
Abstract
A novel solid acid catalyst with recoverability, named as Fe3O4@SiO2@TAD-G2-SO3H, was successfully synthesized by immobilizing sulfonic acid groups on triazine dendrimer-modified magnetic nanoparticles. This nanomaterial structure and composition were thoroughly characterized using various analytical techniques, including thermogravimetric analysis (TGA), elemental analysis, Fourier transform infrared spectroscopy (FT-IR), energy-dispersive X-ray spectroscopy (EDX), elemental mapping, acid–base titration, X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and vibrating sample magnetometry (VSM). The acid-decorated magnetic dendrimer was served as a highly effective catalyst for the synthesis of tetrahydrobenzo[c]acridin-8(9H)-one and benzo[h]indeno[1,2-b]quinoline-8-one derivatives. The reaction proceeded smoothly under mild conditions through the one-pot condensation of aromatic aldehydes, 1-naphthylamine, and either dimedone or 1,3-indanedione, affording the desired products in high yields ranging from 90 to 96%. The catalyst was easily separated from the reaction mixture by employing a magnetic field, allowing for its recycling up to five times with slight loss in its activity (only 10%). Nearly, quantitative recovery of catalyst (up to 95%) could be obtained from each run. So, this catalyst facilitates the reaction progress and simplifies the purification process. Other remarkable features of this method are operational simplicity, excellent yields, mild condition, and a wide range of substrate applicability.
Similar content being viewed by others


Introduction
The development of efficient and environmentally friendly synthesis methods for complex organic molecules is crucial in modern organic chemistry. Traditionally, organic synthesis involved multiple steps, each requiring individual reactions and purification processes. This approach consumed significant amounts of energy and generated substantial waste. multicomponent reactions (MCRs), on the other hand, enable the simultaneous assembly of multiple reactants into complex molecules in a single step. The development and utilization of MCRs has revolutionized the field of organic and medicinal chemistry1. The ability to combine multiple starting materials in a one-pot reaction greatly streamlines the synthesis process, minimizes energy consumption and waste generation, saves time and resources, and enhance overall synthetic efficiency. In addition, MCRs often exhibit high atom economy, meaning that a large proportion of the starting materials are incorporated into the final product without generating unnecessary byproducts2.
Acridine derivatives have attracted considerable attention for their potential pharmacological activity. These compounds have shown promising results in various areas of medicine, including anticancer, antimalarial, antimicrobial, anti-inflammatory, and calcium antagonist activities3,4,5,6,7,8. Their ability to inhibit DNA (deoxyribonucleic acid) replication and induce apoptosis has made them attractive candidates for the development of novel chemotherapeutic agents9.
The wide range of biological activities exhibited by quinoline derivatives has garnered significant interest among organic chemists. Consequently, numerous synthetic pathways have been devised to facilitate the synthesis of quinoline-fused heterocycles. Indenoquinoline derivatives exhibit various pharmacological effects, including antitumor, antimalarial, anti-inflammatory, steroid reductase and acetylcholinesterase inhibitory, and anticancer activities10,11,12. These properties render them highly promising candidates for the development of diverse therapeutic agents. Several synthetic strategies and catalysts, such as succinimide-N-sulfonic acid, tribromomelamine, cellulose-based magnetic nanocomposite, benzyltriethylammonium chloride, tetramethylguanidinium carboxylate ionic liquid, L-Proline, KO2 with 18-crown-6 ether, and Cu/MCM-41 have been developed for creating substituted acridine and quinolone derivatives13,14,15,16,17,18,19,20,21,22. However, many of the existing literature reports involve the use of organic solvents, multiple steps, harsh conditions, and laborious work-up procedures. Therefore, there is a growing demand for more efficient and sustainable methods that minimize the use of hazardous chemicals and simplify experimental procedures.
Dendrimers are unique materials that possess polymer-like properties, characterized by their intricate tree-like branching structures. These structures, combined with their reactive terminal functional groups, branched and regular three-dimensional framework, and internal cavities capable of accommodating targeted ions, have propelled dendrimers to the forefront of various fields such as biomedical research, environmental studies, nanotechnology, supramolecular chemistry, contaminant removal, chemical sensors, catalysis, and drug delivery applications23. Compared to traditional polymers, dendrimers exhibit exceptional physical and chemical properties owing to their three-dimensional structures, well-defined shape, uniform size distribution, remarkable flexibility, and multifunctional nature. These highly branched and nano-sized macromolecules offer hundreds of potential binding sites for active species, thus showcasing their immense potential in binding to specific molecules which opens up a world of possibilities for targeted drug delivery, efficient contaminant removal, and enhanced catalytic processes24,25,26.
The advancements in nanotechnology have opened up a world of possibilities for various fields, including medicine, miRNA (microRNA) detection, environmental science, environmental remediation, bio-sensing, and materials engineering. Furthermore, the development of active and intelligent nanoscale systems has paved the way for new materials with enhanced properties and functionalities27,28. Magnetic nanoparticles (MNPs) have emerged as versatile supports for active molecules, finding extensive applications in catalysis and pollutant removal29,30. In recent years, magnetic nanoparticles have taken center stage as the primary materials for creating dendritic structures, giving rise to magnetic-core dendrimers (MNDs)31,32,33,34. These nanoparticles can be easily recovered from aqueous solutions using an external magnet, eliminating the need for additional processes like centrifugation. The magnetic core is effectively shielded by a silica shell, which greatly enhances its stability. Moreover, the surface of these particles can be functionalized by attaching diverse active agents through reactions with the silanol groups. This process ensures the protection of the magnetic core and also allows for the incorporation of different functional groups onto the particle surface, thereby expanding its potential applications28,35,36,37,38.
