
Abstract
In this, a three-component reaction for the preparation of novel tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives from N,N′-(sulfonylbis(1,4-phenylene))bis(3-oxobutanamide), aldehydes and 1H-tetrazol-5-amine is reported. The application of Fe3O4@SiO2-(PP)(HSO4)2 (A) as a catalyst afforded the desired products (a1–a18) in high yields in DMF as solvent as well as under solvent-free conditions.
Similar content being viewed by others


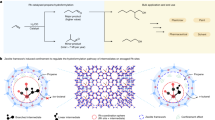
Introduction
Fused poly-heterocyclic systems have long been considered essential cores in the synthesis of drugs and natural products. The wide potential applications of fused heterocycles; especially in drug discovery, have encouraged chemists to synthesize them1. On the other hand, any compound with a tetrazole unit is a suitable candidate for interesting pharmaceutical applications. Many compounds bearing a tetrazole moiety are known as xanthine oxidase2, antitubercular agents3, antimicrobial agents4, and antinociceptive active compounds5.
According to literature reports, fused heterocycles bearing a tetrazole core are potent compounds; especially in the field of synthetic drugs, and various methods are developed for the incorporation of tetrazoles into fused heterocycles. Some of such effective synthetic routes include C–H carbonylative annulation of N,1-diaryl-1H-tetrazol-5-amines6, Ugi 4-component reaction7, diazotization of 1-benzyloxy-5-aminotetrazoles and 1-phenethyl-5-aminotetrazoles8, three-component reaction of 4-chloro-3-formylcoumarins, sodium azide, alkyl/aryl acetonitriles9, [3 + 2]cyclization of azidotrimethylsilane with quinoxalin-2(1H)-ones10, and so-on. Additionally, there is a simple procedure comprised of the multi-component reaction of active methylene compounds such as acetoacetic esters, diverse aldehydes, and 5-amino tetrazole, which is promoted by acid/base catalysts. The targeted products, which are a series of tetrazolopyrimidines, are known for their biological potentials as analgesic materials11, antimicrobial and antioxidant compounds12, anticancer agents13, and antitumor materials14. Different reports on the synthesis of tetrazolopyrimidines using (1,2,3-triazolium-N-butyl sulfonic acid phosphotungstate)15, HMTA-BAIL@MIL-101(Cr)16, Fe2O3@SiO2-(CH2)3NHC(O)(CH2)2PPh217, nano-Fe3O4@SiO2-NH-gallic acid18, and Mg–Al LDHs cross-linked poly triazine-urea-sulfonamide organic–inorganic hybrids have been published19. (MNCs) are believed to be effective alternatives for various toxic liquid acids and expensive solid catalysts. MNCs could be considered green catalysts as they can be recovered by a magnet and reused several times. Accordingly, a wide range of catalytic reactions have been reported in the literature including multi-component preparation of indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione derivatives using nano Fe2O3@SiO2-SO3H20, synthesis of 3-(9-methyl-9H-carbazol-3-yl)-2-arylthiazolidin-4-one derivatives using NiFe2O4@SiO2 grafted alkyl sulfonic acid21, preparation of 14-aryl-14H-dibenzo[a,j]xanthene derivatives using Fe3O4@SiO2 functionalized sulfonic acid22, preparation of chromeno[4,3-d]pyrido[1,2-a]pyrimidine derivatives using NiFe2O4@SiO2 grafted di(3-propylsulfonic acid) nanoparticles23, synthesis of anticancer drugs24, Heck and Suzuki reactions catalyzed by palladium nanoparticles stabilized on the amino acids-functionalized Fe3O425, reduction of organic pollutants by Fe3O4@CMC-Cu nano-catalyst26, and so-on.
Today, the main challenge in the use of catalytic systems is their ability to be recycled or not. In the absence of easy and practical recycling of catalysts, various environmental problems are created, which require a high cost to solve. Connecting functional groups such as –SO3H, –COOH, –NH, etc. to magnetic cores, in addition to creating heterogeneous catalytic systems, increases the possibility of easy and low-cost recycling and minimizes catalyst losses and, as a result, environmental problems. In addition, the easy recycling of the catalyst leads to a reduction in the production cost of the products. The main challenge in using magnetic particles is the very low potential of these particles in connecting to different groups and atoms. To solve this problem, magnetic particles are usually coated with polymer or silica layers, and core–shell structures like Fe3O4@SiO2 are created. The new structures have a high binding ability and at the same time increase the stability of the magnetic particles. Thus, the development of new magnetically separable catalysts is a great demand for synthetic chemists27,28,29,30,31,32,33,34.
In this study and the continuation of our research35,36,37,38,39,40,41,42,43,44, we intend to use a magnetic nano-catalyst for the three-component condensation of N,N′-(sulfonylbis(1,4-phenylene))bis(3-oxobutanamide), 1H-tetrazol-5-amine, and aromatic aldehydes for the synthesis of tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives. To achieve this aim, in this work Fe3O4@SiO2-(PP)(HSO4)2 (A) as an efficient magnetic hybrid nano-catalyst was prepared, characterized by FT-IR, XRD, FE-SEM, EDX, TGA-DTA, and VSM techniques, and was used for the catalytic synthesis of tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives.
Materials and methods
The complete procedures, material characterization, and instruments can be found in the supplementary data file attached to this paper.
Fe3O4 and Fe3O4@SiO2 nanoparticles were prepared according to our previous work22,27
General procedure
Method 1 In a 50 mL balloon equipped with a condenser, N,N'-(sulfonylbis(1,4-phenylene))bis(3-oxobutanamide) (1 mmol), 1H-tetrazol-5-amine (2 mmol), and benzaldehyde (2 mmol), and A (0.025g, 0.05 mmol H+) were mixed in DMF (20 mL) and the mixture was mechanically stirred at 100 °C under ultrasonic irradiation for the time depicted in Table 2. After the reaction was completed (TLC following), the solvent was evaporated under reduced pressure and the solid was recrystallized from ethanol to afford the desired products.
