
Abstract
Characterisation of genomic variation among corals can help uncover variants underlying trait differences and contribute towards genotype prioritisation in coastal restoration projects. For example, there is growing interest in identifying resilient genotypes for transplantation, and to better understand the genetic processes that allow some individuals to survive in specific conditions better than others. The coral species Pocillopora acuta is known to survive in a wide range of habitats, from reefs artificial coastal defences, suggesting its potential use as a starter species for ecological engineering efforts involving coral transplantation onto intertidal seawalls. However, the intertidal section of coastal armour is a challenging environment for corals, with conditions during periods of emersion being particularly stressful. Here, we scanned the entire genome of P. acuta corals to identify the regions harbouring single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) that separate intertidal colonies (n = 18) from those found in subtidal areas (n = 21). Findings revealed 74,391 high quality SNPs distributed across 386 regions of the P. acuta genome. While the majority of the detected SNPs were in non-coding regions, 12% were identified in exons (i.e. coding regions). Functional SNPs that were significantly associated with intertidal colonies were found in overrepresented genomic regions linked to cellular homeostasis, metabolism, and signalling processes, which may represent local environmental adaptation in the intertidal. Interestingly, regions that exhibited CNVs were also associated with metabolic and signalling processes, suggesting P. acuta corals living in the intertidal have a high capacity to perform biological functions critical for survival in extreme environments.
Similar content being viewed by others

Introduction
Many coral reef ecosystems worldwide are threatened by both impacts of climate change and unprecedented rates of urbanization1,2. Coastal development has resulted in extensive transformation of natural shorelines through the installation of artificial structures, such as seawalls and other defences, to prevent coastal inundation3. As the intertidal surfaces of seawalls are typically species-poor, there is a growing interest in enhancing the ecological value of these structures via transplantation of reef-building corals4,5,6. However, the seawall environment in the intertidal zone is generally suboptimal for hard coral establishment7,8,9. In particular, the conditions during periods of emersion, with associated exposure to desiccation risk and high light levels and temperatures, can be especially detrimental for corals10,11.
Spatial structuring of hard coral communities is generally attributed to the surrounding environment in which presence or absence of species reflects their tolerance limits12,13. This is commonly exhibited as depth ranges, but also applies to salinity, wave energy, and sediment tolerances9,14,15,16. It is assumed that organisms living in extreme environments are more stress-resistant17,18. While previous reports have documented rich coral communities thriving in intertidal nearshore habitats8,9,14, few studies have attempted to determine how these intertidal corals survive such stressful conditions. In particular, Zhang et al.19 have shown that selective sweeps occur among intertidal coral populations in which gene ontology categories associated with regulation of apoptosis were significantly enriched.
Generally, intertidal species exhibit various massive morphological forms (e.g. massive, encrusting, foliose) but branching corals, including acroporids and pocilloporids, also exist in these areas9,15,20. In particular, the pocilloporid species Pocillopora acuta can inhabit a relatively wide range of habitats, including the upper subtidal and lower intertidal zones20,21,22,23. In Singapore, this species has been observed growing well in atypical coral habitats such as the base of seawalls and the sides of marina pontoons24. However, they exhibit among-genotype variation in physiological and molecular responses to changing conditions4,25,26,27. For instance, a previous experiment showed this species can withstand 2 h of emersion, but only certain genotypes were able to survive longer emersion periods4. This indicates substantial intraspecific variability in stress tolerance in P. acuta that is likely driven by genetic variation among genotypes.
To date, most studies on genome-wide DNA polymorphisms in corals aimed to determine population structure and genetic connectivity28,29,30,31 while the structural and functional effects of these variants are rarely examined. Nevertheless, some research has highlighted genomic signatures of adaptation in corals to challenging conditions [e.g. 32,33,34. In particular, Bay and Palumbi33 showed temperature adaptation in Acropora hyacinthus colonies living in a backreef pool that experiences highly-variable temperature. They found signals of selection on coding regions that interact with heat-shock proteins, as well as those that involved in antioxidant defense and apoptosis. Another study on Acropora corals by Cooke et al.32 showed selective sweeps for colonies found in inshore habitats of the Great Barrier Reef. The sweep loci are associated with osmotic regulation and skeletal development, which were linked to challenging water quality conditions (e.g. hyposaline) surrounding inshore coral populations.