Developing strategies to control the arrangement of different functional groups and active sites on the surface of nanoparticles remains a crucial task for effective catalytic applications39,40,41,42. One approach to enhance this control is to introduce a polymer or dendritic shell onto the nanoparticle surface. This not only improves the placement of functional groups but also enhances the dispersion of the nanoparticles in organic solvents, which further enhances their catalytic performance. Notably, star-like dendritic structures exhibit exceptional properties, boasting a multitude of functional groups that significantly increase the number of active sites on the nanoparticle surface43. The advantages of using dendritic magnetic nanostructures as catalysts are numerous. Their high surface area and surface functionality provide active sites for catalytic reactions. Additionally, their magnetic properties allow for easy separation, recyclability, simplifies the purification process and saves us from the tedious task of removing impurities. These nanostructures have shown exceptional catalytic activity and selectivity in various organic transformations, making them promising candidates for enhancing reaction efficiency44,45,46,47,48.
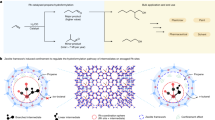
Considering the unique properties of both magnetic nanoparticles (MNPs) and dendrimers, this research focuses on the preparation of dendrimer-decorated magnetic nanoparticles (Fe3O4@SiO2@Den-SO3H) and their application in the one-pot synthesis of benzo[c]acridine-8(9H)-one and benzo[h]indeno[1,2‐b]quinoline‐8‐one derivatives, two structurally diverse and biologically active compounds of interest (Fig. 1).

synthesis of benzo[c]acridin-8(9H)-ones and benzo[h]indeno[1,2-b] quinolin-8-ones in the presence of Fe3O4@SiO2@TAD-G2-SO3H.
Experimental
Cyanuric chloride, tetrabutylammonium bromide, ethanolamine, chlorosulfonic acid, dioxane were commercially supplied by Merck and Acros companies. FeCl3.(H2O)6, FeCl2.(H2O)4, KOH, NaOH, HCl, and ethanol were commercially supplied by neutron chemical company (Iran). All the materials and reagents were used without any further purification. Fourier-transform infrared spectroscopy (FT-IR) measurements were conducted using a KBr pellet and an Alpha Bruker Fourier transform (FT-IR) spectrophotometer, covering the range of 400–4000 cm−1. The crystallinity of the nanomaterials was assessed using. A Philips Xpert X-ray powder diffraction (XRD) instrument, employing Cu-Kα radiation with a wavelength of λ = 0.1545 was applicated to evaluate the crystallinity of nanomaterials. To examine the surface morphology and elemental distribution, a MIRA 3-XMU field emission scanning electron microscope (FE-SEM) equipped with energy dispersive X-ray analysis (EDX) was employed. Thermogravimetric analysis (TGA) was performed on a Mettler TA4000 system under a nitrogen atmosphere, within the temperature range of 25–800 °C, with a heating rate of 15 °C/min. The magnetic properties of the nanoparticles were investigated using a 730 vibrating sample magnetometer (VSM) at room temperature.
Preparation of Fe3O4@SiO2-TzCl2 nanoparticles
The Fe3O4@SiO2-TzCl2 nanoparticles were prepared based on our previous report34.
Preparation of first generation dendritic magnetic nanoparticles (Fe3O4@SiO2@TAD-G1).
The Fe3O4@SiO2-TzCl2 (1 g) was dispersed in 20 mL of dioxane for a duration of 20 min. Subsequently, K2CO3 (5 mmol, 0.7 g) and ethanolamine (10 mmol, 0.7 g) were introduced to the mixture and subjected to reflux for a period of 24 h. The resulting solid was collected using an external magnet, rinsed with ethanol, and then dried under vacuum at a temperature of 50 °C.
Synthesis of second generation dendritic magnetic nanoparticles (Fe3O4@SiO2@TAD-G2).
Fe3O4@SiO2@TAD-G1 (1 g) was poured into a round-bottom flask and dispersed in dichloromethane (50 mL) for 10 min. Then, Cyanuric chloride (10 mmol, 1.8 g), tetrabutylammonium bromide (TBAB) (0.001 g), and 1 mL of NaOH (10 M) were added to the suspension and stirred for 2 h at 5 °C in an ice water bath. Afterward, the mixture was stirred at room temperature for 24 h. The resulting black precipitate was isolated using an external magnet, washed with dichloromethane, and dried. Next, the obtained particles (1 g) were mixed with K2CO3 (10 mmol, 1.4 g) and ethanolamine (20 mmol, 1.2 g) in dioxane (50 mL). The suspension was refluxed for 24 h. The resulting precipitate was isolated, washed with ethanol, and dried under vacuum at 50 °C.
Preparation of acid-decorated dendritic nanoparticles (Fe3O4@SiO2@TAD-G2-SO3H)
Fe3O4@SiO2@TAD-G2 (1 g) was dispersed in 40 mL of dichloromethane and stirred for 20 min. Subsequently, a solution of chlorosulfonic acid (2 mL) in 5 mL of dichloromethane was meticulously prepared and slowly added to the mixture over the course of 1 h. The resulting solid was collected using an external magnet, thoroughly washed with dichloromethane, and subsequently dried under vacuum at 50 °C.
Acidity of Fe3O4@SiO2@TAD-G2-SO3H
The concentration of sulfonic acid groups was determined by quantitatively estimating it using back titration with HCl (0.01 N). Initially, 2 mL of KOH (0.01 N) was added to 0.02 g of the Fe3O4@SiO2@TAD-G2-SO3H nanoparticles, and the mixture was stirred for a duration of 30 min. Subsequently, the catalyst was separated and thoroughly washed with deionized water. To determine the excess amount of KOH, it was titrated with HCl (0.01 N) in the presence of phenolphthalein as an indicator. To ensure precision, three separate titrations were conducted, and the average value was calculated to ascertain the acid content of Fe3O4@SiO2@TAD-G2-SO3H. The findings revealed that this nanostructure possessed an acid content of 2.5 mmol g−1.
General procedure for the synthesis of benzo[c]acridin-8(9H)-ones (5a–i):
In a 25 mL round bottom flask, an aromatic aldehyde (1 mmol), dimedone (1 mmol, 0.14 g), and 1-naphthylamine (1 mmol, 0.143 g) were combined in 5 ml of ethanol/water (1:1). This mixture was stirred in the presence of Fe3O4@SiO2@TAD-G2-SO3H (0.03 g) at a temperature of 70 °C. To monitor the progress of the reaction, thin-layer chromatography (TLC) with a solvent mixture of n-hexane and ethyl acetate (2:1) was used. Once the reaction was completed, the catalyst separated from the reaction medium using a magnet. Next, the solvent was evaporated and the product recrystallized using ethanol. Finally, the product was dried in an oven at 50 °C.
