
Abstract
Natural killer (NK) cells destroy tissue that have been opsonized with antibodies. Strategies to generate or identify cells with increased potency are expected to enhance NK cell-based immunotherapies. We previously generated NK cells with increased antibody-dependent cell mediated cytotoxicity (ADCC) following treatment with kifunensine, an inhibitor targeting mannosidases early in the N-glycan processing pathway. Kifunensine treatment also increased the antibody-binding affinity of Fc γ receptor IIIa/CD16a. Here we demonstrate that inhibiting NK cell N-glycan processing increased ADCC. We reduced N-glycan processing with the CRIPSR-CAS9 knockdown of MGAT1, another early-stage N-glycan processing enzyme, and showed that these cells likewise increased antibody binding affinity and ADCC. These experiments led to the observation that NK cells with diminished N-glycan processing capability also revealed a clear phenotype in flow cytometry experiments using the B73.1 and 3G8 antibodies binding two distinct CD16a epitopes. We evaluated this “affinity profiling” approach using primary NK cells and identified a distinct shift and differentiated populations by flow cytometry that correlated with increased ADCC.
Similar content being viewed by others


Introduction
Natural killer cells are early responders that destroy virally infected cells and malignant tissue1. Accordingly, the occurrence of genetic polymorphisms that impaired NK cell gene expression correlated with poorer prognosis in the treatment of solid tumors2. Low NK cell activity also correlated with increased cancer risk3 and increased risk of recurrence4,5. Improving the NK cell response, either through enhancing NK cell responses or identifying features of NK cells with superior cytotoxic potential, is widely expected to improve treatment efficacy and durability.
Engineered NK cells are being explored to complement the body’s natural response in part because contemporary treatment regimens impair NK cell responses6. As of this writing, we are aware of four current clinical trials involving “high-affinity” NK (haNK) cells paired with a therapeutic monoclonal antibody (mAb) to improve antibody-dependent cell-mediated cytotoxicity (ADCC) and target cell destruction (Refs.7,8 and clinicaltrials.gov). Both these engineered NK cells and endogenous NK cells are activated by antibody-mediated clustering of Fc γ Receptor IIIa (FcγRIIIa/CD16a). CD16a and ADCC are important components of treatments with many mAbs, including rituximab and trastuzumab9,10.
The affinity of Fc-mediated antibody binding to CD16a affects both ADCC potency and therapeutic efficacy. The haNK cells utilize the tighter binding CD16a V158 variant. NK cells expressing the F158 variant exhibit lower responses to mAbs11,12,13. Antibody engineering to increase the affinity likewise increases cell response14. Our laboratory identified a new strategy to increase antibody-binding affinity. The composition of carbohydrate moieties attached to the CD16a asparagine residues (N-glycans) affected affinity, with minimally remodeled N-glycans termed “oligomannose” types providing higher antibody-binding affinity15,16 (for a review of protein N-glycosylation, see17). Notably, both lower-affinity highly remodeled “complex-type” glycans and high affinity oligomannose-type N-glycans appeared at the key CD16a N162 glycosylation site on NK cells isolated from healthy donors18. Furthermore, the level of high and low affinity CD16a forms differed dramatically among different donors.
NK cell glycan processing likewise affects both antibody binding at the cell surface and effector function. YTS-CD16a NK cells, treated with kifunensine, exhibited both increased CD16a affinity and ADCC potency19. YTS cells are derived from the YT cell line isolated from a 15 year old patient; YTS-CD16a cells are engineered to express the CD16a V158 allotype20,21. The ADCC increases were observed using either rituximab with Raji B cells or trastuzumab with SK-BR3 cells. Primary human NK cells also demonstrated increased ADCC upon treatment with kifunensine that blocks a specific mannose trimming step and produces primarily oligomannose-type N-glycans19. It is possible that an off-target kifunensine effect, rather than N-glycan biosynthesis, is responsible for increased NK cell ADCC. Here we evaluate this possibility by creating an NK clone with reduced N-glycan processing.