Method 2 a mixture of N,N′-(sulfonylbis(1,4-phenylene))bis(3-oxobutanamide) (1 mmol), 1H-tetrazol-5-amine (2 mmol), and benzaldehyde ( 2 mmol), and A (0.025g, 0.05 mmol H+) was heated at 100 °C under ultrasonic irradiation for the time depicted in Table 2. After the reaction was completed (TLC following), the was cooled and recrystallized from ethanol to afford the desired products.
Scaleup procedure
Different experiments were performed by increasing the scale of starting materials up to 20 × and 30 ×. All experiments proceeded successfully and the desired product was achieved in high yields. (20 ×: method 1: 3.5 h, 85%, method 2: 2.9 h, 88%; 30 ×: method 1: 4 h, 87%, method 2: 3 h, 84%).
Selected spectral data
N,N'-(Sulfonylbis(1,4-phenylene))bis(5-methyl-7-phenyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a1): 1H NMR (400 MHz, DMSO-d6): δ = 2.38 (s, 6H, CH3), 6.66 (s, 2H), 7.25 (t, J = 7.8 Hz, 4H), 7.28–33 (m, 6H), 7.38 (d, J = 8.0 Hz, 4H), 7.68 (d, J = 8.0 Hz, 4H), 8.97 (s, 2H), 10.12 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 19.7, 60.3, 97.7, 120.3, 124.8, 127.6, 128.1, 128.9, 130.8, 134.1, 135.7, 147.8, 151.2, 159.7 ppm; Elemental analysis: Found: C, 59.58; H, 4.23; N, 23.07; S, 4.44%; C36H30N12O4S; requires: C, 59.50; H, 4.16; N, 23.13; S, 4.41%.

Preparation of tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives using Fe3O4@SiO2-(PP)(HSO4)2 (A).
N,N′-(Sulfonylbis(1,4-phenylene))bis(5-methyl-7-(p-tolyl)-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a2): 1H NMR (400 MHz, DMSO-d6): δ = 2.26 (s, 6H, CH3), 2.36 (s, 6H, CH3), 6.59 (s, 2H), 7.06 (d, J = 7.8 Hz, 4H), 7.17 (d, J = 7.8 Hz, 4H), 7.38 (d, J = 8.0 Hz, 4H), 7.66 (d, J = 8.0 Hz, 4H), 8.78 (s, 2H), 10.18 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 19.9, 21.1, 59.3, 98.6, 120.7, 124.8, 126.7, 127.9, 130.8, 134.4, 135.8, 136.9, 148.1, 151.3, 160.3 ppm; Elemental analysis: Found: C, 60.35; H, 4.49; N, 22.28; S, 4.23%; C38H34N12O4S; requires: C, 60.47; H, 4.54; N, 22.27; S, 4.25%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(4-methoxyphenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a3): 1H NMR (400 MHz, DMSO-d6): δ = 2.37 (s, 6H, CH3), 3.82 (s, 6H, OCH3), 6.47 (s, 2H), 6.94 (d, J = 7.8 Hz, 4H), 7.03 (d, J = 7.8 Hz, 4H), 7.38 (d, J = 8.0 Hz, 4H), 7.66 (d, J = 8.0 Hz, 4H), 8.78 (s, 2H), 10.18 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 19.8, 55.4, 59.0, 98.6, 118.7, 120.1, 123.4, 124.7, 130.4, 134.2, 135.8, 148.0, 151.4, 155.7, 160.0 ppm; Elemental analysis: Found: C, 58.09; H, 4.44; N, 21.42; S, 4.16%; C38H34N12O6S; requires: C, 58.01; H, 4.36; N, 21.36; S, 4.07%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(4-chlorophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a4): 1H NMR (400 MHz, DMSO-d6): δ = 2.41 (s, 6H, CH3), 6.69 (s, 2H), 7.34–7.38 (m, 8H), 7.43 (d, J = 7.8 Hz, 4H), 7.67 (d, J = 8.2 Hz, 4H), 8.96 (s, 2H), 10.25 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.6, 62.3, 98.9, 120.4, 124.7, 128.4, 129.2, 130.6, 134.2, 136.1, 144.3, 148.2, 151.1, 160.3 ppm; Elemental analysis: Found: C, 54.38; H, 3.61; N, 21.08; S, 3.97%; C36H28Cl2N12O4S; requires: C, 54.34; H, 3.55; N, 21.13; S, 4.03%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(4-bromophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a5): 1H NMR (400 MHz, DMSO-d6): δ = 2.42 (s, 6H, CH3), 6.72 (s, 2H), 7.38 (d, J = 8.0 Hz, 4H), 7.41 (d, J = 7.8 Hz, 4H), 7.63 (d, J = 7.8 Hz, 4H), 7.69 (d, J = 8.0 Hz, 4H), 8.91 (s, 2H), 10.22 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.5, 62.1, 98.6, 120.1, 124.6, 128.6, 129.4, 130.7, 134.8, 136.4, 146.3, 148.4, 151.0, 160.2 ppm; Elemental analysis: Found: C, 48.85; H, 3.17; N, 18.96; S, 3.64%; C36H28Br2N12O4S; requires: C, 48.88; H, 3.19; N, 19.00; S, 3.62%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(5-methyl-7-(4-nitrophenyl)-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a6): 1H NMR (400 MHz, DMSO-d6): δ = 2.42 (s, 6H, CH3), 6.85 (s, 2H), 7.39 (d, J = 8.3 Hz, 4H), 7.68–72 (m, 8H), 8.28 (d, J = 7.9 Hz, 4H), 8.95 (s, 2H), 10.31 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.5, 63.6, 100.6, 120.9, 124.6, 128.7, 129.8, 130.6, 134.8, 136.7, 138.4, 148.5, 151.2, 160.5 ppm; Elemental analysis: Found: C, 52.88; H, 3.41; N, 24.04; S, 3.85%; C36H28N14O8S; requires: C, 52.94; H, 3.46; N, 24.01; S, 