General procedure for the synthesis of benzo[h]indeno[1,2-b]quinolin-8-ones (6a–h):
In a 25 mL round bottom flask, an aromatic aldehyde (1 mmol), 1,3-indanedione (1 mmol, 0.146 g), 1-naphthylamine (1 mmol, 0.143 g), and Fe3O4@SiO2@TAD-G2-SO3H (0.03 g) was combined in 5 mL of a 1:1 mixture of ethanol/water. The mixture was stirred in the presence of 5 mL of a 1:1 mixture of ethanol and water at a temperature of 70 °C. Thin-layer chromatography (TLC) with a solvent mixture of n-hexane and ethyl acetate in a ratio of 3:1 was used to monitor the progress of the reaction. Once the reaction was completed, the catalyst was separated from the reaction medium using a magnet, and the solvent was evaporated. Finally, the product was recrystallized using ethanol and dried in an oven at 50 °C.
Spectral data for selected products
7-(4-chlorophenyl)-10,10-dimethyl-7,10,11,12-tetrahydrobenzo[c]acridin-8(9H)-one (5b)
In a 25 mL round bottom flask, 4-chlorobenzaldehyde (1 mmol, 0.141 g), 1,3-indanedione (1 mmol, 0.146 g), 1-naphthylamine (1 mmol, 0.143 g), and Fe3O4@SiO2@TAD-G2-SO3H (0.03 g) was combined in 5 mL of a 1:1 mixture of ethanol/water. The mixture was stirred at a temperature of 70 °C. Once the reaction was completed, the catalyst was separated from the reaction medium using a magnet, and the solvent was evaporated. Finally, the product was recrystallized using ethanol and dried in an oven at 50 °C. Melting point: 268–270 °C. IR: 3335 (NH), 2957 (CHAr), 2930 (CHAl), 1606 (C = O), 1573 (C = C), 1492 (C = C), 1261 (C–N), 847 cm−1. 1HNMR (300 MHz, DMSO-d6): δ = 0.95 (s, 3H, CH3), 1.05 (s, 3H, CH3), 2.04 (d, J = 16.0 Hz, 1H, CH2), 2.20 (d, J = 16.0 Hz, 1H, CH2), 2.60 (d, J = 16.8 Hz, 1H, CH2), 2.71 (d, J = 16.8 Hz, 1H, CH2), 4.99 (s, 1H, CH), 6.83–7.52 (m, 10H, ArH), 9.29 (s, 1H, NH) ppm.
7-(4-nitrophenyl)-10,10-dimethyl-7,10,11,12-tetrahydrobenzo[c]acridin-8(9H)-one (5d)
IR: 3318 (NH), 2957 (CHAr), 2870 (CHAl), 1597 (C = O), 1570 (C = C), 1518 (-NO2), 1491 (C = C), 1370, 1344 (-NO2), 1261 (C–N), 857 \({{\text{cm}}}^{-1}\); 1HNMR (300 MHz, DMSO-d6): δ = 0.97 (s, 3H, CH3), 1.08 (s, 3H, CH3), 2.04 (d, J = 16.4 Hz,1H, CH2), 2.22 (d, J = 16.4 Hz, 1H, CH2), 2.70 (d, J = 16.8 Hz, 1H, CH2), 2.77 (d, J = 16.8 Hz,1H, CH2) 5.18 (s, 1H, CH), 7.55–8.07 (m, 11H, ArH), 9.41 (s, 1H, NH) ppm.
7-(3-nitrophenyl)-10,10-dimethyl-7,10,11,12-tetrahydrobenzo[c]acridin-8(9H)-one (5e)
IR: 3312 (NH)(CHAr), 2955 (CHAr), 2870 (CHAl), 1589 (C = O), 1527 (-NO2), 1494 (C = C), 1388, 1347(-NO2), 1261 (C–N), 807, 756, 729,\({{\text{cm}}}^{-1}\); 1HNMR (300 MHz, DMSO-d6): δ = 0.97(s, 3H, CH3), 1.08 (s, 3H, CH3), 2.07 (d, J = 16.4 Hz, 1H, CH2), 2.22 (d, J = 16.4 Hz, 1H, CH2), 2.27 (d, J = 16.8 Hz, 1H, CH2), 2.75 (d, J = 16.8 Hz, 1H, CH2) 5.42 (s, 1H, CH), 7.69–8.07 (m, 10H, ArH), 9.41 (s, 1H, NH) ppm.
7-(3-hydroxyphenyl)-10,10-dimethyl-7,10,11,12-tetrahydrobenzo[c]acridin-8(9H)-one (5h)
IR: 3395 (NH), 3298 (OH), 2957 (CHAr), 2928 (CHAl), 1606 (C = O), 1555 (C = C), 1487 (C = C), 1256 (C–N), 761,699,\({{\text{cm}}}^{-1}\); 1HNMR (300 MHz, DMSO-d6): δ = 0.94 (s, 3H, CH3), 1.05 (s, 3H, CH3), 2.04 (d, J = 16.0 Hz, 1H, CH2), 2.22(d, J = 16.0 Hz, 1H, CH2), 2.58 (d, J = 16.8 Hz, 1H, CH2), 2.70 (d, J = 16.8 Hz, 1H, CH2) 4.86 (s, 1H, CH), 6.39–6.97 (m, 10H, ArH), ), 9.05 (s, 1H, OH), 9.22 (s, 1H, NH) ppm.