Expression levels of genes are known to be influenced by many factors including the structural location and nature of DNA polymorphisms within the genome, which in turn can affect the associated functions that control various biological processes35. Generally, such inferences are obtained by identifying single nucleotide polymorphisms (SNPs), however, more recently, copy number variation (CNV) of putative genes has also emerged as an important genetic mechanism that can ultimately influence an organism's adaptive capacity to environmental change36. For example, some hypoxia-associated genes were identified to exhibit large copy variation among colonies of the massive Porites lutea, which is known to tolerate highstress environments such as the intertidal zone37. Although contributions of CNVs and SNPs to phenotypic variation are largely independent, they may act in concert with each other38,39. While knowledge about the effect of both types of variation co-existing in the same region is limited, it has been shown that co-existent CNVs can magnify gene-trait association power of SNPs40.
Studies aiming to identify genomic variations and their corresponding biological functions are fundamental to uncovering variants underlying trait differences among and within-coral species. Findings can also uncover genes that are potentially under selection which can suggest adaptation to certain environments41. Here, we hypothesize that (1) there is significant genomic variation, in the form of single nucleotide polymorphisms (SNPs), that differentiates intertidal and subtidal P. acuta colonies and, (2) identified regions harbouring SNPs also exhibit CNVs. Lastly, we aim to characterize the functional relevance of these genomic variations (i.e. SNPs and CNVs) in the context of the intertidal/subtidal environments they were sampled from.
Materials and methods
Sample collection
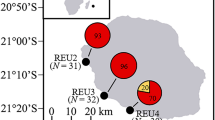
Coral nubbins (~ 2 cm size) from different colonies of Pocillopora acuta found on the intertidal seawalls at Pulau Hantu (n = 8), Kusu (n = 5) and Sentosa (n = 5) (Fig. 1) were collected in duplicate during low tide periods. Colonies of P. acuta were identified following Poquita-Du et al.42. Specifically, these corals were collected at the base of the seawalls and are submerged during high tide up to a depth of 2 to 3 m, depending on the location43. For P. acuta colonies in subtidal areas (~ 6 m depth), duplicate fragments were collected from different colonies at Kusu (n = 12) and another site: Raffles Lighthouse (n = 9) underwater using SCUBA as they are fully-submerged even during low tide. Collection sites and number of samples depended on the presence of P. acuta and colonies having approximately the same maximum diameter size (~ 20 cm).

Map showing all sites within Southern Islands of Singapore where P. acuta colonies were collected from intertidal (Pulau Hantu, Kusu Island, Sentosa Island) and subtidal (Kusu, Raffles Lighthouse) areas.
All 78 coral nubbins (samples hereafter) of P. acuta (39 colonies × 2 replicates) were immediately fixed in RNAlater and kept in an icebox while being transported to the laboratory. All samples were kept in a − 80 °C freezer until DNA extraction (conducted within a week of collection).
DNA isolation, whole genome sequencing and data quality check
Genomic DNA (gDNA) was extracted using Qiagen DNeasy Blood and Tissue DNA extraction kit following the manufacturer’s protocol with an extra step of RNA removal using RNase solution (Promega). The procedures for library preparation and sequencing followed Azenta Life Sciences (China). Briefly, the library preparation was carried out using ND607 VAHTS Universal Library Kit V3 following the manufacturer’s protocol. For each sample, 100 ng genomic DNA was randomly fragmented to < 500 bp by sonication (Covaris S220). The fragments were treated with End Prep Enzyme Mix for end repairing, 5’ phosphorylation and dA-tailing in one reaction, followed by a T-A ligation to add adaptor to both ends. Size selection of adaptor-ligated DNA was then performed using beads and fragments of ~ 470 bp (with approximate insert size of 350 bp) were recovered. Each sample was then amplified by polymerase chain reaction (PCR) using P5 and P7 primers, with both primers carrying sequences which can anneal with flow cell to perform bridge PCR and, P5/P7 primer carrying index allowing for multiplexing. The PCR products were subsequently purified using magnetic beads, validated using an Qsep 100 (Bioptic, Taiwan, China), and quantified by Qubit 3.0 Fluorometer (Invitrogen, Carlsbad, California, USA).
The libraries with different indices were multiplexed and paired-end (pe) sequencing was performed on the Illumina NovaSeq 6000 platform according to the manufacturer’s instructions (Illumina, San Diego, California, USA) to generate 150 bp pe reads (30 × coverage). Image analysis and base calling were conducted using the NovaSeq Control Software (NCS) + OLB _ GAPipeline-1.6 on the NovaSeq instrument. Adapter sequences, PCR primers, content of N bases more than 10% and bases of quality lower than 20 were removed with cutadapt (V1.9.1)44.