The variability of CD16a processing by NK cells, revealing an abundance of high affinity glycoforms for some donors, suggests that ADCC activation is regulated by N-glycan processing in the NK cell. Approaches to identify higher activity forms provides an ability to enrich these forms to evaluate in a treatment regimen, particularly where NK cell activity is reduced. The prior studies utilized rigorous cell isolation, protein purification and mass-spectrometry (MS)-based glycopeptide analysis to identify compositional variability. These methods, though sensitive, and not practical for screening NK cell variability in large populations due to the requirement of high numbers of cells (> 2 × 107; Ref.15). Furthermore, MS-based methods do not directly characterize a functional parameter like the antibody-binding affinity of CD16a. Based on published studies, we anticipate that NK cells exhibit variability of antibody-binding affinity by CD16a, and this affinity affects ADCC potency. Here we demonstrate a method to distinguish NK cells with different antibody-binding affinities using flow cytometry, and show that NK cell CD16a affinity changes correlate with changes in NK cell ADCC.
Results
Polyclonal IgG decreases the ADCC of NK cells treated with kifunensine
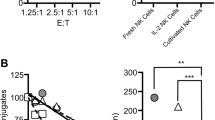
We previously reported that NK cells with minimally processed oligomannose-type N-glycans exhibited increased affinity for IgG1 Fc, and increased antibody-dependent cell-mediated cytotoxicity (ADCC)15,16,19. These assays, like many ADCC assays, were performed in a buffered medium that recapitulates the salinity and pH of the blood, but notably lacks serum proteins including polyclonal IgG (hereafter referred to as “blocking” IgG). We examined the impact of blocking IgG on the ADCC of YTS-CD16a cells to determine if serum IgG affects ADCC potency in this cell-based assay. We observed a slight, non-significant decrease in ADCC in the presence of 0.5 and 2 mg/mL blocking IgG (Fig. 1a, Fig. S1a). As previously reported, YTS cells which do not express CD16a receptor showed no ADCC with rituximab (RTX)19, nor IgG blocking following kifunensine treatment (p > 0.9999) (Fig. 1b, Fig. S1b).

Polyclonal IgG reduced the ADCC (by TR-F) of kifunensine-treated YTS-CD16a cells. (A) YTS-CD16a cells show a slight trend in ADCC decrease only upon IgG blocking at high concentration (2 mg/mL), (B) YTS cells show no ADCC or IgG blocking due to lack of CD16a receptor, (C) Kifunensine-treated (Kif., 20 µM) YTS-CD16a cells show significant decrease in ADCC with IgG blocking at high concentration (2 mg/mL), (D) Kifunensine-treated (Kif., 100 µM) YTS-CD16a cells show significant decrease in ADCC with IgG blocking at both low and high concentrations (0.5 and 2 mg/mL). For all panels, data points represent the mean for three independent experiments collected on three different days at 20:1 E:T ratio, each with three replicates, ± SD by one-way ANOVA. ns, not significant; RTX, rituximab (20 µg/mL); **p < 0.01; ****p < 0.0001.
We next evaluated the impact of increased IgG-binding affinity on the cell surface by treating YTS-CD16a cells with kifunensine. ADCC was significantly reduced at the high concentration of blocking IgG (2 mg/mL; p = 0.0087) at all target-to-effector (T:E) ratios used in this study for the 20 μM kifunensine treatment (Fig. 1c, Fig. S1c). The 100 μM kifunensine treatment similarly reduced ADCC at the higher blocking IgG concentration (2 mg/mL; p < 0.0001), but also with the lower concentration of blocking IgG (0.5 mg/mL; p = 0.0004; Fig. 1d, Fig. S1d). We confirmed the impact of kifunensine treatment on N-glycan processing using LC-NSI-MS/MS and MALDI-MS and found the increased abundance of minimally processed oligomannose N-glycans on the NK cell surface (Fig. 2, Figs. S2, S7).

