alterations in several cases between primary tumors and the corresponding metastatic tumors.
Results
Patients’ characteristics
The demographic features of 52 patients with PPC are described in Table 1. Their median age at the time of sample collection was 68 years (range, 36‒84 years), 43 (83%) were male, and 47 (90%) were smokers. The median primary lesion diameter was 4.6 cm (range, 1.5–10.3 cm). Their pathological stages were as follows: stage I, n = 15 (29%); stage II, n = 17 (33%); stage III, n = 15 (29%); and stage IV, n = 5 (10%). Forty-nine patients underwent surgical resection, and 44 (90%) underwent lobectomy.
Genomic alterations in PPC
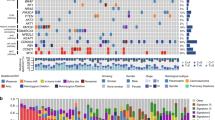
The study is summarized in Supplementary Fig. 1. From 4 hospitals, 78 specimens were collected from 52 consecutive patients with PPC in this study. Four FFPE specimens from 3 patients (all were male and current smokers) were excluded from the DNA analysis because of low quality, and 74 specimens from 49 patients were subjected to DNA sequence analyses to detect genomic alterations. Among the 74 specimens, whole-exome sequencing of 12 fresh-frozen tissue samples from 12 patients and target-capture sequencing of 62 FFPE samples from 37 patients were performed. Sixty-six samples were from the primary tumors, and the other eight samples were obtained from the metastatic regions. The genomic DNAs of the sarcomatoid and epithelial components of tumors of 34 samples of 17 patients were individually analyzed (Fig. 1a).

a This case comprises adenocarcinoma (green area) and sarcomatoid (yellow area) components (hematoxylin and eosin staining). Magnified images of the epithelial (E) and sarcomatoid (S) components are shown. b Mutations in KRAS were detected in 13 patients (27%). Other activating mutations were detected in EGFR (8%), HRAS (4%), NRAS (2%), BRAF (2%), and MAP2K1 (4%). MET exon 14 skipping (4%) and EML4-ALK fusion (2%) were detected using RNA sequencing. These driver mutations were mutually exclusive.
An overview of the mutations in the primary lesions is presented in Fig. 1b. We placed emphasis on determining the genomic alterations associated with lung cancer detected in previous comprehensive genomic studies17. TP53 was the most frequently mutated gene, detected in 35 (71%) patients. KRAS mutations (13 patients, 27%) were the most prevalent oncogenic mutations (G12A—2 cases, G12C—5 cases, G12D—1 case, G12R—1 case, G12S—1 case, and G12V—3 cases), followed by EGFR (8%), HRAS (4%), MAP2K1 (4%), PIK3CA (4%), NRAS (2%), and BRAF (2%). Other recurrent mutations were identified in PTPRD (22%), ARID2 (14%), and NF1 (12%). Comprehensive mutation list is shown in Supplementary Data 1.
RNA sequencing of the remaining samples identified an EML4-ALK fusion in one patient (2%) and MET exon 14 skipping in four (8%) (Fig. 1b). In the sample harboring the EML4-ALK fusion, Sanger sequencing of the EML4-ALK cDNA revealed that exon 13 of EML4 was ligated to exon 20 of ALK with an insertion of 24 base pairs corresponding to ALK intron 19 and six base pairs of unknown origin (Supplementary Fig. 2a). This fusion produces an in-frame transcript with strong oncogenic transforming potential indicated by the focus formation assay (Supplementary Fig. 2b).
We further searched for genomic alterations using the Memorial Sloan Kettering-Cancer Center (MSKCC) cohort data18 (http://www.cbioportal.org) (Supplementary Fig. 3). Among 17 cases of PPC, TP53 and KRAS mutations were detected in 10 (59%) and 5 (29%) samples, respectively. These mutation rates were similar to the results of our cohort.
Comparison of genomic alterations between epithelial and sarcomatoid components
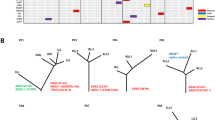
The genomic alterations in epithelial and sarcomatoid components were compared in 17 PPC cases. The mean number of shared nonsynonymous mutations detected in both components was 5.5 (range, 0‒20), whereas the mean numbers of private nonsynonymous mutations (detected in one component) were 2.7 (range, 0‒9) and 1.8 (range: 0‒7) in epithelial and sarcomatoid components, respectively (Fig. 2a). The recurrent nonsynonymous mutations are listed in Fig. 2b. Notably, oncogenic KRAS and EGFR mutations were shared by both components. Mutations of ARID2, ASPM, NF1, and PIK3CG were detected in the sarcomatoid components, while EPHB1 mutations were observed only in the epithelial components. A phylogenetic tree of each tumor was generated using the LICHeE method (Supplementary Fig. 4)19.