7-(2,4-dichlorophenyl)-10,10-dimethyl-7,10,11,12-tetrahydrobenzo[c]acridin-8(9H)-one (5i)
IR: 3324 (NH), 3067 and 2956 (CHAr), 2870 (CHAl), 1607 (C = O), 1588 (C = C), 1496 (C = C), 1259 (C–N), 861, 566,\({{\text{cm}}}^{-1}\); 1HNMR (300 MHz, DMSO-d6): δ = 0.97 (s, 3H, CH3), 1.03 (s, 3H, CH3), 2.00 (d, J = 16.0 Hz, 1H, CH2), 2.17 (d, J = 16.0 Hz, 1H, CH2), 2.22 (d, J = 16.4 Hz, 1H, CH2), 2.70 (d, J = 16.4 Hz,1H, CH2) 5.48 (s, 1H, CH), 7.14–7.56 (m, 9H, ArH), 9.22 (s, 1H, NH) ppm.
7-(4-Chlorophenyl)-8H-benzo[h]indeno[1,2-b]quinolin-8-one (6b)
IR: 3060 (CHAr), 2923 (CHAr), 1710 (C = O), 1603 (C = N), 1576 and 1484 (C = C), 1337 (C–N), 840 cm−1. 1HNMR (300 MHz, DMSO-d6): δ = 7.45–8.07 (m, 14H, ArH) ppm.
7-(3-Nitrophenyl)-8H-benzo[h]indeno[1,2-b]quinolin-8-one (6e)
IR: 3060 (CHAr), 2923 (CHAr), 1710 (C = O), 1656 (C = C), 1613 (C = N), 1527 (C = C), 1345 (C–N), 759, 712 cm−1. 1HNMR (300 MHz, DMSO-d6): δ = 7.36–8.08 (m, 14H, ArH) ppm.
7-(4-Nitrophenyl)-8H-benzo[h]indeno[1,2-b]quinolin-8-one (6f)
IR: 3057 (CHAr), 1712 (C = O), 1662 and 1580 (C = C), 1613 (C = N), 1524 (NO2), 1394, 1344 (NO2), 1264 (C–N), 859 cm−1. 1HNMR (300 MHz, DMSO-d6): δ = 7.25–7.96 (m, 14H, ArH) ppm.
7-(3-Hydroxyphenyl)-8H-benzo[h]indeno[1,2-b]quinolin-8-one (6g)
IR: 3406 (OH), 3051 and 2955 (CHAr), 1707 (C = O), 1608 (C = N), 1574 (C = C), 1324 (C–N), 753, 698 cm−1. 1HNMR (300 MHz, DMSO-d6): δ = 7.35–8.04 (m, 14H, ArH), 9.36 (s, 1H, OH) ppm.
7-(2,4-Dichlorophenyl)-8H-benzo[h]indeno[1,2-b]quinolin-8-one (6h)
IR: 3058 (CHAr), 1715 (C = O), 1607 (C = N), 1654 and 1576 (C = C), 1469, 1338, 851, 569 cm−1. 1HNMR (300 MHz, DMSO-d6): δ = 6.82–7.56 (m, 13H, ArH) ppm.
Results and discussion
In this study, sequential reactions were employed to introduce dendrimers onto solid supports (divergent strategy). This was achieved by attaching a linker onto the surface of magnetic nanoparticles and the triazine-based poly-aminoethanol (TAD) dendrimers was successfully grown on the surface of silica-coated magnetite nanoparticles via sequential reactions (Fe3O4@SiO2@TAD-G2-SO3H, Fig. 2). Initially, magnetite (Fe3O4) nanoparticles were easily prepared via the chemical co-precipitation of Fe2+ and Fe3+ ions in basic solution. Next, a layer of silica was placed on the magnetic iron nanoparticles, via Stober method, to prevent their aggregation and oxidation. Then, the (3-aminopropyl)triethoxysilane (APTMS) as a linker was grafted to the surface of Fe3O4@SiO2 particles under nitrogen atmosphere to produce the Fe3O4@SiO2@PrNH2 nanostructure49. In the subsequent stages, TAD dendrons (functional arms) up to generation two (G2) were constructed through a sequential and progressive nucleophilic aromatic substitution of ethanolamine on the aromatic triazine ring34,50. Ultimately, the nanoparticles were decorated with acidic groups through a reaction of active –NH and –OH groups with chlorosulfonic acid.

Synthetic pathway for construction of dendrimer-coated magnetite nanoparticles (Fe3O4@SiO2@TAD-G2-SO3H).
Figure 3, shows the FT-IR spectra of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2 -PrNH2, Fe3O4@SiO2-TzCl2, Fe3O4@SiO2@TAD-G1, Fe3O4@SiO2-Tz-BEA-TzCl4, Fe3O4@SiO2@TAD-G2, Fe3O4@SiO2@TAD-G2-SO3H in the wavenumber range of 400–4000 cm−1. The FT-IR spectrum of the bare magnetic Fe3O4 nanoparticles exhibited the characteristic Fe–O absorption band in the range of 580 cm−1 (Fig. 3a). The bands observed at 1101, 900–800, and 450 cm−1, correspond to asymmetric stretching, symmetric stretching, in-plane bending of the Si-O-Si and Si–O bonds, respectively (Fe3O4@SiO2 spectrum, Fig. 3b). The band in the region of 1300 cm−1 is related to the stretching bond S = O (Fig. 3h). The broad band in the range 3200–3500 cm−1 appertained to the stretching vibration mode of Si–OH bonds and the weak band at 1630 cm−1 related to the twisting vibration mode of H–O-H adsorbed in the silica layer. The presence of alkyl groups has been corroborated by the weak symmetric and asymmetric stretching vibrations at 2980 and 2895 cm−1 (Fig. 3c–h). Also, the bands corresponding to C = N, C = C, and C–N appeared at 1350–1600 cm−1 (Fig. 3d–h)34,51. The N–H bending vibration is overlapped with O–H vibration. Therefore, these results confirm the successful bonding of different functional groups at each fabrication step.

FT-IR spectra of Fe3O4 (a), Fe3O4@SiO2 (b), Fe3O4@SiO2 -PrNH2 (c), Fe3O4@SiO2-TzCl2 (d), Fe3O4@SiO2@TAD-G1 (e), Fe3O4@SiO2 -Tz-BEA-TzCl4 (f), Fe3O4@SiO2@TAD-G2 (g), Fe3O4@SiO2@TAD-G2-SO3H (h).