Minimally-processed N-glycans are found on NK cells with greater ADCC. Relative intensity of the ten most abundant N-glycans identified from YTS-CD16a cells using NSI-MS/MS. (A) Wild-type cells, (B) the MGAT1 knockdown clone E1, (C) cells treated with kifunensine (Kif., 20 µM), (D) cells treated with kifunensine (Kif., 100 µM). Pie charts represent the distribution of all N-glycan types identified. Cartoons indicate one possible glycan configuration consistent with the composition, isobaric species were not distinguished. Monosaccharide residues in the cartoons are defined by the CFG convention. The code for the glycan composition on the x-axes reports the numbers of the following residues as identified by MS (HexNAc-hexose-deoxyhexose-Neu5Ac).
We previously reported that two treatments that increase the antibody affinity for the NK cell surface, namely removing antibody fucosylation and restricting NK cell N-glycan processing, additively increased ADCC19. We next examined whether the impact of blocking IgG similarly reduced the ADCC of kifunensine-treated NK cells when using an afucosylated antibody (RTX-fuc). As expected, untreated YTS-CD16a cells showed decreased ADCC, albeit not significant (from 20.91 to 17.84%, p = 0.5926) with 2 mg/mL blocking IgG present (Fig. S3). YTS-CD16a cells treated with kifunensine, however, showed a significant ADCC decrease with RTX-fuc and blocking IgG (from 39.92 to 13.86%, p < 0.0001). Taken together, these data suggest that increased affinity of CD16a after kifunensine treatment enhances the blocking effect of IgG.
To determine if the observed sensitivity to blocking IgG decreased the effector function of primary human NK cells, we isolated NK cells from three healthy donors. Kifunensine treatment enhanced ADCC of the primary NK cells from two donors in the absence of blocking IgG. NK cells from NK121 and NK123 showed significant ADCC reduction (minimum of p = 0.0017) upon incubation with 2 mg/mL blocking IgG following kifunensine treatment (Fig. 3). The ADCC value becomes negative as a result of subtracting the target cell killing in the absence of antibody; in contrast to fresh primary cells these NK cells are cultured for 3 days in a medium lacking human serum and human antibodies which are presumed to block NK cell activity. Donor NK122 showed no increased specific lysis in the presence of the rituximab antibody and thus no sensitivity to blocking IgG upon kifunensine treatment. It is common to find variability in the effector function of primary NK cells from different donors22. It is worth mentioning that the NK cell donors used for this particular experiment came from one specific age range of the population, specifically 31–32 year old white males so it is possible that donors with different ages or genders may respond differently.

IgG blocks ADCC (by TR-F) of primary NK cells. (A-C) IgG blocking (2 mg/mL) of ADCC following treatment with kifunensine (Kif., 20 µM) for primary NK cells isolated from three donors at 20:1 E:T ratio. Means are indicated with a horizontal black line ± SD. RTX, rituximab (20 µg/mL). For all panels, one-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
MGAT1 knockdown introduces sensitivity to blocking IgG
To confirm that the blocking IgG effect was due to N-glycan processing and not due to an off-target effect of kifunensine, we generated a YTS-CD16a clone deficient in MGAT1 activity. MGAT1 catalyzes the first step in the synthesis of highly processed hybrid and complex-type glycans23,24. Thus, inhibiting MGAT1 activity is expected to generate cells that express predominantly oligomannose-type N-glycans. We used an established CRISPR-CAS9-GFP strategy that targets the human MGAT1 N-terminal domain25 (Fig. 4a). After multiple bulk-sorting rounds, the GFP + population was increased to 95.6%, from which single cells were sorted and allowed to develop into stable cell populations (Fig. 4b). We selected one for further study, which was designated clone E1. Analyses of Western blots indicated significant MGAT1 reduction for the E1 clone and reduced staining with the fluorescently-labeled Phaseolus vulgaris hemagglutinin (L-PHA) that binds to complex N-glycans, downstream products of MGAT126, specifically β1,6GlcNAc branches (Fig. 4c,d, Fig. S4).