a Bar chart of the average numbers of shared and unshared nonsynonymous mutations between the epithelial and sarcomatoid components. b Individual repeatedly detected mutations in each component. Activating mutations were shared between components, while ARID2, ASPM, NF1, and PIK3CG mutations were only detected in sarcomatoid components. The number of patients who harbor the mutations of indicated genes is shown in parentheses. c Average numbers of nonsynonymous mutations in primary and metastatic tumors, revealing more in metastatic tumors relative to primary tumors. d Individual mutations detected in primary and metastatic tumors. The number of patients who harbor the mutations of indicated genes is shown in parentheses. Error bars, standard error of the mean.
Comparison of genomic alterations between primary and the corresponding metastatic tumors
We determined the differences in genomic alterations between primary and the metastatic tumors of six cases. Notably, a higher number of nonsynonymous mutations were observed in the metastatic tumors (mean, 17.3; range, 3‒28) compared with those in primary tumors (mean, 11.5; range, 1‒33) (Fig. 2c). The oncogenic mutation in KRAS, NRAS, or MAP2K1 was identified in one case each, which was detected in the primary and metastatic region. TP53 mutations were detected in both tumors of five cases. PDE4DIP, ROBO1, and NOTCH4 mutations were observed only in metastatic tumors, whereas mutations specific to primary tumors were undetectable (Fig. 2d). Phylogenetic trees of these six cases were generated using the LICHeE method (Supplementary Fig. 5).
RNA expression profiles in PPC, pathological stages I and II
Even patients with early-stage PPC face a significant risk of recurrence after undergoing complete surgical resection1,2. Therefore, reliable prognostic biomarkers are desirable to identify such patients. For this purpose, association between the RNA expression of each gene and recurrence-free survival (RFS) of patients with pathological stage I or II PPC who underwent complete surgical resection was assessed using a univariate Cox proportional hazards regression model. Among the 32 patients with stage I or II in our cohort, 25 patients were analyzed using FFPE samples and the others were analyzed using fresh frozen samples. Because the difference in starting material can cause difference in the representation of RNA expression, survival analysis was performed using only FFPE samples from 25 patients. As a result, we found that the expression of 15 genes significantly correlated with RFS (q < 0.05, Fig. 3a). The patients were then divided into high and low groups according to the average expression level of each gene, and pairwise comparisons of RFS were performed using the log-rank test. High expression of CAPN14, LIN7A, LNX1, or PDGFRA significantly correlated with shorter RFS (p < 0.05, Fig. 3b). In contrast, high expression of the other 11 genes correlated with longer RFS (Supplementary Fig. 6).

a The expression level of each gene was calculated and normalized using the DESeq2 package. The association between the RNA expression of each gene and recurrence-free survival (RFS) of 25 patients with pathological stage I or II PPC was assessed using univariate Cox proportional hazard regression model, showing the expression levels of the 15 genes strongly correlated with RFS (q < 0.05). The midline in each box represents the median, and the lower and upper boundaries indicate the first and third quartiles, respectively. Whiskers represent the 95% confidence intervals of the mean values. b Kaplan–Meier analysis of RFS of 25 patients according to the average RNA level of each gene demonstrated that strong expression of four genes was associated with shorter RFS (p < 0.05).
Comparison of RNA levels between epithelial and sarcomatoid components
We next compared the RNA levels of 30 FFPE specimens from each of epithelial and sarcomatoid components of 15 cases. Hierarchical clustering using the most variable 100 genes revealed that the epithelial and sarcomatoid components of respective cases were clustered next to each other, suggesting that the differences among the patients were greater than those between the epithelial and sarcomatoid components (Fig. 4a). Gene Set Enrichment Analysis (GSEA) of the gene sets “SHEDDEN_LUNG_CANCER_POOR_SURVIVAL_A6” and “SHEDDEN_LUNG_CANCER_GOOD_SURVIVAL_A4” revealed that they were enriched in the sarcomatoid or epithelial component, respectively (Fig. 4b). GSEA further identified specific and significant enrichment of gene sets related to the cell cycle in the sarcomatoid group. When we searched for biomarkers that distinguished between epithelial and sarcomatoid components using the Wald test, we found that ACE2, AQP3, BCAS1, BNIPL, FHDC1, MUC21, PARM1, PGC, SCGB3A2, and SFTA) were significantly expressed only in the epithelial group (q < 0.05, Fig. 4c).