Sequence alignment and SNP detection
An annotated draft genome of P. acuta45 was used as the reference genome assembly in this study. While the P. acuta genome assembled by Vidal-Dupiol et al.45 was based on Illumina short-read sequencing, the quality is acceptable (BUSCO completeness of 89.4% and 91.4% based on Metazoa and Eukaryota datasets, respectively) and it was the only available P. acuta genome during the analysis. In support, the colony used to generate the P. acuta genome by Vidal-Dupiol et al.45was collected from the same region (i.e. Indonesia) not far from where our samples were collected (Singapore), therefore, it is an ideal reference genome for our study.
Sequence alignment was performed by individually mapping pe reads for all samples against the reference assembly using Burrowers-Wheeler Aligner (BWA, v 0.7.13) maximal exact match (mem) algorithm46,47. The reference assembly was indexed prior to mapping with sample sequences using the function ‘bwa index’. Unmapped and duplicate mapped reads were subsequently removed using SAMtools (v 1.10)48. Initial identification of genotype likelihoods at each genomic position, variant calling and subsequent normalization of variants were performed using bcftools v1.1149. The outputs were filtered to retain SNPs using Plink50. Further, SNPs that showed extended linkage disequilibrium (LD) in which a correlated (r2 = 0.5) pair of SNPs within a window of 50 kb were removed from the data set. In addition, markers that showed extensive deviation from Hardy–Weinberg equilibrium (HWE; p < 0.001), a missing rate of 1%, and low minor allele frequency (MAF) of 5% were excluded.
Examining population structure and admixture proportions
To examine the genetic divergence between P. acuta colonies from two tidal environments, the fixation index (FST) was estimated in Plink using the final output of SNPs generated above. Principal component analysis (PCA) was also performed in Plink and plotted in R51 to visualize the genetic structure of P. acuta colonies from different sites and tidal environment. Additionally, we assessed the proportions of ancestry for up to five potential K values, with computations of cross validation (CV) errors, using Admixture v1.3.052 and visualized using custom python scripts.
Association analysis and annotation of SNPs
Identification of specific regions harbouring SNPs that were significantly associated with intertidal vs. subtidal P. acuta colonies was performed using Plink53. Allele frequencies of quality-filtered SNPs were compared between intertidal and subtidal colonies using Fisher’s exact test to generate p values. SNPs with association p values of ≤ 0.01 were considered significantly associated with intertidal P. acuta colonies50,54,55. Results were visualized with a Manhattan plot using ggplot2 and qqman packages in R51,56,57.
Analysis for the distribution of all SNPs across genomic regions and structural annotation were performed using snpEff58. The output of the annotation run included translated protein sequences which were filtered to regions where SNPs were significantly associated with intertidal colonies using the sequence processing tool, ‘mothur’59. The output list of protein sequences obtained from the previous step were then aligned against the most updated Swissprot/Uniprot database using NCBI Blast (‘blastp’ command). Gene ontology (GO) enrichment analysis was subsequently performed using BiNGO (V 3.0.5; https://www.psb.ugent.be/cbd/papers/BiNGO/Home.html) available in Cytoscape (V 3.9.1, http://www.cytoscape.org/)60,61 by performing Hypergeometric test with Benjamin and Hochberg False Discovery Rate (FDR) multiple testing correction (significance level = 0.05) on the set of genes from the blastp output.
Orthology and copy number variation
Orthogroups were predicted to help determine whether genomic variations that differentiate intertidal and subtidal P. acuta colonies were also driven by CNVs. OrthoFinder v2.5.4, a tool that implements the phylogenetic orthology inference method62,63, was used to predict orthogroups. Sequences from protein-coding regions harbouring all variants, including SNPs, insertions, and deletions, for both tidal environments (i.e. intertidal and subtidal) were used for the orthology analysis (default settings). Orthogroups that exhibited CNVs, particularly those that were present in both environments, were identified and the corresponding protein sequences belonging to these orthogroups were retrieved for alignment process. Using the BLAST built-in tool (blastp) in CLC Genomics Workbench (V 10.0.1), protein sequences from the different orthogroups were aligned against the most recent version of the UniProt SwissProt protein database (accessed 14 April 2022). BLAST hits that showed the highest number of HSP (high-scoring segment pair) were used to search for corresponding gene IDs and putative functions on UniProtKB64.
Results
Sequencing and alignment
Illumina sequencing resulted in an average of 56.1 million pe raw reads per sample (Supplementary data). After removal of low quality and adapter reads, 55.5 million pe reads per sample were retained, with 38.76% GC content and Q phred scores of 97% Q20. For each sample, an average of ~ 96% of quality-filtered sequences mapped to the P. acuta draft genome45.