3.93%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(5-methyl-7-(3-nitrophenyl)-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a7): 1H NMR (400 MHz, DMSO-d6): δ = 2.42 (s, 6H, CH3), 6.83 (s, 2H), 7.32 (t, J = 7.8 Hz, 2H), 7.39 (d, J = 8.2 Hz, 4H), 7.56 (d, J = 7.8 Hz, 2H), 7.70 (d, J = 7.9 Hz, 4H), 8.21 (d, J = 7.9 Hz, 2H), 8.39 (s, 2H), 8.90 (s, 2H), 10.27 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.4, 63.8, 100.1, 120.6, 124.4, 127.6, 128.1, 129.4, 130.4, 131.2, 134.8, 136.9, 138.2, 148.7, 151.0, 160.8 ppm; Elemental analysis: Found: C, 52.99; H, 3.53; N, 24.02; S, 3.81%; C36H28N14O8S; requires: C, 52.94; H, 3.46; N, 24.01; S, 3.93%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(3-chlorophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a8): 1H NMR (400 MHz, DMSO-d6): δ = 2.39 (s, 6H, CH3), 6.63 (s, 2H), 7.26 (t, J = 7.8 Hz, 2H), 7.31 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 8.2 Hz, 4H), 7.44 (d, J = 7.8 Hz, 2H), 7.49 (s, 2H), 7.68 (d, J = 7.9 Hz, 4H), 8.92 (s, 2H), 10.17 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.1, 61.3, 98.7, 120.1, 124.5, 127.2, 128.1, 129.2, 129.3, 130.4, 134.5, 136.4, 144.2, 148.0, 151.3, 160.4 ppm; Elemental analysis: Found: C, 54.41; H, 3.63; N, 21.06; S, 4.07%; C36H28Cl2N12O4S; requires: C, 54.34; H, 3.55; N, 21.13; S, 4.03%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(3,4-dichlorophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a9): 1H NMR (400 MHz, DMSO-d6): δ = 2.39 (s, 6H, CH3), 6.66 (s, 2H), 7.33 (d, J = 7.8 Hz, 2H), 7.37 (d, J = 8.3 Hz, 4H), 7.45 (d, J = 7.8 Hz, 2H), 7.51 (s, 2H), 7.67 (d, J = 8.3 Hz, 4H), 8.88 (s, 2H), 10.14 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.3, 61.6, 98.6, 120.2, 124.6, 128.7, 129.3, 130.1, 130.5, 134.7, 136.6, 144.3, 144.8, 148.5, 151.4, 160.6 ppm; Elemental analysis: Found: C, 50.08; H, 3.09; N, 19.40; S, 3.66%; C36H26Cl4N12O4S; requires: C, 50.01; H, 3.03; N, 19.44; S, 3.71%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(2,4-dichlorophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a10): 1H NMR (400 MHz, DMSO-d6): δ = 2.39 (s, 6H, CH3), 6.69 (s, 2H), 7.32 (d, J = 7.9 Hz, 2H), 7.37 (d, J = 8.1 Hz, 4H), 7.46 (d, J = 7.9 Hz, 2H), 7.53 (s, 2H), 7.68 (d, J = 8.1 Hz, 4H), 8.76 (s, 2H), 10.23 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.3, 61.9, 100.2, 120.6, 124.7, 128.4, 129.1, 129.5, 130.1, 134.7, 136.7, 144.1, 144.6, 148.2, 151.3, 160.4 ppm; Elemental analysis: Found: C, 49.98; H, 3.07; N, 19.43; S, 3.64%; C36H26Cl4N12O4S; requires: C, 50.01; H, 3.03; N, 19.44; S, 3.71%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(3,5-dichlorophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a11): 1H NMR (400 MHz, DMSO-d6): δ = 2.39 (s, 6H, CH3), 6.68 (s, 2H), 7.37 (d, J = 8.2 Hz, 4H), 7.47 (s, 4H), 7.50 (s, 2H), 7.68 (d, J = 8.2 Hz, 4H), 8.85 (s, 2H), 10.19 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.0, 62.3, 99.2, 120.4, 124.5, 129.3, 130.4, 130.7, 134.7, 136.4, 144.3, 148.2, 151.1, 160.2 ppm; Elemental analysis: Found: C, 50.06; H, 3.13; N, 19.51; S, 3.78%; C36H26Cl4N12O4S; requires: C, 50.01; H, 3.03; N, 19.44; S, 3.71%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(2-chlorophenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a12): 1H NMR (400 MHz, DMSO-d6): δ = 2.37 (s, 6H, CH3), 6.61 (s, 2H), 7.25–7.28 (m, 4H), 7.32 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 8.0 Hz, 4H), 7.43 (d, J = 7.8 Hz, 2H), 7.68 (d, J = 8.0 Hz, 4H), 8.90 (s, 2H), 10.19 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.1, 61.7, 98.2, 120.1, 124.4, 127.1, 127.6, 128.1, 128.4, 128.9, 134.4, 136.5, 145.2, 148.4, 151.3, 160.6 ppm; Elemental analysis: Found: C, 54.38; H, 3.67; N, 21.17; S, 4.11%; C36H28Cl2N12O4S; requires: C, 54.34; H, 3.55; N, 21.13; S, 4.03%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(7-(furan-2-yl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a13): 1H NMR (400 MHz, DMSO-d6): δ = 2.34 (s, 6H, CH3), 5.81 (s, 2H), 6.49 (d, J = 6.8 Hz, 2H), 6.56 (t, J = 6.9 Hz, 2H), 7.38–7.41 (m, 6H), 7.68 (d, J = 8.2 Hz, 4H), 8.55 (s, 2H), 10.09 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 19.7, 56.7, 97.2, 104.6, 111.3, 119.7, 123.4, 124.5, 127.9, 131.4, 134.1, 148.1, 151.2, 159.7 ppm; Elemental analysis: Found: C, 54.36; H, 3.65; N, 23.71; S, 4.45%; C32H26N12O6S; requires: C, 54.39; H, 3.71; N, 23.78; S, 4.54%.