a Heat map of the z-scores of the top 100 genes differentially expressed between the epithelial and sarcomatoid components. b Gene Set Enrichment Analysis revealed that the “SHEDDEN_LUNG_CANCER_POOR_SURVIVAL_A6” set was significantly enriched in the sarcomatoid group, whereas the “SHEDDEN_LUNG_CANCER_GOOD_SURVIVAL_A4” set was enriched in the epithelial group. Cell cycle-related gene sets were significantly enriched in the epithelial group. c Heat map of the z-scores of 10 genes that were differentially expressed between epithelial and sarcomatoid components. All genes were more strongly expressed in the epithelial group. FDR, false discovery rate; NES, normalized enrichment score.
PD-L1 expression and TMB
When we used IHC to determine the levels of PD-L1 among 56 FFPE specimens compared with those of CD274 mRNA from the RNA-seq dataset, we found a significant correlation with the latter (Pearson correlation coefficient r = 0.63; p < 0.001) (Fig. 5a). Using the cut-off thresholds 1% and 50%, according to previous studies20,21, we found that the level of CD274 mRNA was significantly upregulated in the high PD-L1 expression group (≥50%) (p < 0.05, Fig. 5b). There was not a significant correlation between the TMB and PD-L1 level in the same specimens (r = −0.21; p = 0.13) (Supplementary Fig. 7).

a Normalized CD274 counts significantly correlated with the PD-L1 level (Pearson correlation coefficient r = 0.62; p < 0.001). Separated and unseparated samples are indicated in blue and orange, respectively. b Bar chart of the normalized CD274 counts according to PD-L1 expression. CD274 expression was significantly upregulated in the high PD-L1 expression group (≥50%). In each box, the midline represents the median, and the lower and upper boundaries represent the first and third quartiles, respectively. The whiskers indicate the minimum and maximum values. c The correlation between the CD274 normalized counts in the epithelial and sarcomatoid components was evaluated. Strong correlations in the expression of CD274 were observed between the two components (r = 0.95; p < 0.001). d The correlation between the epithelial and sarcomatoid components with TMB was evaluated. There were strong correlations of the TMB score with each of the two components (r = 0.82; p < 0.001). e PD-L1 levels were compared between the epithelial and sarcomatoid components. Several cases exhibited higher PD-L1 expression in the sarcomatoid component compared with that of the epithelial component. f Representative images of immunohistochemical analysis of PD-L1 expression in pulmonary pleomorphic carcinoma. High levels of PD-L1 in the sarcomatoid component (yellow area) were detected, while PD-L1 expression was infrequent in the epithelial component (green area). g The Pearson correlation coefficient between the normalized PELI2 counts and PD-L1 expression was −0.64, the highest absolute value of all tested genes. Separated and unseparated samples are blue and orange, respectively.
The levels of CD274 mRNA and PD-L1, as well as the TMB, were compared between the epithelial and sarcomatoid components of 15 PPC cases. There was a significant correlation between the level of CD274 mRNA or the TMB score between the two components (r = 0.95; p < 0.001 or r = 0.82; p < 0.001, respectively) (Fig. 5c and d). In contrast, several cases exhibited higher levels of PD-L1 in the sarcomatoid components compared with those in the epithelial components (Fig. 5e and f), indicating that post-transcriptional processes may regulate the levels of PD-L1. We therefore searched for genes whose expression levels correlated with those of PD-L1. The strongest positive correlation was observed for the levels of CD274 mRNA, supporting the validity of this approach. The strongest negative correlation was observed for the gene encoding Pellino E3 ubiquitin protein ligase family member 2 (PELI2, r = −0.64) (Fig. 5g). Notably, the correlation coefficient of the association of PELI2 and PD-L1 was stronger than that between CD274 mRNA and PD-L1 (absolute r = 0.82 vs 0.60) when the analysis separately considered the epithelial and sarcomatoid components.
Discussion
To the best of our knowledge, this is the first relatively large-scale (n = 49) study to conduct a comprehensive evaluation of the molecular profiles of PPCs. The macrodissection analysis of the epithelial and sarcomatoid components of PPCs found that both components harbored similar genomic alterations, including activating driver mutations. Further, the expression level of PD-L1 was more frequently higher in the sarcomatoid components, and the levels of CD274 mRNA and the TMB were highly concordant between the two components.
Here, we show that 57% (28/49) of the PPC samples harbored activating mutations, which is consistent with the results of other studies17. Mutations within members of the RAS gene family were particularly frequent (16/49, 33%), suggesting that therapies targeting the RAS pathway may be effective for PPC. For example, ongoing clinical trials targeting KRAS G12 mutations are showing promise for patients with NSCLC22. Further investigations of the abilities of RAS-targeted inhibitors to improve the treatment outcomes of patients with PPC are required.
In the present study, our cohort included four patients (8%) with MET exon 14 skipping. This frequency was higher than that observed in studies of patients with lung adenocarcinoma (0.9–2.2%)23,24, although it was consistent with other reports (3–22%) on PPC25,26. Capmatinib and tepotinib are selective inhibitors of the receptor MET, which was recently approved by the United States Food and Drug Administration (FDA) for patients with NSCLC with MET exon 14 skipping; therefore, these drugs would be promising for PPC with MET exon 14 skipping.