SNP detection and annotation
Overall, there were 29,619,409 variants obtained from the variant detection analysis, with average genotyping rate of approximately 99%. These variants consisted of 25,773,468 SNPs, 1,345,237 insertions, and 2,500,704 deletions. Following a series of stringent filtering to obtain independent and high-quality SNPs, only 74,391 SNPs were retained. These SNPs were detected in varying locations within 386 genomic regions, of which 87.31% were categorized as modifiers (Fig. 2a), which usually are variants affecting non-coding genes. The SNPs were found in different regions within the P. acuta genome (Fig. 2b), including ~ 29% within introns and only ~ 12% in functional regions (i.e. exons).

Structural annotation showing percentage of SNP effects by impact categories (a) and distribution of SNPs in different genomic regions (b).
Population structure and admixture proportions
Results from both PCA and Admixture (Figs. 3a and 4, respectively) showed no structuring based on site and tidal environments. FST values for all identified SNPs across various genomic positions (mean Global FST = 0.02) were generally low (Fig. 3b), indicating low genetic differentiation between P. acuta colonies found in the intertidal and subtidal environment.

Genetic structure of Pocillopora acuta based on (a) Principal component analysis at the colony level and, (b) genetic divergence among colonies between the two tidal environments, showing estimates of FST for each identified SNP across various genomic positions.

Proportion of ancestry calculated using Admixture, showing up to five potential K values and the corresponding CV errors in brackets.
Association analysis
Out of 386 regions that harbour SNPs, 134 of them were found to be significantly associated with intertidal colonies (p values < 0.01; Fig. 5). A complete inventory of all the regions with SNPs and their specific locations are provided in the Supplementary Data.

Manhattan plot showing allele frequencies of quality-filtered SNPs from different chromosomes. SNPs that were highly associated with intertidal colonies (p values of < 0.01) are indicated by the red horizontal line.
Gene ontology enrichment analysis for intertidal-associated SNPs
While the majority of detected SNPs highly associated with intertidal P. acuta colonies were observed in introns, some were found in exons and hence predicted to be functional regions. However, only a small portion (~ 28%) of these intertidal-associated SNPs could be annotated based on homologs from well-studied taxa which were used to search for GO terms under biological processes categories. Significant overrepresentation of GO was observed for genes that are associated with cellular homeostasis, metabolism, and signalling processes (Fig. 6; also refer to Supplementary data). Due to the interdependency of GO categories, whole branches for each GO hierarchy are shown in Fig. 6, but only coloured nodes that are farthest down the network were considered relevant (refer to60). In particular, the overrepresentation of categories—cellular homeostasis, metabolism, and signalling process—reflect the presence of genes associated with responses to glucose stimulus, fatty-acid beta-oxidation, and regulation of mitogen-activated protein kinase (MAPK) and nuclear factor-kappa B (NF-kappaB) cascades.

Gene ontology enrichment analysis of genes significantly associated with intertidal P. acuta colonies, showing a gene network with significantly overrepresented genes (shaded nodes; cut-off p value 0.05) under the biological processes category.
Orthology and copy number variation
OrthoFinder assigned 93.4% of the total number of genes (11,911 out of 12,755) into 2665 orthogroups, 570 of which were single copy orthogroups. Only genes that showed copy number differences > 2 between the two tidal environments are summarized in Table 1, with corresponding protein matches and putative functional profiles. The complete set of orthogroups that exhibit CNVs can be found in the Supplementary data.
Discussion
The environmental conditions surrounding intertidal areas are generally suboptimal for coral survival8,11, but several species have been observed to establish at the lower intertidal section of seawalls in Singapore including the common branching coral, Pocillopora acuta9,15. Here, we identified genomic regions harbouring SNPs that distinguish intertidal from subtidal P. acuta colonies. We also identified the presence of CNVs in all regions harbouring variants. The functional relevance of these regions, particularly coding regions, was further examined to gain insights into the possible involvement of these SNPs and CNVs in biological processes. Our results revealed that the genomic regions differentiating intertidal and subtidal colonies are mainly associated with cellular homeostasis, metabolism, and signalling processes, suggesting these are likely signatures of adaptation to extreme environments.
To account for population stratification and genetic ancestry, we examined the genetic structure and proportions of ancestry among P. acuta colonies studied here and found that there is no structuring based on site and tidal environment. While the five population divisions inferred when K = 5 had the lowest CV error, they do not coincide with the site or environmental groupings, implying that the limited structuring observed was not associated with these a priori subdivisions. Our observations align with findings in Afiq-Rosli et al.28, which also showed no clear among-site separation for P. acuta corals collected from reef sites within the Southern Islands of Singapore, including the collection sites in the present study [i.e., Hantu, Kusu, Satumu (Raffles Lighthouse)].