N,N'-(Sulfonylbis(1,4-phenylene))bis(5-methyl-7-(2-oxo-2H-chromen-4-yl)-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide) (Scheme 1, Product a14): 1H NMR (400 MHz, DMSO-d6): δ = 2.39 (s, 6H, CH3), 6.45 (s, 2H), 6.81 (s, 2H), 6.98 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.1 Hz, 4H), 7.43 (t, J = 8.2 Hz, 2H), 7.69 (d, J = 8.2 Hz, 4H), 7.76 (t, J = 8.2 Hz, 2H), 7.92 (d, J = 8.1 Hz, 2H), 8.91 (s, 2H), 10.29 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.7, 63.7, 98.1, 102.3, 106.6, 119.7, 124.5, 127.8, 128.6, 129.3, 131.4, 133.7, 134.7, 138.9, 148.2, 151.2, 155.6, 161.7, 173.8 ppm; Elemental analysis: Found: C, 58.43; H, 3.55; N, 19.49; S, 3.68%; C42H30N12O8S; requires: C, 58.47; H, 3.50; N, 19.48; S, 3.72%.
7-(9-Ethyl-9H-carbazol-2-yl)-N-(4-((4-(7-(9-ethyl-9H-carbazol-3-yl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamido)phenyl)sulfonyl)phenyl)-5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide (Scheme 1, Product a15): 1H NMR (400 MHz, DMSO-d6): δ = 0.97 (t, J = 6.4 Hz, 6H), 2.37 (s, 6H, CH3), 3.49 (q, J = 6.4 Hz, 4H), 6.21 (s, 2H), 6.84 (d, J = 8.0 Hz, 2H), 7.02 (s, 2H), 7.18 (t, J = 8.0 Hz, 2H), 7.24–7.27 (m, 4H), 7.36–7.40 (m, 6H), 7.67 (d, J = 8.1 Hz, 4H), 7.82 (d, J = 8.1 Hz, 2H), 8.89 (s, 2H), 10.11 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 15.3, 20.8, 34.9, 64.7, 98.6, 107.1, 108.6, 111.8, 112.3, 114.9, 115.6, 123.5, 126.7, 127.4, 17.8, 128.6, 129.3, 134.4, 136.4, 137.1, 137.8, 148.2, 151.4, 162.9 ppm; Elemental analysis: Found: C, 65.07; H, 4.69; N, 20.45; S, 3.38%; C52H44N14O4S; requires: C, 64.99; H, 4.61; N, 20.40; S, 3.34%.
5-Methyl-N-(4-((4-(5-methyl-7-(9-methyl-9H-carbazol-2-yl)-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamido)phenyl)sulfonyl)phenyl)-7-(9-methyl-9H-carbazol-3-yl)-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide (Scheme 1, Product a16): 1H NMR (400 MHz, DMSO-d6): δ = 2.37 (s, 6H, CH3), 3.43 (s, 6H), 6.23 (s, 2H), 6.85 (d, J = 8.2 Hz, 2H), 7.04 (s, 2H), 7.18 (t, J = 8.2 Hz, 2H), 7.23–7.27 (m, 4H), 7.35–7.39 (m, 6H), 7.67 (d, J = 8.0 Hz, 4H), 7.81 (d, J = 8.2 Hz, 2H), 8.96 (s, 2H), 10.18 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 20.6, 34.1, 64.7, 97.9, 107.0, 108.3, 111.2, 112.4, 114.7, 115.1, 123.9, 126.6, 127.4, 17.7, 128.6, 129.2, 134.4, 136.3, 136.8, 137.4, 148.2, 151.1, 162.2 ppm; Elemental analysis: Found: C, 64.29; H, 4.37; N, 21.06; S, 3.38%; C50H40N14O4S; requires: C, 64.37; H, 4.32; N, 21.02; S, 3.44%.
Results and discussion
Fe3O4@SiO2-(PP)(HSO4)2 (A): preparation and characterization
First, the preparation of Fe3O4@SiO2-functionalized propylpiperazine-1,4-diium dihydrogensulfate (A) is reported. Catalyst (A) was prepared in three steps, as shown in Scheme 2. First, piperazine was reacted with (3-chloropropyl)trimethoxysilane to form intermediate A1. Et3N was then used to trap the HCl gas. Next, A1 was reacted with Fe3O4@SiO2 nanoparticles to form A2. The final step was the acidification of (A2) to form Fe3O4@SiO2-(PP)(HSO4)2 (A) as the final product. The successful grafting of the organic part to Fe3O4@SiO2 nanoparticles was investigated by FT-IR analysis. The FT-IR spectra of catalyst A, Fe3O4, intermediate A2, and Fe3O4@SiO2 nanoparticles, could be seen in Fig. 1. The FT-IR spectrum of Fe3O4 nanoparticles shows distinctive peaks below 600 cm−1 related to the Fe–O bonds (stretching vibration). However, in the FT-IR spectrum of Fe3O4@SiO2 nanoparticles, in addition to the peak corresponding to the F-O bond (below 600 cm−1), there are distinctive peaks as Si–O, Si–OH, and Si–O–Si at 1622, 1028, 915, and 871 cm−1 (stretching and bending vibrations) respectively. The FT-IR spectrum of intermediate A2 shows distinctive peaks at 3694 (N–H), 2988, 2944 (C–H), 1584 (Si–O), 1373, 1221, 1129, 1037, 892, 674, and 490 (Fe–O) cm−1 ascribed to the vibration of C–H, C–C, Si–O-Si, Fe–O and C–N bonds. Compared to the FT-IR spectra of A2, some changes are observed in the spectra of sample A. The peaks located at 3706 and 3669cm−1 are related to the N–H vibration bonds. The sulfonic acid groups have a broad peak at 3000–3600 cm−1.

Preparation of Fe3O4@SiO2-(PP)(HSO4)2 (A).

FT-IR spectra of intermediate A2 and Fe3O4@SiO2-(PP)(HSO4)2 (A) (Down), Fe3O4 and Fe3O4@SiO2 nanoparticles (Up).