Our present comparison of the epithelial and sarcomatoid components of 17 patients revealed that 11 (65%) carried the same driver mutations in both components. These patients may benefit from therapies specifically targeting EGFR, MET, and BRAF mutations. Moreover, TP53 and KEAP1 alterations were detected in both components of numerous cases, suggesting that these alterations occurred early during tumor progression. These results are consistent with studies that analyzed intratumoral heterogeneity of patients with NSCLC27,28.
In contrast, private alterations, which likely arose during tumor progression, were identified in 16 patients (94%). Further, private mutations in ARID2, ASPM, NF1, and PIK3CG were detected in the sarcomatoid components of multiple cases. ARID2, NF1, and PIK3CG mutations are associated with poor tumor differentiation and the epithelial–mesenchymal transition29,30,31. Thus, private alterations may contribute to the transition to a sarcomatoid phenotype associated with poor prognosis. Moreover, the private alterations in ASPM and ARID2, whose functions are to regulate the cell cycle31,32, may partly explain the enrichment of a gene set related to the cell cycle in the sarcomatoid component.
Primary and metastatic tumors exhibit a high concordance of genomic alterations, including oncogenic mutations of KRAS and TP53. Two studies of various primary tumors show that matched metastatic regions exhibit a high degree of similarity with respect to genomic alterations33,34. Notably, several alterations occur only in the metastatic tumors of PPC, which may be acquired during disease progression. For example, NOTCH4 mutations promote the metastasis of melanoma cells35. The identification of the molecular mechanism of metastasis may lead to the prevention and treatment of metastasis.
The expression of multiple genes may be related to a high risk of recurrence after curative surgery, including patients with early-stage PPC. Here we show that the strong expression of CAPN14, LIN7A, LNX1, and PDGFRA was significantly associated with poor prognosis after surgery. LNX1, which is strongly expressed in soft tissue sarcoma36, contributes to tumor growth by destabilizing p5337. Moreover, strong expression of PDGFRA may serve a significant indicator of poor disease-specific survival. PDGFRA regulates mesenchymal cell activity in the tumor microenvironment through mechanisms including vascular reorganization, proliferation, and pericyte recruitment38,39. Therefore, FDA-approved drugs targeting PDGFRA, such as regorafenib for colorectal carcinoma and pazopanib for renal carcinoma40,41, may inhibit tumor progression of PPC with high PDGFRA expression. Few studies evaluated the relationship between cancer progression and LIN7A or CAPN14 expression.
Here we show that PD-L1 was expressed at significantly higher levels in the sarcomatoid components of several PPCs compared with the epithelial components. For example, PD-L1 levels differ among intratumoral the components of PPC14. PD-1 expression serves as a predictive biomarker for immune checkpoint inhibitors (ICI)12,13. Therefore, treatment with ICIs may eradicate the sarcomatoid component, which is regarded as resistant to chemotherapy. In contrast to the intratumoral heterogeneity in PD-L1 expression, we found that both components had similar TMBs, and thus a TMB score in a portion of a tumor likely represents the TMB of the entire tumor.
The correlation of CD274 mRNA levels with those of PD-L1 is the subject of at least two studies42,43. Notably, in the present study, we found that compared to CD274 expression, PELI2 expression was more strongly associated with PD-L1 expression. PELI2 encodes a member of the E3 ubiquitin ligase family (PELI2) that plays regulatory roles in immune pathways44, including promotion of the ubiquitination of IRAK145. IRAK1 promotes the induction of PD-L1 expression by associating with MyD88 and TRAF6 through the IFN-γ and TLR signaling pathways46,47. Further research investigating a direct connection of PD-L1 with PELI2 is warranted.
Three limitations of this study must be considered. First, the different tumor components were separately investigated in approximately one-third of samples. Therefore, it was difficult to draw definite conclusions regarding the significance of the comparison of epithelial and sarcomatoid components within PPC. To our knowledge, however, the present study is the first study to separately analyze the PPC components, and thus the results may help to evaluate intratumoral heterogeneity and tumor evolution, which may affect the selection of treatment options. Second, this study lacked sufficient information regarding therapeutic efficacy of molecular targeted drugs and ICIs. Therapeutic efficacy data was not included because half of the tumors did not recur after surgery, and ICI or MET inhibitors have only recently been approved in Japan. Therefore, therapeutic efficacy of these drugs for PPC and its association with biomarkers, such as TMB, PD-L1, or PELI2 expression, should be confirmed in future clinical studies. Third, association between the RNA expression of each gene and RFS was evaluated using univariate analysis in the study because of the sample size; thus, the implications of the findings are limited. Other parameters, which affect RFS, such as lymphovascular invasion, and spread through alveolar spaces, should be included and assessed using multivariate analysis in large future cohort studies.