We found enrichment of genomic regions with SNPs that are linked to cellular homeostasis for intertidal P. acuta colonies. In particular, overrepresentation of responses to glucose stimulus was detected, likely due to corals relying on their endosymbionts for most of their daily energy budget through translocation of photosynthetic products (i.e. glucose)65. While photosynthesis is critical for corals to survive, it can contribute to the production of reactive oxygen species (ROS) which are known to be toxic for coral hosts and therefore need to be regulated to achieve homeostasis in the coral holobiont for efficient cellular functioning66,67. This is especially challenging for intertidal corals as they are exposed to high solar radiation and elevated temperatures for longer periods of time, which can destabilize the photosynthetic electron-transport chain of the endosymbiotic dinoflagellates, resulting in increased production of ROS67,68,69. Relatedly, there was a remarkably high number of gene copies coding for Protein O-mannosyl-transferase in the intertidal corals (Table 1). While the role of this protein is not known for corals, the function of O-mannosylation has been described for Saccharomyces cerevisae where it plays a role in solubilization of misfolded proteins during stressful conditions70. When there is an accumulation of misfolded proteins, the unfolded protein response (UPR) is triggered, however, prolonged UPR activation can cause production of ROS71. It is widely known that high levels of ROS can cause DNA damage which is detrimental to many organisms including corals67,72,73. Thus, high numbers of copies of genes coding for Protein O-mannosyl-transferase, which are protective against cell damage, may be beneficial for intertidal corals which are frequently exposed to extreme conditions.
Further, significant overrepresentation of genes associated with signalling pathways, MAPK and NF-kappaB were also detected in regions with SNPs associated with intertidal colonies. These pathways have been linked previously to oxidative stress in corals73,74. For instance, a significant activation of c-Jun N-terminal kinase (JNK, one of the major subgroups of MAPK gene family) has been shown to repress oxidative stress in Stylophora pistillata corals exposed to ultraviolet radiation (UVR) and thermal stress75. Similarly, genes associated with signal-transduction also displayed a relatively high number of copies for intertidal corals, particularly genes linked to Palmitoyltransferase specific to casein kinase 1 (AKR1) as well as calcium-dependent cell–cell adhesion via plasma (stan) and calcium ion export across the plasma membrane (Atp2b3). This suggests a highly coordinated signalling response to enable intertidal corals to adapt to extreme environments.
As with many other animals, metabolic activities in corals are known to be dynamic, responding to diel variations in environmental conditions such as light availability. For example, during daytime the coral’s diffusive boundary layer (DBL) can experience hyperoxia due to high photosynthetic activity76. Further, intense solar radiation can cause photoinhibition to endosymbionts, reducing their capacity to provide photosynthetically-fixed carbon to the coral host65. Our results show significant enrichment of metabolic processes associated with fatty acid beta-oxidation for intertidal corals. Fatty acids are important components of lipids in a wide range of living organisms, including corals77. Storage lipids play a crucial role in a coral’s metabolism, functioning as reserves to generate energy for survival during periods of adverse conditions78,79. A coral’s lipid stores can comprise up to ~ 40% of its total biomass, representing a substantial quantity of energy reserves80,81. The high occurrence of SNPs in metabolism-associated regions observed here implies a relatively high utilization of lipids for intertidal P. acuta corals, corroborating recent findings on metabolic plasticity of this species to withstand extreme environment82.
Overall, there is clear genomic variation within the species P. acuta living in two distinct tidal zones, suggesting population differentiation associated with these distinct environments. The genome-wide SNPs and CNVs reported here provide important insights into the biological functions (cellular homeostasis, metabolism, and signalling) that contribute to the capacity of P. acuta to withstand extreme conditions in the intertidal zone, but additional research on the expression of quantitative trait loci which involve direct association tests is required to confirm this. For instance, assessing lipid levels in colonies inhabiting two tidal environments can provide insights into the role of lipid metabolism in local acclimatization to different conditions. It is important to mention that this study is limited by the absence of P. acuta corals in the subtidal or intertidal area for some sites, thus, there is an unbalanced representation of the two tidal environments. Nevertheless, the genomic resources generated in this study can help facilitate discovery of functional variants associated with traits (e.g. stress tolerance) which can contribute towards active conservation efforts such as coral transplantation and selective breeding.
Data availability
Raw sequence reads and metadata are deposited at NCBI under BioProject PRJNA812628. Supplementary data supporting this article can be found online at https://zenodo.org/doi/10.5281/zenodo.6318484.