FE-SEM images (Fig. 2) were used to investigate the surface morphology of the as-prepared Fe3O4@SiO2-(PP)(HSO4)2 (A). As observed in the images, the sample has a homogeneously spherical morphology with an average diameter of less than 100 nm.

FE-SEM images of Fe3O4@SiO2-(PP)(HSO4)2 (A).
To further characterize Fe3O4@SiO2-(PP)(HSO4)2 (A), the samples were subjected to XRD analysis to determine the crystalline phases. Figure 3 shows the XRD patterns of Fe3O4 and (A). The XRD pattern of Fe3O4 nanoparticles demonstrates prominent peaks at 30.6, 35.4, 44.3, 53.9, 57.4, 64.5, and 75.0 [2Ɵ°], indicating a cubic structure for Fe3O4 [Reference code: 00-001-1111]. The XRD pattern of sample (A) shows a similar pattern with a shoulder located in the 10–30 (2Ɵ°) range, which may be due to the amorphous phase of silica. Furthermore, the peaks related to the Fe3O4 phase have lower intensities due to the integration of organic parts and the SiO2 phase.

XRD patterns of Fe3O4, fresh, and recovered Fe3O4@SiO2-(PP)(HSO4)2 (A) catalyst.
Fe3O4@SiO2-(PP)(HSO4)2 (A): thermal stability and chemical composition
The chemical composition of Fe3O4@SiO2-(PP)(HSO4)2 (A) was determined by EDX analysis (Fig. 4). The EDX analysis indicates the presence of Fe (32.84%), Si (15.01%), S (6.89%), N (2.21%), C (6.71%), and O (39.04%), confirming the integration of the organic part and sulfate group into Fe3O4@SiO2. The presence of Fe, Si, S, C, N, and O elements indicates the formation of Fe3O4@SiO2-(PP)(HSO4)2 (A).

EDX analysis of Fe3O4@SiO2-(PP)(HSO4)2 (A).
Next, the thermal behavior of (A) was investigated by TGA-DTA analysis (Fig. 5). The sample is stable up to 200 °C and shows four different mass losses due to the removal of the adsorbed water (50–200 °C), removal of SOX gases (200–320 °C), decomposition of the organic part by the removal of CO2, H2O, and NOx gases (300–550 °C), and the formation of SiO2 phase (500–800 °C).45 Accordingly, the ratio of inorganic to organic parts is nearly 2/1, which is close to the ratio of the initial substrates.

TGA-DTA analysis of Fe3O4@SiO2-(PP)(HSO4)2 (A).
Determination of active sites
The sample has an acidic nature and thus, the determination of H+ values is important to investigate the role and determine the conditions for the application of the sample as a catalyst. The values of H+ were determined by EDX analysis, TGA method, and barium sulfate (BaSO4) titration-precipitation test. The obtained results are shown in Table 1.
The sulfur values of the sample were determined through the sulfur element percent in the results of EDX analysis (S, 6.89%). Similarly, the amount of S atoms could be determined by the values of SOx removal using TGA. The BaSO4 method involves titration by barium chloride solution. Accordingly, the H+ capacities of the sample were found to be 2.15, 2.71, and 2.03 mmol H+/g by EDX, TGA, and BaSO4 tests, respectively.
To assure the desirable performance and facile separation of the nano-catalyst, by a magnetic field, VSM analysis was used. Figure 6 shows the plotted results of VSM analysis performed at 25°C within the magnetic field of − 10,000 to 10,000 Oe. According to the hysteresis curves shown in Fig. 6, the functionalization of the Fe3O4 decreased the VSM characteristic values including saturation magnetization (Ms), remanence magnetization (Mr), and coercivity field (Hc) (Table 2). However, Fe3O4 exhibited a considerable magnetic nature.

VSM analysis of (A), Fe3O4, and Fe3O4@SiO2 samples.
In organic–inorganic hybrids, such as those used here, the organic part hurts the saturation magnetization. The organic chain has a diamagnetic effect and accordingly, the sample (A) shows lower magnetic saturation than Fe3O4 and Fe3O4@SiO2 samples. In addition, our observations confirm the easy recovery of the catalyst by an external magnet.
Preparation of tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives
Reaction condition optimization
The prepared sample (A) was then used in the synthesis of tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives to act as a catalyst. Initially, the reaction of N,N'-(sulfonylbis(1,4-phenylene))bis(3-oxobutanamide), 1H-tetrazol-5-amine, and benzaldehyde was chosen as a model for the synthesis of 5-methyl-N-(4-((4-(5-methyl-7-phenyl-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidine-6-carboxamido)phenyl)sulfonyl)phenyl)-7-phenyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxamide (a1). To determine the optimal conditions in the synthesis of compound (a1), the model reaction was studied using different solvents, catalyst dosages, and temperatures (Table 3). According to the results obtained, the reaction did not proceed at low temperatures. In addition, non-polar, less polar, and polar solvents with boiling point less than 100°C such as hexane, dichloromethane (CH2Cl2), chloroform (CHCl3), and ethyl acetate (EtOAc) were not suitable for the reaction. In aqueous media, no products were formed. Upon increasing the reaction temperature up to 100 °C, the reaction yields in tetrahydrofuran (THF) and toluene were 56 and 20%, respectively. Notably, the reaction had an acceptable yield in dimethyl formamide (DMF, 79%). To obtain better product yields, ultrasonic irradiation (US) was used. A high product yield (95%) was obtained under ultrasonic irradiation using DMF solvent. Notably, under solvent-free conditions and ultrasonic irradiation, the desired product (a1) was formed in a high yield (91%) at 100 °C.
Next, the reaction was investigated using different dosages of the catalyst. In the absence of the catalyst, no product was formed. The results revealed that 0.5 g of the catalyst gave the highest yield of the product (a1). Thus, DMF solvent and solvent-free conditions were selected as the two best media for the reaction while the suitable catalyst dosage was determined as 0.05 g, as it provided the highest yields at reasonable reaction times (Table 3).