In conclusion, both epithelial and sarcomatoid components shared activating driver mutations, suggesting that these truncal mutations can be identified by testing either component, and that matched targeted therapy may be effective for PPC patients with druggable mutations. Moreover, there is a significant enrichment for MET exon 14 alterations in PPC, indicating that PPC tumors with negative DNA-based testing for a driver mutation need additional examination by RNA-based testing. Finally, the combination of cytotoxic chemotherapies and ICIs may represent an option for PPC cases without any druggable mutations when they harbor a sarcomatoid component that expresses high levels of PD-L1.
Methods
Samples
Tumor specimens were obtained from 52 patients with PPC (surgically resected samples from 49 patients and autopsy tumor samples from three patients) at four Japanese hospitals from 2005 through 2016. However, three cases were excluded because of poor DNA quality. All surgically resected samples were from chemotherapy-naïve patients. Pathological diagnoses were performed by the pathologists A. Ushiku, T. Hayashi, and S. Morita, according to the 4th edition of the World Health Organization Classification of Lung Tumors. Pathologic tumor-node-metastasis (TNM) staging was based on the 8th American Joint Committee on Cancer guidelines. The follow-up endpoint was December 31, 2017. The Institutional Review Board (IRB) of the National Cancer Center, Japan approved this study (research project number: 2015-202). Written informed consent was obtained from all participants except those we were unable to contact due to a loss to follow-up or death at registration. For these latter cases, the Institutional Review Board at each participating institution granted permission for the use of existing tissue samples for research purposes. None of the samples used in this study were obtained from patients who had opted out of study participation.
Genomic DNA extraction and sequencing
Genomic DNA was extracted from 12 fresh-frozen tissue samples of 12 patients using a QIAamp Fast DNA Tissue Kit (Qiagen, Hilden, Germany) and sheared using a Covaris LE220 (Covaris, Woburn, MA, USA). Adjacent normal lung fresh-frozen tissue samples were also extracted from each patient as a source of matched normal DNA. Whole-exome sequencing libraries were prepared from 1 µg of genomic DNA using the Agilent SureSelect Human All Exon Kit v6 (Agilent Technologies, Santa Clara, CA, USA) and sequenced using a HiSeq 2500 (Illumina, San Diego, CA, USA) with the paired-end option. Genomic DNA was extracted from 66 formalin-fixed, paraffin-embedded (FFPE) samples of 40 patients using a GeneRead DNA FFPE Kit (Qiagen). FFPE samples from adjacent normal lung tissue were also extracted from each patient as a source of matched normal DNA. The genomic DNA was separately extracted from 18 PPC samples of cores taken from the sarcomatoid and epithelial components (Fig. 1a). Genomic DNA was fragmented using a KAPA HyperPlus Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA), and 750 ng of each sample was subjected to target fragment enrichment using a custom target-capturing panel (SureSelectXT Custom Kit, Agilent Technologies). The Todai OncoPanel (TOP) included all exons of 465 cancer-relevant genes48. The target capture libraries were sequenced using a HiSeq 2500 with the paired-end option. Raw.fastq files were analyzed using FastQC v0.11.3, and the sequencing reads were mapped to the human reference genome GRCh38 using BWA, Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), and NovoAlign (http://www.novocraft.com/products/novoalign/). Samples in which <80% of bases were covered at a depth of 100× were considered low quality and excluded from the analyses. Somatic mutations were called using MuTect (http://www.broadinstitute.org/cancer/cga/mutect) and SomaticIndelDetector (http://www.broadinstitute.org/cancer/cga/node/87). Mutations were excluded if the variant allele frequency (VAF) was <10%, or the number of variant reads was <10. False-positive calls were discarded through visual inspection. Further, the heterogeneity and evolutionary trajectory between primary tumors and paired metastatic tumors were evaluated using the LICHeE method19, which was developed in 2015 to construct phylogenetic trees for multiple tumors according to the VAFs of somatic single-nucleotide variants. Using the TOP, the TMB was calculated as the total number of nonsynonymous and synonymous mutations divided by the length of the total target region (3.12 Mb).