MGAT1 knockdown affects YTS-CD16a NK cell effector function and IgG binding sensitivity to CD16a receptor. (A) Schematic diagram of the dual CRISPR-Cas9 plasmid system and strategy used in this study to generate MGAT1 knockdown clones with the YTS-CD16a cell line, (B) Representative flow cytometry plots of the multiple rounds of bulk-sorting used in this study to increase GFP + YTS-CD16a cell population, (C) Western blot and densitometry of the MGAT1 knockdown clone E1, (D) Representative flow cytometry histograms showing decreased L-PHA lectin staining of MGAT1 knockdown clone E1, (E) ADCC (by TR-F) of the MGAT1 knockdown YTS-CD16a clone E1 and WT cells. Data shown include three independent experiments collected on different days, each with three replicates, by two-way ANOVA, (F) The MGAT1 knockdown clone E1 is likewise sensitive to blocking IgG (2 mg/mL) in ADCC (by TR-F) by two-way ANOVA. Data shown include three independent experiments collected on different days, each with three replicates. RTX, rituximab (20 µg/mL). For panels E and F, ***p < 0.001; ****p < 0.0001.
Mass spectra of N-glycans released from the MGAT1 knockdown E1 whole cell lysate are consistent with reduced but not absent, MGAT1 activity. LC-NSI-MS/MS revealed increased abundance of hybrid-type N-glycans (43%) and decreased complex-type N-glycans (22%) when compared to the wild-type YTS-CD16A cells with predominant complex-type (42%) N-glycans (Fig. 2, Figs. S2, S7). Interestingly, E1 retained a comparable percentage of oligomannose-type N-glycans (35%) as the unmodified parent cell line (34%). E1 cells showed the fucosylated paucimannose form as the most abundant species, followed by the Man8 oligomannose-type N-glycan. There was more abundance in the hybrid-type N-glycans and less abundant presence of sialylated bi- and tri-antennary complex-type N-glycans. It is not surprising that the E1 clone is not completely lacking MGAT1 expression or activity because lymphocytes are reportedly highly sensitive to MGAT1 depletion27,28. It is interesting that DNA sequence analysis of the region surrounding the intended mutation site showed both mutated and wild type sequences (57% mutant heterozygous), likely indicating at least one mutation reducing MGAT1 expression that lies outside the site targeted by the CRISPR guide RNA.
We next assessed the cytotoxic capability of the MGAT1 knockdown on YTS-CD16a cells with ADCC assays. The E1 clone showed increased ADCC at the target-to-effector cell ratios of 1:10 (from 26 to 38%; p = 0.0003) and 1:20 (30 to 48%; p < 0.0001) when compared to WT YTS-CD16a (Fig. 4e). E1 was likewise sensitive to blocking IgG, with significant reductions in contrast to the parent YTS-CD16a cell line (p < 0.0001; Fig. 4f).
IgG affinity profiling of NK cells
As noted, N-glycan processing decreases CD16a-mediated IgG binding affinity on the surface of YTS-CD16a cells compared to untreated cells19. Based on this, we examined if the sensitivity of CD16a to blocking IgG can be increased with a higher concentration of kifunensine treatment (100 μM). After 3 days of kifunensine treatment of YTS-CD16a cells at different concentrations (20 and 100 μM), cells were blocked with 10% human serum and stained for CD16a by two antibodies recognizing discrete epitopes: 3G8 and B73.1 (Fig. 5a). As expected, we noticed a reduction in staining with 3G8 for kifunensine treated YTS-CD16a cells upon human serum blocking due to the retention of IgG that competes for the 3G8 epitope19. We also noticed increased B73.1 staining in the kifunensine treated cells (Fig. 5a), despite the slight twofold decrease in affinity noted using surface plasmon resonance (Fig. S5). We observed no significant difference in CD16a staining with these antibodies between kifunensine treatment at 20 or 100 μM. This is consistent with the highly similar N-glycan composition determined by MS for these two treatments (Fig. 2c,d).