Under the optimized conditions, the scope of the reaction was expanded using various aromatic and aliphatic aldehydes. The results are shown in Table 3. Accordingly, when aliphatic aldehydes were used, no product was formed. However, different aromatic aldehydes were found to be appropriate substrates in the reaction. The electronic effects of the substituents on the aromatic ring in the aromatic aldehydes are expected to affect the reaction rate. Based on the results obtained, electron-donating substituents increased the reaction rate, contrary to electron-withdrawing groups (Table 4).
Scheme 3 shows a plausible proposed reaction mechanism for the synthesis of compounds a1–a18. As suggested, the Brønsted acid catalyst activates the carbonyl groups. The reaction starts with the reaction of NH2 group with the activated carbonyl groups to form an enamine active compound (Intermediate I1). The next step is the reaction of I1 with the activated aldehyde to form I2. Intermediate I2 undergoes cyclization and enamine formation to yield the final products.

Proposed mechanism for the synthesis of tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives using Fe3O4@SiO2-(PP)(HSO4)2 (A).
Finally, an external magnet could be used to recover the catalyst, which was then washed with ethanol, dried, and used again. The preparation of (a1) was chosen for the recovery test. The recovery experiments showed acceptable results after 10 catalytic runs (Fig. 7). The XRD pattern of the recovered catalyst confirmed the stability of the catalyst during the reaction (Fig. 1). In addition, after each run, the recovered catalyst was tested using titration by barium chloride solution. The results indicated good catalyst stability and no clear leaching was observed.

Recovery of A (Methods 1,2) and leaching test results in the synthesis of (a1).
Conclusion
In this work, tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives were prepared using Fe3O4@SiO2-(PP)(HSO4)2 (A) as a catalyst. The TGA-DTA analysis indicated the stability of this organic–inorganic hybrid up to 200 °C. In addition, the ratio of the inorganic to organic parts was 2/1, which was close to that of the initial substrates. Using the barium chloride titration test, the H+ capacity of the sample was determined to be 2.03 mmol H+/g. The XRD pattern of the fresh and recovered samples (A) confirmed the stability of the catalyst. The results showed promising potential and easy recovery of magnetic nano-catalysts. The obtaining of reasonably high yields in short reaction times and readily available starting materials make this protocol potentially useful in organic synthesis.
Data availability
The spectral data, which could support our findings, are available as a supplementary material attached to this article.
References
Gwynn, P.E., 2009. Synthesis of Fused Heterocycles. Chemistry of Heterocyclic Compounds: A Series of Monographs. 47, 1. eISBN: 9780470188835.
Zhang, T.-J. et al. Design, synthesis and biological evaluation of N-(3-(1H-tetrazol-1-yl)phenyl)isonicotinamide derivatives as novel xanthine oxidase inhibitors. Eur. J. Med. Chem. 183, 111717. https://doi.org/10.1016/j.ejmech.2019.111717 (2019).
Szulczyk, D. et al. Development of (4-methoxyphenyl)-1H-tetrazol-5-amine regioisomers as a new class of selective antitubercular agents. Eur. J. Med. Chem. 186, 111882. https://doi.org/10.1016/j.ejmech.2019.111882 (2020).
Szulczyk, D. et al. Design and synthesis of novel 1H-tetrazol-5-amine based potent antimicrobial agents: DNA topoisomerase IV and gyrase affinity evaluation supported by molecular docking studies. Eur. J. Med. Chem. 156, 631–640. https://doi.org/10.1016/j.ejmech.2018.07.041 (2018).
Rajasekaran, A. & Thampi, P. P. Synthesis and antinociceptive activity of some substituted-{5-[2-(1,2,3,4-tetrahydrocarbazol-9-yl)ethyl]tetrazol-1-yl}alkanones. Eur. J. Med. Chem. 40, 1359–1364. https://doi.org/10.1016/j.ejmech.2005.07.013 (2005).
Chandrasekhar, A., Ramkumara, V. & Sankararaman, S. Palladium catalyzed carbonylative annulation of the C(sp2)–H bond of N,1-diaryl-1H-tetrazol-5-amines and N,4-diaryl-4H-triazol-3-amines to quinazolinones. Org. Biomol. Chem. 16, 8629–8638. https://doi.org/10.1039/C8OB02516A (2018).
Nixey, T., Kelly, M., Semin, D. & Hulme, C. Short solution phase preparation of fused azepine-tetrazoles via a UDC (Ugi/de-Boc/Cyclize) strategy. Tetrahedron Lett. 43, 3681–3684. https://doi.org/10.1016/S0040-4039(02)00636-6 (2002).
Ha, H.-J. et al. Synthesis of tricyclic tetrazoles by cascade diazotization/intramolecular radical C-H heteroarylation of arenes. J. Org. Chem. 88, 2714–2725. https://doi.org/10.1021/acs.joc.2c02187 (2023).
Borah, P., Naidu, P. S. & Bhuyan, P. J. Synthesis of some tetrazole fused pyrido[2,3-c]coumarin derivatives from a one-pot three-component reaction via intramolecular 1,3-dipolar cycloaddition reaction of azide to nitriles. Tetrahedron Lett. 53, 5034–5037. https://doi.org/10.1016/j.tetlet.2012.07.060 (2012).
Yang, Q. et al. [3+2] Cyclization of azidotrimethylsilane with quinoxalin-2(1H)-ones to synthesize tetrazolo[1,5-a]quinoxalin-4(5H)-ones. Adv. Synth. Catal. 360, 4509–4514. https://doi.org/10.1002/adsc.201801076 (2018).
Gein, V. L., Prudnikova, A. N., Kurbatova, A. A. & Rudakova, I. P. Analgesic activity and acute toxicity of dihydrotetrazolo[1,5-a]pyrimidine derivatives. Pharm. Chem. J. 55, 228–230. https://doi.org/10.1007/s11094-021-02403-2 (2021).