RNA sequencing
Total RNA was isolated from 12 fresh-frozen tissue samples of 12 patients using RNA-Bee (Tel-Test, Gainesville, FL, USA) and purified using an RNeasy Mini Kit (Qiagen). After poly(A)-RNA selection, the library was prepared using 1 µg of each sample and an NEBNext Ultra Directional RNA Library Prep Kit (NEB, Ipswich, MA, USA) and sequenced using a HiSeq 2500 with the paired-end option. Total RNA was isolated from 66 FFPE samples of 40 patients using an RNeasy FFPE Kit (Qiagen) and purified using an RNeasy Mini Kit. RNAs from 18 patients were separately extracted from cores taken from the sarcomatoid and epithelial components. RNA quality was calculated using a 2200 TapeStation (Agilent Technologies). The synthesis of cDNAs and library preparation were performed using a TruSeq RNA Access Library Prep Kit (Illumina) and 300 ng of each sample. The libraries were sequenced using a HiSeq 2500 with the paired-end option. Raw.fastq files were analyzed using FastQC v0.11.3, and read mapping to the reference genome GRCh38 was performed using BWA, Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), and NovoAlign (http://www.novocraft.com/products/novoalign/). We excluded samples that did not meet the post-sequencing quality control criteria for a good-quality RNA-seq experiment, namely >50% of housekeeping gene regions (ACTB, B2M, GAPDH, HPRT1, HSP90AB1, PPIA, RPL13A, RPLP0, TFRC, and UBC) with >100× coverage. Gene fusions were detected using the deFuse pipeline (https://bitbucket.org/dranew/defuse)49. MET exon 14 skipping was detected by generating reference sequences of the 3ʹ junction of MET exon 13 and the 5ʹ junction of exon 15 and counting split reads that supported 60-mers of the junction.
RNA expression analysis
The expression level of each gene was calculated and normalized using the DESeq2 package (https://bioconductor.org/packages/release/bioc/html/DESeq2.html). Genes were excluded from our analyses if the maximum number of normalized counts of all samples was <20, or if ≥4 samples had normalized counts = 0. Heat maps of the expression data were created using the pheatmap package (https://cran.r-project.org/web/packages/pheatmap). Ward’s clustering method and correlation distances were used to generate hierarchical clusters of genes and samples from the heatmaps.
Gene-set enrichment analysis (GSEA)
The log fold-change in the expression level of each gene between the epithelial and sarcomatoid components was calculated using the DESeq2 package. All genes were ranked in descending order according to log fold-change values and analyzed using GSEA version 2.2.0. GSEA PreRanked software was used to calculate the normalized enrichment scores (NES) and false discovery rates (FDRs) of gene sets obtained from the MSigDB database, which are publicly available at http://www.broadinstitute.org/gsea/msigdb50. A gene set was considered significantly enriched if its NES had an FDR q-value < 0.01.
Immunohistochemistry (IHC)
FFPE sections (4-µm thick) were subjected to IHC using an antibody directed against the extracellular domain of human PD-L1 (clone 22C3; Dako, Glostrup, Denmark) with 1:50 dilution. The slides were stained using a Dako Autostainer Link 48 platform with an automated staining protocol validated for the PD-L1 IHC 22C3 pharmDx assay. The pathologists A. U. and T. H. used a light microscope to score the percentage of positive tumor cells in each sample.
Detection of EML4-ALK
We obtained the sequences of complete EML4-ALK transcripts from clinical specimens by subjecting total RNA extracted from fresh-frozen samples to reverse transcription with SuperScriptTM IV VILO (Thermo Fisher Scientific, Waltham, MA, USA) followed by PCR using PrimeSTAR HS DNA polymerase (Takara Bio, Shiga, Japan) for 35 cycles at 98 °C for 10 s, 60 °C for 5 s, and 72 °C for 4 min. The primers used were 5ʹ-GCTTGAATTCACTCTGTCGGTCCGCTGAATGAAG-3ʹ (sense) and 5ʹ-GAATACGCGTTCCCAAGGAAGAGAAGTGAGTGTG-3ʹ (antisense). The PCR products were sequenced using the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and analyzed using a 3730 ABI capillary electrophoresis system.
Focus formation assay
The cDNAs encoding GFP, ALK, or EML4-ALK were each inserted into the pcx4 retroviral plasmid. The recombinant plasmids were transduced together with packaging plasmids (Takara Bio) into human embryonic kidney (HEK) 293 T cells to produce recombinant retroviral particles. 3T3 cells grown in 6-well plates were infected with ecotropic recombinant retroviruses in the presence of 4 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO, USA) for 24 h. 3T3 cells expressing various mutant proteins were cultured for 2 weeks in Dulbecco’s modified Eagle’s medium-F12 supplemented with 5% bovine calf serum, 2 mmol/L glutamine, and 1% penicillin/streptomycin (all from Thermo Fisher Scientific). The cells were then stained with Giemsa solution to detect foci. HEK 293 T cells and 3T3 cells were purchased from the American Type Culture Collection (Manassas, VA, USA).