The crystallinity of Fe3O4 (a), and Fe3O4@SiO2@TAD-G2-SO3H (b) was analyzed using X-ray powder diffraction patterns (Fig. 4). By comparing the XRD patterns of Fe3O4 and Fe3O4@SiO2@TAD-G2-SO3H, it can be confirmed that the ferrite core remains stable without undergoing any phase changes during the functionalization process52. The XRD patterns of the prepared Fe3O4@SiO2@TAD-G2-SO3H show distinct diffraction peaks at 2θ = 30.2°, 35.4°, 43.7°, 56.6°, and 63.0°, corresponding to the (220), (311), (400), (422), (511), and (440) Miller planes of Fe3O4 nanoparticles. These XRD patterns confirm that the material structure has a cubic spinel phase (Fig. 4a,b) and match with the standard Fe3O4 sample (JCPDS file No. 19–0629)53,54. The presence of the SiO2 layer is confirmed by the appearance of a broad peak at 2θ = 18° to 25° (Fig. 4b). To determine the average crystallite size of Fe3O4@SiO2@TAD-G2-SO3H, the Scherrer equation (D = Kλ/βcosθ) was employed. In this equation, λ is the x-ray Cu wavelength, β is the width of the X-ray peak on the 2θ axis which measured as the line broadening at half the maximum intensity, θ is Bragg angle, and K is the so-called Scherrer constant. The crystallite size calculated from the width of the peak at 2θ = 35.4° (311), is approximately 102 nm41.

XRD patterns of Fe3O4 (a), JCPDS file No. 19–0629), and Fe3O4@SiO2@TAD-G2-SO3H (b).
The size and morphology of Fe3O4@SiO2@TAD-G2-SO3H are investigated by field emission scanning electron microscopy (FE-SEM). As shown in Fig. 5, the Fe3O4@SiO2@TAD-G2-SO3H nanostructure possesses nearly spherical morphology with an average size of 100–130 nm.

The FE-SEM images and histogram for particles size of Fe3O4@SiO2@TAD-G2-SO3H.
The results obtained from X-ray energy diffraction spectroscopy (EDX) of Fe3O4@SiO2@TAD-G2-SO3H magnetic nanoparticles (Fig. 6) confirm the presence of elements S, C, N, Si, O, Fe in the hybrid nanostructure. The higher intensity of the peak corresponding to the elements Si and S compared to the Fe peak indicates that the Fe3O4 nanoparticles have been covered by SiO2 layer and –SO3H functionalities on the surface of the nanostructure. The terminal ends of the catalyst have a lot of –SO3H groups, causing the sulfur have a higher peak intensity. These results provide evidence for the successful synthesis of the core–shell nanostructure (Fe3O4@SiO2@TAD-G2-SO3H). Moreover, the EDX map analysis reveals a good distribution of various elements including Fe (a), N (c), S (d), O (e), and Si (f) throughout the nanostructure.

EDX spectrum and EDX and map analysis for Fe3O4@SiO2@TAD-G2-SO3H.
Thermal gravimetric studies were conducted within the temperature range of 25–800 °C under a N2 atmosphere, to evaluate the thermal stability of Fe3O4@SiO2@TAD-G2-SO3H (Fig. 7). A weight loss of approximately 4–6% is observed in the temperature range of 25 to 250 °C, which is attributed to the loss of residual solvent and water on the sample surface (physically absorbed) process. Furthermore, a significant weight loss of approximately 40% was observed in 250–650 °C, which can be attributed to the degradation of dendrimer branches and organic moieties, followed by a phase change in the silica nanoparticle groups. The remaining weight (˃650 °C) is attributed to inorganic support which was not decomposed.

TGA analyses of Fe3O4@SiO2@TAD-G2-SO3H.
The magnetization curves of Fe3O4 (a), Fe3O4@SiO2@TAD-G2-SO3H (b) were measured using the VSM technique at room temperature, within the range of ± 8000 Oe (Fig. 8). The S-like magnetization curves, the coincidence of the hysteresis loop, and the low remanence and coercivity confirm the superparamagnetic properties of these hybrid materials. As seen in Fig. 6, the saturation level for the Fe3O4 sample is 40 emu g−1, While it is 15 emu g−1 for Fe3O4@SiO2@TAD-G2-SO3H nanostructure. This decrease in magnetic property indicates the presence of a surface coating on the Fe3O4 nanoparticles, confirming the core–shell structure and the grafting of the dendritic branches to the magnetic nanoparticles.

VSM analysis of Fe3O4 nanoparticles (a), and Fe3O4@SiO2@TAD-G2-SO3H nanostructure (b).
After the characterization of the Fe3O4@SiO2@TAD-G2-SO3H catalyst, its catalytic activity was evaluated for the synthesis of benzo[c]acridin-8(9H)-one derivatives (Fig. 1). For this purpose, the reaction of 4-chlorobenzaldehyde (1 mmol), dimedone (1 mmol) and 1-naphthylamine (1 mmol) was used as a model and conducted under different reaction parameters including the catalyst amount, solvent, and temperature (Table 1). The best result was achieved when the reaction took place in the presence of 30 mg of the catalyst, in a polar solvent mixture of H2O/EtOH (1:1), and at a temperature of 70 °C (Table 1, entry 9). In continuation, the catalytic activity of Fe3O4@SiO2@TAD-G2-SO3H catalyst was evaluated for the synthesis of benzo[h]indeno[1,2-b]quinolin-8-ones. Interestingly, the best result was achieved in H2O/EtOH (1:1) at 70 °C, using 30 mg of the catalyst. After obtaining the best conditions for the reaction, various arylaldehydes including electron-withdrawing and electron-releasing groups were subjected to the reactions. All substituted arylaldehydes effectively reacted and afforded the corresponding products in excellent yields without any side product (Table 2).