Raju, C., Madhaiyan, K., Uma, R., Sridhar, R. & Ramakrishna, S. Antimicrobial and antioxidant activity evaluation of tetrazolo[1,5-a]pyrimidines: A simple diisopropylammonium trifluoroacetate mediated synthesis. RSC Adv. 2, 11657–11663. https://doi.org/10.1039/C2RA21330C (2012).
Wu, L., Liu, Y. & Li, Y. Synthesis of spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine hybrids as potential anticancer agents. Molecules 23, 2330. https://doi.org/10.3390/molecules23092330 (2018).
Wu, L. Q. Synthesis and biological evaluation of novel 1,2-naphthoquinones possessing tetrazolo[1,5-a]pyrimidine scaffolds as potent antitumor agents. RSC Adv. 5, 24960–24965. https://doi.org/10.1039/C5RA00711A (2015).
Taib, L. A. & Keshavarz, M. A fascinating click strategy to novel 1,2,3-triazolium based organic-inorganic hybrids for highly accelerated preparation of tetrazolopyrimidines. Polyhedron 213, 115630. https://doi.org/10.1016/j.poly.2021.115630 (2022).
Abdollahi-Basir, M. H., Mirhosseini-Eshkevari, B., Zamani, F. & Ghasemzadeh, M. A. Synthesis of tetrazolo[1,5-a]pyrimidine-6-carbonitriles using HMTA-BAIL@MIL-101(Cr) as a superior heterogeneous catalyst. Sci. Rep. 11, 5109. https://doi.org/10.1038/s41598-021-84379-3 (2021).
Maleki, A., Rahimi, J., Demchuk, O. M., Wilczewska, A. Z. & Jasiński, R. Green in water sonochemical synthesis of tetrazolopyrimidine derivatives by a novel core-shell magnetic nanostructure catalyst. Ultrason. Sonochem. 43, 262–271. https://doi.org/10.1016/j.ultsonch.2017.12.047 (2018).
Maleki, A., Niksefat, M., Rahimi, J. & Azadegan, S. Facile synthesis of tetrazolo[1,5-a]pyrimidine with the aid of an effective gallic acid nanomagnetic catalyst. Polyhedron 167, 103–110. https://doi.org/10.1016/j.poly.2019.04.015 (2019).
Shekarlab, N., Ghorbani-Vaghei, R. & Alavinia, S. Nickel (II) coordination on cross-linked poly triazine-urea-sulfonamide grafted onto Mg-Al LDHs: As a green catalytic system for the synthesis of tetrazolo[1,5-a] pyrimidines. J. Organometal. Chem. 949, 121971. https://doi.org/10.1016/j.jorganchem.2021.121971 (2021).
Ghashang, M., Guhanathan, S. & Mansoor, S. S. Nano Fe2O3@SiO2-SO3H: Efficient catalyst for the multi-component preparation of indeno[2′,1′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione derivatives. Res. Chem. Intermed. 43, 7257–7276. https://doi.org/10.1007/s11164-017-3073-6 (2017).
Hajiarab, R., Mohammad Shafiee, M. R. & Ghashang, M. Access to a library of 3-(9-methyl-9H-carbazol-3-yl)-2-arylthiazolidin-4-one derivatives using NiFe2O4@SiO2 grafted alkyl sulfonic acid as an efficient catalyst. Polycycl. Aromat. Compound. 43, 2032–2043. https://doi.org/10.1080/10406638.2022.2039231 (2023).
Khalaj, M., Taherkhani, M., Samadi Kazemi, M., Kalhor, M. & Talebian Dehkordy, G. New nanoparticles of Fe3O4@SiO2 functionalized sulfonic acid magnetic properties and catalytic investigation on the multi-component preparation of some multi-component preparation of some organic compounds. Polycycl. Aromat. Compound. 42, 7354–7367. https://doi.org/10.1080/10406638.2021.1998155 (2022).
Khalaj, M., Taherkhani, M. & Kalhor, M. Preparation of some chromeno[4,3-d]pyrido[1,2-a]pyrimidine derivatives by ultrasonic irradiation using NiFe2O4@SiO2 grafted di (3-propylsulfonic acid) nanoparticles. New J. Chem. 45, 10718. https://doi.org/10.1039/D1NJ01676H (2021).
Khan, S. et al. Magnetic nanocatalysts as multifunctional platforms in cancer therapy through the synthesis of anticancer drugs and facilitated Fenton reaction. J. Adv. Res. 30, 171–184. https://doi.org/10.1016/j.jare.2020.12.001 (2021).
Naeim, M., Naghipour, A., Fakhri, A. & Sayadi, M. Palladium nanoparticles stabilized on the amino acids-functionalized Fe3O4 as the magnetically recoverable nanocatalysts for Heck and Suzuki reactions. Inorg. Chim. Acta 542, 121109. https://doi.org/10.1016/j.ica.2022.121109 (2022).
Ahmad, I., Abbasi, A., Mangla, D. & Ikram, S. Unraveling the catalytic efficacy of copper(II)-anchored carboxymethyl cellulose as a magnetically recyclable nanocatalyst for effective reduction of organic pollutants. Catal. Commun. 181, 106724. https://doi.org/10.1016/j.catcom.2023.106724 (2023).
Khalaj, M. & Zarandi, M. A Cu (II) complex supported on Fe3O4@SiO2 as a magnetic heterogeneous catalyst for the reduction of environmental pollutants. RSC Adv. 12, 26527. https://doi.org/10.1039/D2RA04787J (2022).
Zhang, M., Liu, Y.-H., Shang, Z.-R., Hu, H.-C. & Zhang, Z.-H. Supported molybdenum on graphene oxide/Fe3O4: An efficient, magnetically separable catalyst for one-pot construction of spiro-oxindole dihydropyridines in deep eutectic solvent under microwave irradiation. Catal. Commun. 88, 39–44. https://doi.org/10.1016/j.catcom.2016.09.028 (2017).
Gao, G., Di, J.-Q., Zhang, H.-Y., Mo, L.-P. & Zhang, Z.-H. A magnetic metal organic framework material as a highly efficient and recyclable catalyst for synthesis of cyclohexenone derivatives. J. Catal. 387, 39–46. https://doi.org/10.1016/j.jcat.2020.04.013 (2020).