Statistical analysis
Univariate Cox regression analysis was performed to evaluate the correlation between the expression level of each gene and recurrence-free survival (RFS) of patients with pathological stages I and II PPC. Only genes with q-value < 0.05 were considered candidates in the correlation analysis. Recurrence-free survival (RFS) curves were generated using the Kaplan–Meier method and compared using the log-rank test. P < 0.05 indicates a significant difference. The log fold-change in the expression level of each gene between the epithelial and sarcomatoid components was evaluated using the Wald test, and differences with q < 0.05 indicate a significant difference. The correlation between the normalized count of each gene and the level of PD-L1 in each sample was calculated using Pearson’s correlation, and statistical significance was defined as q < 0.05. Furthermore, the correlations of the levels of PD-L1 with the levels of CD274 mRNA, which encodes PD-L1, and the TMB were evaluated using the Mann–Whitney test, and p < 0.05 indicates a significant difference. Statistical analyses were performed using the R platform (version 3.5.1; https://www.r-project.org/) and associated packages.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
We have deposited the raw sequencing data under accession number JGAS000297 in the Japanese Genotype-Phenotype Archive (http://trace.ddbj.nig.ac.jp/jga), which is hosted by the DNA Data Bank of Japan.
Code availability
No unreported custom computer code or algorithm was used to generate the results of this manuscript. The references of all source codes are included within Materials and Methods.
References
Yuki, T. et al. Pleomorphic carcinoma of the lung: a surgical outcome. J. Thorac. Cardiovasc. Surg. 134, 399–404 (2007).
Chang, Y. L., Lee, Y. C., Shih, J. Y. & Wu, C. T. Pulmonary pleomorphic (spindle) cell carcinoma: peculiar clinicopathologic manifestations different from ordinary non-small cell carcinoma. Lung Cancer 34, 91–97 (2001).
Travis, W. D. et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015).
Bae, H. M. et al. Palliative chemotherapy for pulmonary pleomorphic carcinoma. Lung Cancer 58, 112–115 (2007).
Mochizuki, T. et al. Pleomorphic carcinoma of the lung: clinicopathologic characteristics of 70 Cases. Am. J. Surg. Pathol. 32, 1727–1735 (2008).
Chang, Y. L., Wu, C. T., Shih, J. Y. & Lee, Y. C. EGFR and p53 status of pulmonary pleomorphic carcinoma: implications for EGFR tyrosine kinase inhibitors therapy of an aggressive lung malignancy. Ann. Surg. Oncol. 18, 2952–2960 (2011).
Kaira, K. et al. Pulmonary pleomorphic carcinoma: a clinicopathological study including EGFR mutation analysis. J. Thorac. Oncol. 5, 460–465 (2010).
Ushiki, A. et al. Genetic heterogeneity of EGFR mutation in pleomorphic carcinoma of the lung: response to gefitinib and clinical outcome. Jpn J. Clin. Oncol. 39, 267–270 (2009).
Zhang, J. et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 (2014).
Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017).
Miyoshi, T. et al. Genomic profiling of large-cell neuroendocrine carcinoma of the Lung. Clin. Cancer Res. 23, 757–765 (2017).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Kim, S. et al. Programmed death-1 ligand 1 and 2 are highly expressed in pleomorphic carcinomas of the lung: comparison of sarcomatous and carcinomatous areas. Eur. J. Cancer 51, 2698–2707 (2015).
Yarchoan, M., Hopkins, A. & Jaffee, E. M. Tumor mutational burden and response rate to PD-1 Inhibition. N. Engl. J. Med. 377, 2500–2501 (2017).
Goodman, A. M. et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 16, 2598–2608 (2017).
Pao, W. & Hutchinson, K. E. Chipping away at the lung cancer genome. Nat. Med. 18, 349–351 (2012).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Popic, V. et al. Fast and scalable inference of multi-sample cancer lineages. Genome Biol. 16, 91 (2015).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Román, M. et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol. Cancer 17, 33 (2018).
Liu, S. Y. et al. The unique characteristics of MET exon 14 mutation in Chinese patients with NSCLC. J. Thorac. Oncol. 11, 1503–1510 (2016).
Li, S. et al. Comprehensive characterization of oncogenic drivers in Asian lung adenocarcinoma. J. Thorac. Oncol. 11, 2129–2140 (2016).
Liu, X. et al. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J. Clin. Oncol. 34, 794–802 (2016).
Awad, M. M. et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-Met overexpression. J. Clin. Oncol. 34, 721–730 (2016).
Nahar, R. et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat. Commun. 9, 216 (2018).
de Bruin, E. C. et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256 (2014).
Redig, A. J. et al. Clinical and molecular characteristics of NF1-mutant lung cancer. Clin. Cancer Res. 22, 3148–3156 (2016).