A plausible mechanism is shown in Fig. 9. The carbonyl group of the aldehyde is initially protonated using the acidic catalyst. Subsequently, dimedone is enolized in acidic condition and undergoes condensation with activated aldehyde, leading to the formation of an intermediate (I). In the next step, the intermediate (I) is subjected to nucleophilic attack by 1-naphthylamine and followed by tautomerization, resulting in the production of structure (II). The intramolecular cyclization of compound (II) and elimination of water ultimately yields the benzoacridine (4).

A proposed mechanism for the synthesis of benzo[c]acridin-8(9H)-one in the presence of Fe3O4@SiO2@TAD-G2-SO3H nanostructure.
The catalytic activity of Fe3O4@SiO2@TAD-G2-SO3H MNPs for the model reaction was also compared with that reported in the literature (Table 3). These data confirm that the Fe3O4@SiO2@TAD-G2-SO3H nanostructure is a suitable catalyst for synthesizing the desired corresponding product. The absence of the need for extraction with organic solvents not only simplifies the synthesis process but also reduces the environmental impact of the reaction. Overall, these results highlight the potential of this nanostructure as a green and efficient catalyst for other organic transformations.
Catalyst recovery and reuse
The catalyst retrievability plays a crucial role in industrial applications, as well as in promoting environmentally friendly processes. Therefore, we conducted an investigation into the reusability of the Fe3O4@SiO2@TAD-G2-SO3H catalyst in a sequential reaction involving 4-chlorobenzaldehyde (1 mmol), 1-naphtylamine (1 mmol), and dimedone (1 mmol) in a H2O/EtOH (1:1) mixture at 70 °C. After the reaction was completed, the resulting mixture solidified. To dilute it, 10 mL of hot EtOH was added. The catalyst was easily separated using an external magnet, washed with hot EtOH, dried under vacuum, and reused in the next reaction. Each run allowed for the nearly complete recovery of the catalyst, with up to 95% being obtained. The reused catalyst exhibits high efficiency in the synthesis of benzoacridine, as depicted in Fig. 10. The stability of the catalyst is further considered by the FT-IR spectra of the reused catalyst (Fig. 10), which proves the stability of catalyst in reaction condition.

Reusability of the catalyst, and FT-IR spectra of the fresh and reused nanocatalyst.
Conclusions
A solid acid catalyst (Fe3O4@SiO2@TAD-G2-SO3H) which can be easily retrieved using magnets, was successfully synthesized and characterized. This catalyst was created by growing triazine dendrimer on silica-coated magnetic nanoparticles, while also immobilizing sulfonic acid groups. The prepared dendritic nanostructure (Fe3O4@SiO2@TAD-G2-SO3H) efficiently catalyzed the synthesis of benzo[c]acridin-8(9H)-one and benzo[h]indeno[1,2-b]quinoline-8-one derivatives in excellent yields (90% to 96%). Grafting of dendrimer increases the active sites on Fe3O4 nanoparticles and decreases their agglomeration. The catalyst can be easily separated from the reaction mixture by employing a magnetic field, allowing for its recycling up to five times with slight loss in its activity (10%). Other advantages of this protocol include the use of aqueous ethanol as a mild reaction media, operational simplicity, high to excellent yields, high atom-economy, easy separation of the catalyst, reusability, and elimination of the need for tedious column chromatography during product isolation and purification. These aspects align with the principles of green and sustainable chemistry, which aim to design chemical products and processes that reduce or eliminate hazardous substances and waste generation.
Data availability
All data generated or analysed during this study are included in this article.
References
Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 42, 463–472 (2009).
Toure, B. B. & Hall, D. G. Natural product synthesis using multicomponent reaction strategies. Chem. Rev. 109, 4439–4486 (2009).
Khurana, J. M., Maikap, G. C. & Mehta, S. Reductive coupling of geminal dichlorides by iron (II) oxalate dihydrate. Synthesis 1990, 731–732 (1990).
Gamage, S. A. et al. Structure—activity relationships for substituted bis (acridine-4-carboxamides): A new class of anticancer agents. J. Med. Chem. 42, 2383–2393 (1999).
Yang, P., Yang, Q., Qian, X., Tong, L. & Li, X. Isoquino [4, 5-bc] acridines: Design, synthesis and evaluation of DNA binding, anti-tumor and DNA photo-damaging ability. J. Photochem. Photobiol. B Biol. 84, 221–226 (2006).
Girault, S. et al. Antimalarial, antitrypanosomal, and antileishmanial activities and cytotoxicity of bis (9-amino-6-chloro-2-methoxyacridines): Influence of the linker. J. Med. Chem. 43, 2646–2654 (2000).
Jamalian, A. et al. Synthesis, cytotoxicity and calcium antagonist activity of novel imidazolyl derivatives of 1, 8-acridinediones. J. Iran. Chem. Soc. 8, 983–991 (2011).
Sondhi, S. M. et al. Synthesis, anti-inflammatory and anticancer activity evaluation of some novel acridine derivatives. Eur. J. Med. Chem. 45, 555–563 (2010).
Denny, W. A. Acridine derivatives as chemotherapeutic agents. Current Med. Chem. 9, 1655–1665 (2002).
Tu, S.-J. et al. An efficient one-pot, three-component synthesis of indeno [1, 2-b] quinoline-9, 11 (6H, 10H)-dione, acridine-1, 8 (2H, 5H)-dione and quinoline-3-carbonitrile derivatives from enaminones. Organ. Biomol. Chem. 4, 3664–3668 (2006).
Deady, L. W. et al. Synthesis and antitumor activity of some indeno [1, 2-b] quinoline-based bis carboxamides. Bioorgan. Med. Chem. 8, 977–984 (2000).
Rampa, A. et al. Acetylcholinesterase inhibitors for potential use in Alzheimer’s disease: molecular modeling, synthesis and kinetic evaluation of 11H-indeno-[1, 2-b]-quinolin-10-ylamine derivatives. Bioorgan. Med. Chem. 8, 497–506 (2000).