Xu, L., Zhang, S.-Z., Li, W. & Zhang, Z.-H. Visible-light-mediated oxidative amidation of aldehydes by using magnetic CdS quantum dots as a photocatalyst. Chem. - Eur. J. 27, 5483–5491. https://doi.org/10.1002/chem.202005138 (2021).
Deng, Q., Shen, Y., Zhua, H. & Tu, T. A magnetic nanoparticle-supported N-heterocyclic carbene-palladacycle: An efficient and recyclable solid molecular catalyst for Suzuki-Miyaura cross-coupling of 9-chloroacridine. Chem. Commun. 53, 13063–13066. https://doi.org/10.1039/c7cc06958h (2017).
Di, J.-Q. et al. Copper anchored on phosphorus g-C3N4 as a highly efficient photocatalyst for the synthesis of N-arylpyridin-2-amines. Green Chem. 23, 1041–1049. https://doi.org/10.1039/D0GC03400B (2021).
Wang, Y.-M., Li, W.-J., Wang, M.-M., Zhang, M. & Zhang, Z.-H. Magnetic MoS2 efficient heterogeneous photocatalyst for the α-methoxymethylation and aminomethylation of aromatic ketones. Catal. Sci. Technol. 13, 665–674. https://doi.org/10.1039/D2CY01831D (2023).
Hajiarab, R., Shafiee, M. R. M. & Ghashang, M. Preparation of thiazolidin-4-ones using NiFe2O4@SiO2 grafted propylsulfonic acid as an efficient catalyst. Org. Prep. Proced. Int. 54, 259–267. https://doi.org/10.1080/00304948.2022.2033064 (2022).
Khalaj, M., Mousavi-Safavi, S. M., Farahani, N. & Lipkowski, J. MgO nanopowders catalyzed synthesis of pyrano[4,3-d]thiazolo[3,2-a]pyrimidine derivatives. Appl. Organomet. Chem. 34, e5865. https://doi.org/10.1002/aoc.5865 (2020).
Khalaj, M. Preparation of benzo[4,5]thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6-one derivatives using MgO-MgAl2O4 composite nano-powder. Arab. J. Chem. 13, 6403. https://doi.org/10.1016/j.arabjc.2020.05.041 (2020).
Khalaj, M., Taherkhani, M., Payen, L. & Klein, A. A sulfonic acid polyvinyl pyridinium ionic liquid catalyzes the multi-component synthesis of spiro-indoline-3,5-pyrano[2,3-d]-pyrimidines and –pyrazines. Molecules 28, 3663. https://doi.org/10.3390/molecules28093663 (2023).
Khalaj, M. The preparation of a library of fused nitrogen heterocyclic compounds catalyzed by Zn2SnO4 nanoparticles. Polycycl. Aromat. Compound. 43, 5629. https://doi.org/10.1080/10406638.2022.2105910 (2023).
Kalhor, M., Orouji, Z. & Khalaj, M. 4-Methylpyridinium chloride ionic liquid grafted on Mn@zeolite-Y: Design, fabrication and performance as a novel multi-functional nanocatalyst in the four-component synthesis of pyrazolophthalazine-diones. Microporous Mesoporous Mater. 329, 111498. https://doi.org/10.1016/j.micromeso.2021.111498 (2022).
Maham, M. & Khalaj, M. Synthesis of thiotetrazoles and arylaminotetrazoles using rutile TiO2 nanoparticles as a heterogeneous and reusable catalyst. J. Chem. Res. 38, 502. https://doi.org/10.3184/174751914X14061217395813 (2014).
Sajadi, M., Khalaj, M., Hosseini Jamkarani, S. M., Maham, M. & Kashefi, M. Aluminum(III) hydrogensulfate: An efficient solid acid catalyst for the preparation of 5-substituted 1H-tetrazoles. Synth. Commun. 41, 3053. https://doi.org/10.1080/00397911.2010.516861 (2011).
Khalaj, M. & Ghazanfarpour, M. Regioselective synthesis of 1,4-oxathiane derivatives via multicomponent reaction. Monatsh Chem. 147, 2043. https://doi.org/10.1007/s00706-016-1845-0 (2016).
Khalaj, M., Sadeghpour, M., Mousavi, M. & Lalegani, A. Copper-catalyzed synthesis of thiazolidine derivatives via multicomponent reaction of terminal alkynes, elemental sulfur, and aziridines. Monatsh Chem. 150, 1085. https://doi.org/10.1007/s00706-019-2363-7 (2019).
Taghrir, H. et al. Barium silicate nanoparticles, an efficient catalyst for one-pot green synthesis of α-benzyl amino coumarin derivatives as potential chemotherapeutic agents. RSC Adv. 13, 21127–21137. https://doi.org/10.1039/D3RA00796K (2023).
Kiasat, A. R., Chadorian, F. & Saghanezhad, S. J. Synthesis and characterization of a novel Fe3O4@SiO2/bipyridinium dichloride nanocomposite and its application as a magnetic and recyclable phase-transfer catalyst in the preparation of β-azidoalcohols, β-cyanohydrins, and β-acetoxy alcohols. Compt. Rend. Chimie 18, 1297–1306. https://doi.org/10.1016/j.crci.2015.06.019 (2015).
Acknowledgements
The laboratory support by the Islamic Azad University, Buinzahra Branch is highly acknowledged.
Author information
Authors and Affiliations
Contributions
M.K. conceived of the presented idea. S.M.M. developed the theory and performed the computations. M.G. verified the analytical methods. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khalaj, M., Musavi, S.M. & Ghashang, M. Alkyl ammonium hydrogen sulfate immobilized on Fe3O4@SiO2 nanoparticles: a highly efficient catalyst for the multi-component preparation of novel tetrazolo[1,5-a]pyrimidine-6-carboxamide derivatives. Sci Rep 14, 8870 (2024). https://doi.org/10.1038/s41598-024-59096-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-59096-2
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