Wang, J. et al. MiR-1976 knockdown promotes epithelial-mesenchymal transition and cancer stem cell properties inducing triple-negative breast cancer metastasis. Cell Death Dis. 11, 500 (2020).
Duan, Y. et al. Chromatin remodeling gene ARID2 targets cyclin D1 and cyclin E1 to suppress hepatoma cell progression. Oncotarget 7, 45863–45875 (2016).
Capecchi, M. R. & Pozner, A. ASPM regulates symmetric stem cell division by tuning cyclin E ubiquitination. Nat. Commun. 6, 8763 (2015).
Reiter, J. G. et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 361, 1033–1037 (2018).
Vignot, S. et al. Next-generation sequencing reveals high concordance of recurrent somatic alterations between primary tumor and metastases from patients with non-small-cell lung cancer. J. Clin. Oncol. 31, 2167–2172 (2013).
Fukusumi, T. et al. The NOTCH4- HEY1 pathway induces epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Clin. Cancer Res. 24, 619–633 (2018).
Kresse, S. H., Meza-Zepeda, L. A., Machado, I., Llombart-Bosch, A. & Myklebost, O. Preclinical xenograft models of human sarcoma show nonrandom loss of aberrations. Cancer 118, 558–570 (2012).
Park, R. et al. LNX1 contributes to tumor growth by down-regulating p53 stability. FASEB J. 33, 13216–13227 (2019).
Donnem, T. et al. Prognostic impact of platelet-derived growth factors in non-small cell lung cancer tumor and stromal cells. J. Thorac. Oncol. 3, 963–970 (2008).
Paulsson, J., Ehnman, M. & Östman, A. PDGF receptors in tumor biology: prognostic and predictive potential. Future Oncol. 10, 1695–1708 (2014).
Grothey, A. et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312 (2013).
van der Graaf, W. T. et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379, 1879–1886 (2012).
Goodman, A. M. et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 4, 1237–1244 (2018).
Straub, M. et al. CD274/PD-L1 gene amplification and PD-L1 protein expression are common events in squamous cell carcinoma of the oral cavity. Oncotarget 7, 12024–12034 (2016).
Moynagh, P. N. The roles of pellino E3 ubiquitin ligases in immunity. Nat. Rev. Immunol. 14, 122–131 (2014).
Liu, J. et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 110, 296–304 (2007).
Stickson, S. et al. Roles of the TRAF6 and pellino E3 ligases in MyD88 and RANKL signaling. Proc. Natl Acad. Sci. USA 114, E3481–E3489 (2017).
Schauvliege, R., Janssens, S. & Beyaert, R. Pellino proteins: novel players in TLR and IL-1R signaling. J. Cell Mol. Med 11, 453–461 (2007).
Kohsaka, S. et al. Comprehensive assay for the molecular profiling of cancer by target enrichment from formalin-fixed paraffin-embedded specimens. Cancer Sci. 110, 1464–1479 (2019).
McPherson, A. et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-seq data. PLoS Comput. Biol. 7, e1001138 (2011).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Acknowledgements
The authors would like to thank A. Maruyama-Shiino and H. Tomita for technical assistance. This study was supported by the grants from the Program for Integrated Database of Clinical and Genomic Information under grant number JP19kk0205003, the Leading Advanced Projects for Medical Innovation (LEAP) under grant number JP18am0001001, the Project for Cancer Research And Therapeutic Evolution (P-CREATE) under grant number JP20cm0106502, and the Practical Research for Innovative Cancer Control under grant number JP20ck0106536 from the Japan Agency for Medical Research and Development, AMED.
Author information
Authors and Affiliations
Contributions
S.K. and H.M. conceived the project and designed the study. M.N., S.K. and H.M. developed the methodology. M.N. and S.K. performed the experiments. M.N., T.U., and S.K. analyzed and interpreted the data. A.U., T.H., K.T., S.M., T.M., S.T., T.S., Y.O., F.S., H.K., Y.N., K.S. and J.N. provided administrative, technical, or material support. T.U., S.K., A.U., T.H. and S.M. provided experimental and analytical support. M.N., S.K. and H.M. wrote and edited the manuscript with feedback from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagano, M., Kohsaka, S., Hayashi, T. et al. Comprehensive molecular profiling of pulmonary pleomorphic carcinoma. npj Precis. Onc. 5, 57 (2021). https://doi.org/10.1038/s41698-021-00201-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00201-3
This article is cited by
-
PIK3CA mutations associated with a poor postoperative prognosis in patients with pulmonary pleomorphic carcinoma: a retrospective cohort study
BMC Cancer (2022)
-
Ringed fluorodeoxyglucose uptake predicted poor prognosis after resection of pulmonary pleomorphic carcinoma
Journal of Cardiothoracic Surgery (2022)