IgG sensitivity and blocking can differentiate distinct populations in YTS-CD6a cells based on CD16a epitope staining. (A) Representative flow cytometry plots of untreated and kifunensine (Kif.)-treated (20 and 100 µM) YTS-CD16a cells distinctly showing that the 3G8 binding of CD16a on YTS-CD16a cells (x axis) and B73.1 binding of CD16a (y-axis) is affected by IgG blocking and no significant difference is seen comparing both treatment concentrations. Dashed lines indicate the mean fluorescence intensities in each dimension. (B) Representative flow cytometry plots of untreated and kifunensine (Kif.)-treated (20 µM) YTS-CD16a cells gaging sensitivity in differentiating distinct populations based on N-glycan composition at varying percentages of WT and Kif-treated populations.
We next evaluated whether this “affinity profiling” approach based on differences in 3G8 and B73.1 binding could differentiate untreated vs kifunensine-treated YTS-CD16a populations by mixing various percentages of untreated and kifunensine-treated (20 μM) YTS-CD16a cells, as a potential surrogate for profiling mixed affinities with primary cells. Flow cytometry bivariate plots of the treated YTS-CD16a cells revealed distinct populations with the greater differences following blocking with polyclonal IgG (Fig. 5b). The profile of the treated cells with greater sensitivity to blocking IgG, shifted to the upper left of the plots indicating decreased binding to 3G8 and greater binding of B73.1. Thus, this flow cytometry method provides the ability to distinguish NK cells based upon IgG-binding affinity to CD16a, which we term “affinity profiling”.
We then evaluated whether affinity profiling could differentiate untreated and (20 μM) kifunensine-treated primary NK cells from six healthy donors. All primary NK cells treated with kifunensine showed reduced L-PHA staining when compared to untreated primary cells from same donor indicating a reduction in N-glycan processing at the surface (Fig. S6). Moreover, all primary NK cells treated with kifunensine showed a population shift, and thus differentiated treated and untreated primary cells from same donor (Fig. 6a). Cells from each source demonstrated a clear shift following kifunensine treatment to greater staining with the B73.1 antibody and lower with 3G8 (Fig. 6b). Of these cells from six donors, NK122 showed no cytotoxicity under any conditions evaluated, but NK123 showed a strong increase (Fig. 3). Unfortunately, we were unable to examine the NK124 cells for activity, but NK cells from the NK138 and 140 donors showed increased ADCC following kifunensine treatment (NK139 was increased but fell short of significant at p < 0.05), as well as significantly increased IFNγ production for two of three donors tested (Fig. 7). Interestingly, we only found an increase in CD107a staining as a measure for NK cell degranulation following kifunensine treatment with one donor of three examined (NK140). Thus, the majority of cells in this panel showed both an increase in ADCC and shifts to the upper left in the affinity profiling analyses that is consistent with higher IgG-binding affinity.

IgG affinity profiling of primary human NK cells. (A) Flow cytometry plots of primary NK cell donors showing decreased 3G8 staining for kifunensine (20 µM) cells after incubation with 10% human serum, (B) a plot of the mean fluorescence intensities (MFIs) from (A) showing increased B73.1 staining and decreased 3G8 staining associated with high affinity IgG binding following kifunensine treatment, (C) representative flow cytometry plots of primary NK cell donors of healthy adults and children showing considerable heterogeneity, with high affinity phenotype (44%) observed in adult population and less percentage in children population (20%), (D) MFIs for the B73.1+ and 3G8+ population plotted against a heavy black y = x line with NK cells exhibiting greater antibody-binding affinity expected above this line.

ADCC (by flow cytometry) following treatment with kifunensine for primary NK cells. (A,D,G) Lysis, (B,E,H) CD107a, (C,F,I) hIFNγ production of NK cells from donors NK138, NK139 and NK140 treated with kifunensine (“Kif.”; 20 µM) at 20:1T:E ratio. Means are indicated with a horizontal black line ± SD. RTX, rituximab (20 µg/mL). For all panels, one-way ANOVA, *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant, n.d. not detected.