References
Hoegh-Guldberg, O. et al. Coral reefs under rapid climate change and ocean acidification. Science 318, 1737–1742 (2007).
Heery, E. C. et al. Urban coral reefs: degradation and resilience of hard coral assemblages in coastal cities of East and Southeast Asia. Mar. Pollut. Bull. 135, 654–681 (2018).
Loke, L. H. L., Heery, E. C. & Todd, P. A. Artificial shorelines. In World Seas: an Environmental Evaluation, Vol III: Ecological Issues and Environmental Impacts (ed. Sheppard, C.) 491–504 (Elsevier Limited, 2019).
Pang, H. E. et al. Among-genotype responses of the coral Pocillopora acuta to emersion: implications for the ecological engineering of artificial coastal defences. Marine Environ. Res. 168, 105312 (2021).
Yong, C. L. X. et al. Emersion-associated responses of an intertidal coral and its suitability for transplantation to ecologically engineer seawalls. J. Marine Sci. Eng. 9, 1096 (2021).
Ng, C. S. L. et al. Enhancing the biodiversity of coastal defence structures: Transplantation of nursery- reared reef biota onto intertidal seawalls. Ecol. Eng. 82, 480–486 (2015).
Dandan, S. S. et al. Resilience of coral calcification to extreme temperature variations in the Kimberley region, northwest Australia. Coral Reefs 34, 1151–1163 (2015).
Richards, Z. T. et al. A diverse assemblage of reef corals thriving in a dynamic intertidal reef setting (Bonaparte Archipelago, Kimberley Australia. PLOS One 10, 1–18 (2015).
Lee, Y.-L. et al. Composition and structure of tropical intertidal hard coral communities on natural and man-made habitats. Coral Reefs 40, 685–700 (2021).
Firth, L. B. et al. Biodiversity in intertidal rock pools: Informing engineering criteria for artificial habitat enhancement in the built environment. Marine Environ. Res. 102, 122–130 (2014).
Castrillón-Cifuentes, A. L., Lozano-Cortés, D. F. & Zapata, F. A. Effect of short-term subaerial exposure on the cauliflower coral, Pocillopora damicornis, during a simulated extreme low-tide event. Coral Reefs 36, 401–414 (2017).
Chow, G. S. E. et al. Light limitation selects for depth generalists in urbanised reef coral communities. Marine Environ. Res. 147, 101–112 (2019).
Muir, P. R. et al. Limited scope for latitudinal extension of reef corals. Science 348, 1135–1138 (2015).
Ng, C.S.L., D. Chen, and L.M. Chou, Hard coral assemblages on seawalls in Singapore in Contributions to Marine Science, K.S. Tan, Editor. 2012, National University of Singapore: Singapore. p. 75–79.
Kikuzawa, Y. P. et al. Diversity of subtidal benthic and hard coral communities on sloping and vertical seawalls in Singapore. Mar. Biodivers. 50(95), 1–14 (2020).
Bongaerts, P. et al. Deep reefs are not universal refuges: Reseeding potential varies among coral species. Sci. Adv. 3(2), e1602373 (2017).
Davison, I. R. & Pearson, G. A. Stress tolerance in intertidal seaweeds. J. Phycol. 32, 197–211 (1996).
de Pedro, R. S., Niell, F. X. & Carmona, R. Close but distant: Emersion promotes ecophysiological differentiation between two rhodophytes within an estuarine intertidal zone. J. Exp. Marine Biol. Ecol. 547, 151664 (2022).
Zhang, J. et al. Evolutionary responses of a reef-building coral to climate change at the end of the last glacial maximum. Mol. Biol. Evol. 39, 10. https://doi.org/10.1093/molbev/msac201 (2022).
Shinzato, C. et al. Genome-wide SNP analysis explains coral diversity and recovery in the Ryukyu Archipelago. Sci. Rep. 5, 18211 (2015).
Schmidt-Roach, S. et al. With eyes wide open: a revision of species within and closely related to the Pocillopora damicornis species complex (Scleractinia; Pocilloporidae) using morphology and genetics. Zool. J. Linnean Soc. 170(1), 1–33 (2014).
Baums, I. B. et al. Marginal coral populations: the densest known aggregation of Pocillopora in the Galápagos Archipelago is of asexual origin. Front. Marine Sci. 1, 59 (2014).
Ros, M. et al. Symbiont shuffling across environmental gradients aligns with changes in carbon uptake and translocation in the reef-building coral Pocillopora acuta. Coral Reefs 40, 595–607 (2021).
Chou, L.M., et al., Hidden havens: exploring marine life in Singapore’s Marinas. 2020, Singapore: National Parks Board. 99.