Dutta, A. K., Gogoi, P., Saikia, S. & Borah, R. N, N-disulfo-1, 1, 3, 3-tetramethylguanidinium carboxylate ionic liquids as reusable homogeneous catalysts for multicomponent synthesis of tetrahydrobenzo [a] xanthene and tetrahydrobenzo [a] acridine derivatives. J. Mol. Liquids 225, 585–591 (2017).
Zang, H., Zhang, Y., Zang, Y. & Cheng, B.-W. An efficient ultrasound-promoted method for the one-pot synthesis of 7, 10, 11, 12-tetrahydrobenzo [c] acridin-8 (9H)-one derivatives. Ultrason. Sonochem. 17, 495–499 (2010).
Wang, X.-S. et al. A clean procedure for synthesis of benzo [c] acridine derivatives: reaction of N-arylidenenaphthalen-1-amine with 5, 5-dimethyl-1, 3-cyclohexadione in aqueous medium. Arkivoc 2, 117–123 (2006).
Mamaghani, M. & Larghani, T. H. Ultrasound promoted one-pot three-component synthesis of novel 7-aryl-8 H-benzo [h] indeno [1, 2-b] quinolin-8-ones under solvent-free conditions. J. Chem. Res. 36, 235–237 (2012).
Mansoor, S. S., Ghashang, M. & Aswin, K. Facile one-pot synthesis of a novel series of 7-aryl-8 H-benzo [h] indeno [1, 2-b] quinoline-8-one derivatives catalyzed by tribromomelamine. Res. Chem. Intermed. 41, 6907–6926 (2015).
Ghashang, M., Mansoor, S. S. & Aswin, K. Succinimide-N-sulfonic acid: An efficient and recyclable catalyst for the one-pot synthesis of tetrahydrobenzo [c] acridine-8 (7H)-one derivatives. J. Saudi Chem. Soc. 21, S44–S51 (2017).
Maleki, A. & Nooraie Yeganeh, N. Facile one-pot synthesis of a series of 7-aryl-8H-benzo [h] indeno [1, 2-b] quinoline-8-one derivatives catalyzed by cellulose-based magnetic nanocomposite. Appl. Organomet. Chem. 31, e3814 (2017).
Heravi, M. R. P. & Aghamohammadi, P. l-Proline-catalysed one-pot synthesis of tetrahydrobenzo [c] acridin-8 (7H)-ones at room temperature. Comptes. Rendus. Chimie 15, 448–453 (2012).
Gajaganti, S. & Singh, S. Superoxide Ion Promoted One Pot Multicomponent Synthesis of 1, 4-dihydropyridine derivatives. Mater. Today Proceed. 4, 10653–10657 (2017).
Dhengale, S. D. et al. An efficient and convenient heterogeneous Cu/MCM-41 catalyst for the synthesis of 7, 10, 11, 12-tetrahydrobenzo [c] acridin-8 (9H)-one derivatives. Res. Chem. Intermed. 49, 1581–1600 (2023).
Arshadi, M., Abdolmaleki, M., Eskandarloo, H., Azizi, M. & Abbaspourrad, A. Synthesis of highly monodispersed, stable, and spherical NZVI of 20–30 nm on filter paper for the removal of phosphate from wastewater: batch and column study. ACS Sustain. Chem. Eng. 6, 11662–11676 (2018).
Svenson, S. & Tomalia, D. A. Dendrimers in biomedical applications—reflections on the field. Adv. Drug Deliv. Rev. 64, 102–115 (2012).
Eskandarian, L., Pajootan, E. & Arami, M. Novel super adsorbent molecules, carbon nanotubes modified by dendrimer miniature structure, for the removal of trace organic dyes. Ind. Eng. Chem. Res. 53, 14841–14853 (2014).
Sajid, M., Nazal, M. K., Baig, N. & Osman, A. M. Removal of heavy metals and organic pollutants from water using dendritic polymers based adsorbents: A critical review. Sep. Purif. Technol. 191, 400–423 (2018).
Kuzin, A. et al. Bridging the gap: harnessing liquid nanomachine know-how for tackling harmful airborne particulates. Nanoscale 15, 17727–17738 (2023).
Mohan, B. et al. Nanomaterials for miRNA detection: The hybridization chain reaction strategy. Sens. Diagn. 2, 78–89 (2023).
Shylesh, S., Schünemann, V. & Thiel, W. R. Magnetically separable nanocatalysts: Bridges between homogeneous and heterogeneous catalysis. Angew. Chemie Int. Edition 49, 3428–3459 (2010).
Hedayati, K., Kord, M., Goodarzi, M., Ghanbari, D. & Gharigh, S. Photo-catalyst and magnetic nanocomposites: hydrothermal preparation of core–shell Fe 3 O 4@ PbS for photo-degradation of toxic dyes. J. Mater. Sci. Mater. Electron. 28, 1577–1589 (2017).
Yen, C.-H., Lien, H.-L., Chung, J.-S. & Yeh, H.-D. Adsorption of precious metals in water by dendrimer modified magnetic nanoparticles. J. Hazardous Mater. 322, 215–222 (2017).
Kim, K.-J. & Park, J.-W. Stability and reusability of amine-functionalized magnetic-cored dendrimer for heavy metal adsorption. J. Mater. Sci. 52, 843–857 (2017).
Ahangarani-Farahani, R. & Bodaghifard, M. A. A polyethanolamine nanodendrimer as a magnetic hybrid material for fast adsorption of heavy metal contaminants. J. Mater. Sci. Mater. Electron. 33, 25674–25686 (2022).
Ahangarani-Farahani, R., Bodaghifard, M. A. & Asadbegi, S. Magnetic triazine-based dendrimer as a versatile nanocarrier for efficient antiviral drugs delivery. Sci. Rep. 12, 19469 (2022).
Bodaghifard, A., Faraki, M. Z. & Karimi, R. A. Mild synthesis of mono-, bis-and tris 1, 2-dihydrobenzo [4, 5] imidazo [1, 2-a] pyrimidine derivatives using alkyl disulfamic acid functionalized magnetic nanoparticles. Curr. Organ. Chem. 20, 1648–1654 (2016).