Lastly, we applied the affinity profiling approach NK cells previously collected from a cohort of 16 healthy adults and 20 healthy children29. As expected, CD16 staining for the B73.1 and 3G8 epitopes was correlated for each donor (representative plots in Fig. 6c). However, an overlay of these plots revealed considerable heterogeneity. For example, seven of the 16 adult donors demonstrated affinity profiling results consistent with high affinity, evident from the MFI that was above the y = x line (showing greater B73.1 staining; Fig. 6d). By contrast, only four of 20 child donors revealed an MFI in this region, indicating generally lower affinity in NK cells from children. We did not observe any strong correlations between the affinity profiling and age or sex. It is interesting that we observed adult and child donors within the cohort with shifts in the plot to the lower right (greater 3G8 staining), which was not observed with our donor NK or YTS-CD16a cells. This shift may indicate greater processing, rather than less. These data indicate that the IgG affinity of CD16a in donor NK cells is variable, with shifts towards greater affinity more commonly observed with older donors than with children. By comparison to the functional studies presented above, these data indicate that a significant proportion of adult donors harbor NK cells with increased affinity for IgG.
Discussion
Phenotype profiling of NK cells from healthy donors revealed substantial variability within both donor pools. Increased signal from the B73.1 antibody and decreased signal form the 3G8 antibody identified by flow cytometry correlated with increased ADCC and likely increased CD16a affinity for binding IgG1 through the Fc region. Thus, affinity profiling represents a simple approach to characterize a critical functional attribute of NK cells. This conclusion is supported with a wealth of data using kifunensine treated YTS-CD16a cells and primary NK cells. Furthermore, this conclusion is consistent with previous observations, including the identification of variability in CD16a N-glycan composition, notably at the N162 site that influences IgG Fc binding affinity15,16,18. In contrast to the N162 site, compositions at the remaining four N-glycosylation sites were remarkably similar among different donors, suggesting NK cells regulate the antibody-mediated immune response through modulating N-glycan composition. Lastly, the observation reported here that, on balance, children showed lower IgG affinity compared to adults is consistent with the observation that the abundance of oligomannose N-glycans, providing high IgG affinity on NK cells, increase with age and inflammation30,31,32. One weakness of the current study is a lack of donors with only the F158 allotype (Table S1), and thus it’s possible that these donors show a different response in the affinity profiling experiments. Furthermore, we found no correlations between donor genotype in these samples and NK cell functional or phenotypic differences potentially due to the relatively small sample size.
The phenotypic distinction of different NK cells, as noted, results from differences in IgG binding affinity. The data presented here are consistent with the previous observation that an inhibitor of N-glycan processing increased IgG affinity at the surface of NK cells and provided increased ADCC for both YTS-CD16a and primary NK cells19. Here we have demonstrated that the enhanced NK cells indeed have reduced N-glycan processing and greater IFNγ secretion (for two of three donors). Furthermore, we observed similar IgG binding affinity and ADCC potency in a NK clone with reduced MGAT activity, confirming that N-glycan processing rather than an off-target effect of kifunensine is likely responsible for the functional differences.
YTS-CD16a NK cells with reduced N-glycan processing and increased IgG binding affinity also proved susceptible to inhibition with non-specific serum IgG (Fig. 1). Surprisingly, the ADCC potency of primary NK cells from two donors showed strong inhibition, even in the absence of kifunensine (Fig. 3). It is possible the primary NK cells express factors increasing sensitivity to IgG; YTS cells do not naturally express CD16a, and thus differ from primary cells in multiple key ways29.