Fong, J., Poquita-Du, R. C. & Todd, P. A. Plastic responses in the coral Pocillopora acuta to extreme low-light conditions with and without food provision. Marine Biol. 168, 113 (2021).
Poquita-Du, R. C. et al. Short term exposure to heat and sediment triggers changes in coral gene expression and photo-physiological performance. Front. Marine Sci. 6, 121 (2019).
Poquita-Du, R. C. et al. Gene expression and photophysiological changes in Pocillopora acuta coral holobiont following heat stress and recovery. Microorganisms 8, 1227 (2020).
Afiq-Rosli, L. et al. Barriers and corridors of gene flow in an urbanized tropical reef system. Evolut. Appl. 14, 2502–2515 (2021).
Rosser, N. L. et al. Phylogenomics provides new insight into evolutionary relationships and genealogical discordance in the reef-building coral genus Acropora. Proc. Royal Soc. B Biol. Sci. 284, 20162182 (1846).
Thomas, L. et al. Spatially varying selection between habitats drives physiological shifts and local adaptation in a broadcast spawning coral on a remote atoll in Western Australia. Sci. Adv. 8(17), eabl9185 (2022).
Underwood, J. N. et al. Extreme seascape drives local recruitment and genetic divergence in brooding and spawning corals in remote north-west Australia. Evolut. Appl. 13, 9 (2020).
Cooke, I. et al. Genomic signature in the coral holobiont reveal host adaptations driven by Holocene climate change and reef specific symbionts. Sci. Adv. 6, eabc6318 (2020).
Bay, R. A. & Palumbi, S. R. Multilocus adaptation associated with heat resistance in reef-building corals. Curr. Biol. 24(24), 2952–2956 (2014).
Fuller, Z. L. et al. Population genetics of the coral Acropora millepora: Toward genomic prediction of bleaching. Science 369, eaba4674 (2020).
Jin, Y. K. et al. Genetic markers for antioxidant capacity in a reef-building coral. Sci. Adv. 2, 150084 (2016).
Dorant, Y. et al. Copy number variants outperform SNPs to reveal genotype-temperature association in a marine species. Mol. Ecol. 29(24), 4765–4782 (2020).
Alderdice, R. et al. Deoxygenation lowers the thermal threshold of coral bleaching. Sci. Rep. 12, 18273 (2022).
Momtaz, R. et al. Integrated analysis of SNP, CNV and gene expression data in genetic association studies. Clin. Genet. 93(3), 557–566 (2017).
Goymer, P. Genomic variation: copy number variation doesn’t copy SNPs. Nat. Rev. Genet. 8, 247 (2007).
Liu, J. et al. The coexistence of copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) at a locus can result in distorted calculations of the significance in associating SNPs to disease. Human Genet. 137, 553–567 (2018).
Thomas, L. et al. Spatially varying selection between habitats drives physiological shifts and local adaptation in a broadcast spawning coral on a remote atoll in Western Australia. Sci. Adv. 8, eabl9185 (2022).
Poquita-Du, R. C. et al. New evidence shows that Pocillopora ‘damicornis-like’ corals in Singapore are actually Pocillopora acuta (Scleractinia:Pocilloporidae). Biodiv. Data J. 5, 11407 (2017).
Ng, D., et al., Antagonistic effects of seawalls and urban sedimentation on epilithic algal matrix (EAM)-feeding fishes. Marine Pollut. Bull., 2021. 173.
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17(1), 10 (2011).
Vidal-Dupiol, J., et al., Sequencing, de novo assembly and annotation of the genome of the sceleractinian coral, Pocillopora acuta. BioRxiv, 2020.
Li, H., Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, 2013.
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009).
Danecek, P., et al., Twelve years of SAMtools and BCFtools. GigaScience, 2021. 10(2).
Purcell, S. et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Amer. J. Human Genet. 81(3), 559–575 (2007).
R Core Team, R language and environment for statistical computing. 2022, R Foundation for Statistical Computing: Vienna, Austria.
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Purcell, S. et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Amer. J. Human Genet. 81, 559–575 (2007).
Lee, J. et al. Exact association test for small size sequencing data. BMC Med Genom. 11, 30 (2018).
Llinares-Lo ́Pez, F., et al. Genome-wide detection of intervals of genetic heterogeneity associated with complex traits. Bioinformatics 31, i240–i249 (2015).
Wickham, H. ggplot2: Elegant graphics for data analysis (Springer-Verlag, 2016).
Turner, S., qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. The Journal of Open Source Software, 2018.
Cingolani, P. et al. A program for annotaing and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w118; iso-2; iso-3. Fly 6(3), 80–92 (2012).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75(23), 7537–7541 (2009).