Bodaghifard, M. A. Palladium-melamine complex anchored on magnetic nanoparticles: A novel promoter for CC cross coupling reaction. J. Organ. Chem. 886, 57–64 (2019).
Asadbegi, S., Bodaghifard, M. A. & Mobinikhaledi, A. Poly N, N-dimethylaniline-formaldehyde supported on silica-coated magnetic nanoparticles: A novel and retrievable catalyst for green synthesis of 2-amino-3-cyanopyridines. Res. Chem. Intermed. 46, 1629–1643 (2020).
Hamidinasab, M., Bodaghifard, M. A. & Mobinikhaledi, A. Green synthesis of 1H-pyrazolo [1, 2-b] phthalazine-2-carbonitrile derivatives using a new bifunctional base–ionic liquid hybrid magnetic nanocatalyst. Appl. Organ. Chem. 34, e5386 (2020).
Gheni, S. A., Hmood, H. M., Ahmed, S. M. & Mohammed, M. H. Optimization for kinetic model of oxidative desulfurization of sour naphtha over a natural base zeolite catalyst in a three phase oscillatory baffled reactor. J. Petrol. Res. Studies 13, 91–111 (2023).
Mohammed, A. E. et al. Agricultural waste-based microporous catalysts for oxidative desulfurization of highly sour heavy gas oil. Diamond. Related Mater. 142, 110723 (2024).
Bodaghifard, M. A., Hamidinasab, M. & Ahadi, N. Recent advances in the preparation and application of organic–inorganic hybrid magnetic nanocatalysts on multicomponent reactions. Curr. Organ. Chem. 22, 234–267 (2018).
Geißler, D., Nirmalananthan-Budau, N., Scholtz, L., Tavernaro, I. & Resch-Genger, U. Analyzing the surface of functional nanomaterials—how to quantify the total and derivatizable number of functional groups and ligands. Microchimica Acta 188, 1–28 (2021).
Kainz, Q. M. & Reiser, O. Polymer-and dendrimer-coated magnetic nanoparticles as versatile supports for catalysts, scavengers, and reagents. Accounts Chem. Res. 47, 667–677 (2014).
Bahrami, K. & Arabi, M. S. Copper immobilized ferromagnetic nanoparticle triazine dendrimer (FMNP@ TD–Cu (ii))-catalyzed regioselective synthesis of 1, 4-disubstituted 1, 2, 3-triazoles. New J. Chem. 40, 3447–3455 (2016).
Pan, S., Yan, S., Osako, T. & Uozumi, Y. Batch and continuous-flow Huisgen 1, 3-dipolar cycloadditions with an amphiphilic resin-supported triazine-based polyethyleneamine dendrimer copper catalyst. ACS Sustain. Chem. Eng. 5, 10722–10734 (2017).
Niakan, M., Asadi, Z. & Masteri-Farahani, M. Fe (III)-salen complex supported on dendrimer functionalized magnetite nanoparticles as a highly active and selective catalyst for the green oxidation of sulfides. J. Phys. Chem. Solids 147, 109642 (2020).
Saberi, D., Hashemi, H., Ghanaatzadeh, N., Moghadam, M. & Niknam, K. Ruthenium/dendrimer complex immobilized on silica-functionalized magnetite nanoparticles catalyzed oxidation of stilbenes to benzil derivatives at room temperature. Appl. Organomet. Chem. 34, e5563 (2020).
Karimi, S., Shekaari, H., Halimehjani, A. Z. & Niakan, M. Solvent-free production of 5-hydroxymethylfurfural from deep eutectic substrate reaction mixtures over a magnetically recoverable solid acid catalyst. ACS Sustain. Chem. Eng. 9, 326–336 (2020).
Ahadi, N., Mobinikhaledi, A. & Bodaghifard, M. A. One-pot synthesis of 1, 4-dihydropyridines and N-arylquinolines in the presence of copper complex stabilized on MnFe2O4 (MFO) as a novel organic–inorganic hybrid material and magnetically retrievable catalyst. Appl. Organomet. Chem. 34, e5822 (2020).
Mohan, B. et al. Design and synthesis of two armed molecular receptor for recognition of Gd3+ metal ion and its computational study. Appl. Organomet. Chem. 32, e4502 (2018).
Mohan, B. et al. Selectivity for La3+ ion by synthesized 4-((5-methylfuran-2-yl) methylene) hydrazono) methyl) phenol receptor and its spectral analysis. Spectroch. Acta Part A Mol. Biomol. Spectrosc. 204, 581–589 (2018).
Mohan, B., Ma, S., Kumar, S., Yang, Y. & Ren, P. Tactile sensors: Hydroxyl decorated silver metal-organic frameworks for detecting Cr2O72–, MnO4–, Humic Acid, and Fe3+ Ions. ACS Appl. Mater. Interfaces 15, 17317–17323 (2023).
Zhou, Q. et al. Magnetic polyamidoamine dendrimers for magnetic separation and sensitive determination of organochlorine pesticides from water samples by high-performance liquid chromatography. J. Environ. Sci. 102, 64–73 (2021).
Asadbegi, S., Bodaghifard, M. A., Alimohammadi, E. & Ahangarani-Farahani, R. Immobilization of palladium on modified nanoparticles and its catalytic properties on mizoroki-heck reaction. ChemistrySelect 3, 13297–13302 (2018).
Acknowledgements
We appreciate the financial support of the Research Council of Arak University.
Author information
Authors and Affiliations
Contributions
M.A.B designed the experiments and contributed to interpreting the results. H.A carried out the experiments. H.A and R.A prepared the primary draft of the manuscript. M.A.B supervised the project and revised the final version of the manuscript. All authors approved the final version of the manuscript to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bodaghifard, M.A., Allahbakhshi, H. & Ahangarani-Farahani, R. Efficient synthesis of benzoacridines and indenoquinolines catalyzed by acidic magnetic dendrimer. Sci Rep 14, 8736 (2024). https://doi.org/10.1038/s41598-024-59212-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-59212-2
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