It is not clear to what extent the results of these experiments predict that IgG levels potentially inhibit ADCC in the body. It is known that tight IgG binding affinity of CD16a increases both in vitro ADCC and the patient response to monoclonal antibody therapies where ADCC is a key attribute; patients with the high affinity CD16a V158 allotype generally respond more favorably than those expressing the low affinity F158 variant33,34,35. Thus, the increased CD16a affinity does not inhibit efficacy of the monoclonal antibodies in vivo. This apparent discrepancy may result from a few key differences between the in vitro ADCC assays and antibody-dependent killing by NK cells in vivo. In one example, our ADCC assay is completed in 2 h, in contrast to killing in vivo that may last for days or more. Greater contact time between NK cells and an opsonized target is expected to promote greater CD16a engagement due to high valency of the interaction, and allow dissociation and replacement of the serum IgG that is solely a monovalent interaction. The multivalent interaction is expected to promote greater target killing and thus antibody efficacy.
The ongoing development of engineered NK cells demonstrates the promise of improved therapeutics, and a new landscape for improving ADCC. It would be challenging to modulate the glycan processing of endogenous NK cells for a number of reasons, however, modulating CD16a affinity or CD16a glycan processing through cell engineering prior to infusion is highly feasible. Our prior kinetic analysis of CD16a binding and present ADCC results suggest that the NK cells with high affinity CD16a glycoforms bind strongly and fast, and dissociate slowly from IgG. Thus, mixing engineered NK cells and target-specific IgG prior to infusing has the potential to provide a rapid response, with minimal inhibition from serum IgG.
In conclusion, we developed an affinity profiling strategy to evaluate functionally relevant differences in CD16a affinity for IgG, and identified clear differences among healthy donors. We also determined that N-glycan processing affects ADCC in NK cells, providing a novel pathway towards improving NK cell responses.
Materials and methods
Materials
All materials purchased from Millipore-Sigma unless otherwise noted.
IgG purification
Human IgG (Athens Research & Technology, Inc) was purified using a Superdex 200 size exclusion column as previously described19.
Cell culture
The natural killer cell lymphoblastic leukemia/lymphoma cell lines YTS and YTS-CD16a (V/V allotype) (a gift from Dr. Konrad Krzewski, NIH) cells were grown in RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1 mM non-essential amino acids, and 50 U/mL penicillin/streptomycin. Raji cells were purchased from ATCC (#CCL-86) and were grown in RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, and 50 U/mL penicillin/streptomycin. Raji and YTS cells were grown in suspension at 37 °C, 5% CO2. Trypan blue uptake was assessed to identify non-viable cells when measuring cell density of a culture using a TC-20 automated cell counter (Biorad).
Primary NK cell isolation
NK cells were isolated from leukocyte reduction filters obtained from Shepeard Community Blood Centers (Augusta, GA), after the plateletpheresis procedure was performed on healthy donors. Donors signed consent forms permitting the use of donated blood products for research purposes. We do not directly enroll donors because we obtain deidentified materials from the Shepeard Blood Center, and thus donors were not directly involved in the study. Cell isolation procedures were performed within 24 h of the time the donors completed apheresis procedure. All NK cells were isolated from apheresis filters by negative selection and profiled accordingly as previously described, and viability was assessed by trypan blue, as well as Zombie Violet/Green15,18,19. Donor CD16a genotypes were determined as previously described36. NK cells were cultured in the YTS medium described above but supplemented 5 ng/mL of IL-15. The kifunensine group (20 μM; Cayman Chemicals) was treated for 3 days. Donor information can be found in Table S1. Male donors were selected because they generally provide greater number of NK cells needed for these studies. Primary NK cells were used fresh for functional assays and not cryopreserved.