Maere, S., Heymans, K. & Kuiper, M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 (2005).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
The Uniprot Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids 49(D1), D480–D489 (2020).
Roth, M. S. The engine of the reef: photobiology of the coral-algal symbiosis. Front. Microbiol. 5, 422 (2014).
Laurent, J. et al. The influence of photosynthesis on host intracellular ph in scleractinian corals. J. Exper. Biol. 216, 1398–1404 (2013).
Downs, C. A. et al. Oxidative stress and seasonal coral bleaching. Free Radic. Biol. Med. 33(4), 533–543 (2002).
Lesser, M. P. Elevated temperatures and ultraviolet radiation cause oxidative stress and inhibit photosynthesis in symbiotic dinoflagellates. Limnol. Oceanogr. 41, 271–283 (1996).
Reynolds, J. M. C. et al. Enhanced photoprotection pathways in symbiotic dinoflagellates of shallow water corals and other cnidarians. Proc. Nat. Acad. Sci. USA 105, 13674–13678 (2008).
Lommel, M. & Strahl, S. Protein O-mannosylation: conserved from bacteria to humans. Glycobiology 19(8), 816–828 (2009).
Haynes, C. M., Titus, E. A. & Cooper, A. A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15(5), 767–776 (2004).
Tchernov, D. et al. Apoptosis and the selective survival of host animalsfollowing thermal bleaching in zooxanthellate corals. Proc. Nat. Acad. Sci. USA 108(24), 9905–9909 (2011).
DeSalvo, M. K. et al. Transcriptomic responses to heat stress and bleaching in the elkhorn coral Acropora palmata. Marine Ecol. Progress Series 402, 97–113 (2010).
Pinzón, J.H., et al., Whole transcriptome analysis reveals changes in expression of immune-related genes during and after bleaching in a reef-building coral Royal Society Open Science, 2015.
Courtial, L. et al. The c-Jun N-terminal kinase prevents oxidative stress induced by UV and thermal stresses in corals and human cells. Sci. Rep. 7, 45713 (2017).
Linsmayer, L. B. et al. Dynamic regulation of coral energy metabolism throughout the diel cycle. Sci. Rep. 10, 19881 (2020).
Latyshev, N. A. et al. Fatty acids of reef-building corals. Marine Ecol. Prog. Series 76, 295–301 (1991).
Harriott, V. J. Coral lipids and environmental stress. Environ. Monitor. Assess. 25(2), 131–139 (1993).
Rodrigues, L. J. & Grottoli, A. G. Energy reserves and metabolism as indicators of coral recovery from bleaching. Limnol. Oceanogr. 52, 1874–1882 (2007).
Edmunds, P. J. & Davies, P. S. An energy budget for Porites porites (Scleractinia). Marine Biol. 92, 339–347 (1986).
Grottoli, A. G. et al. Lipids and stable carbon isotopes in two species of Hawaiian corals, Montipora verrucosa and Porites compressa, following a bleaching event. Marine Biol. 145, 621–631 (2004).
Haydon, T. D. et al. Metabolomic signatures of corals thriving across extreme reef habitats reveal strategies of heat stress tolerance. Proc. Royal Soc. B 290, 20221877 (2023).
Acknowledgements
We especially thank the members of the Experimental Marine Ecology Laboratory and Reef Ecology Laboratory at the Department of Biological Sciences, National University of Singapore, for fieldwork and laboratory support. The computational work for this study was partially performed on resources of the National Supercomputing Centre, Singapore (https://www.nscc.sg).
Funding
This research was supported by the National Research Foundation, Prime Minister’s Office, Singapore under its Marine Science Research and Development Programme (Award No. MSRDP-P05), as well as a Singapore Ministry of Education Tier 1 Academic Research Fund (A-0008517-00-00).
Author information
Authors and Affiliations
Contributions
Rosa Celia Poquita-Du, Danwei Huang and Peter Alan Todd conceptualised the study. Rosa Celia Poquita-Du sampled biological material, performed laboratory work and generated the data. Rosa Celia Poquita-Du and Danwei Huang analysed the data. Rosa Celia Poquita-Du led the writing, with major contributions from Danwei Huang and Peter Alan Todd. All authors reviewed and edited manuscript for submission. Peter Alan Todd and Danwei Huang acquired funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poquita-Du, R.C., Huang, D. & Todd, P.A. Genome-wide analysis to uncover how Pocillopora acuta survives the challenging intertidal environment. Sci Rep 14, 8538 (2024). https://doi.org/10.1038/s41598-024-59268-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-59268-0
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