Time-resolved fluorescence-based (TR-F) ADCC assays
The DELFIA EuTDA Cytotoxicity Kit was used and optimized per manufacturer’s instructions (Perkin Elmer). The Eu-TDA DELFIA assay, which uses TR-F, is a non-radioactive alternative that offers similar sensitivity to the 51Cr release assay37, and performed as previously reported19. Briefly, untreated and kifunensine treated effector cells were added to a V-bottom 96-well plate and blocked with purified IgG (0–0.5–2 mg/mL) for 1 h at 37 °C, 5% CO2, and then antibody (rituximab (Selleckchem) or afucosylated rituximab (InvivoGen)) at 0–20 μg/mL was added. Raji cells were labeled with 5 μL of BATDA reagent at 1 × 106 cells/mL in loading buffer (RPMI 1640 medium without phenol red, 10% FBS, 10 mM HEPES) for 30 min at 37 °C, 5% CO2. The cells were washed 5 × 10 mL with PBS, and resuspended at 2 × 105 cells/mL in assay medium (RMPI 1640 medium, 10 mM HEPES). Target Raji cells (10,000 cells in 50 μL) were incubated with effector cells for 2 h at 37 °C, 5% CO2 at specified target-to-effector (T:E) ratios for each experiment in triplicate. The 96-well plate was then centrifuged for 5 min at 100×g, and 20 μL of each supernatant was then combined with 200 μL of europium solution in a separate flat-bottom 96-well plate, followed by a 15 min incubation with orbital shaking at RT. The fluorescent signal was measured with a Neo2 plate-reader (Biotek) with time-resolved fluorescence (TR-F) filter readout parameters. Controls in each experiment monitored Background, Maximum-to-Spontaneous signal ratio as well as Spontaneous Release (%). Specific Lysis (%) was calculated as: [(observed signal − spontaneous signal)/(maximum signal − spontaneous signal)] × 100. Maximum signal was obtained by incubating cells with 10 μL lysis buffer per manufacturer’s instructions. Statistical analysis was performed with Prism 6.09 (GraphPad software).
Flow cytometry-based ADCC assays
Briefly, Raji cells were labeled with CellTrace™ Yellow (CTY) in PBS for 7 min in a bead bath using the CellTrace™ Yellow Cell Proliferation Kit (Invitrogen) as previously described19,38, then 10 mL of assay medium (RPMI 1640 medium, 10% FBS, 2 mM l-glutamine, 50 U/mL penicillin/streptomycin) were added and cells were incubated for an additional 5 min. The cells were then washed 5 × 10 mL with assay medium, and resuspended at 5 × 104 cells/mL in assay medium. Target Raji cells (5000 cells, 100 μL) were incubated for 15 min with 0–20 μg/mL antibody (rituximab; Selleckchem) in a V-bottom 96-well plate, followed by addition of effector cells at 1:10 target-to-effector (T:E) ratios and further incubated for 2 h at 37 °C, 5% CO2 in triplicates. Effector cells were previously treated with kifunensine (20 μM) for 3 days. Cells were then washed with PBS+ azide and stained for viability using Zombie Violet (1:1000, BioLegend #423,114) for 30 min at RT. Cells were washed 3 × 5 min with PBS+ azide, and fixed with 1% PFA for 20 min at RT, followed by 3 × 5 min washes with PBS+ azide. A collection of 2500 events gated for CTY+ cells per sample were acquired on a CytoFLEX instrument (Beckman Coulter), and analysis performed using FlowJo 10 (BD Biosciences). Specific Lysis (%) was calculated as the percentage of dead CTY+ target cells. Controls in each experiment monitored viability upon labeling of target cells with CellTrace reagent, as well as viability upon incubation with antibody as well.
MGAT1 knockdown YTS-CD16a cells
For the CRISPR knock-out generation, a nucleofection strategy was chosen since YTS cells are considered difficult-to-transfect cells. Two plasmids, one containing the Cas9-GFP system and the other the gRNA for human MGAT125 were added to a total of 2 × 106 YTS-CD16a cells suspended in 100 μL Nucleofector solution according to manufacturer’s protocol (Kit V, Lonza #VCA-1003). Plasmids were introduced using program O-017 on the Amaxa Nucleofection II System (Lonza). Cells were then incubated for 24 h, stained with propidium iodide (PI) for 15 min and bulk-sorted based on the following gating strategy: G1 (forward and side scatter population), G2 (singlets), G3 (PI- viable cells), and G4 (GFP+) using a S3 Cell Sorter (Biorad). A second nucleofection of the previously bulk-sorted population was performed after cells had above 90% viability, followed by bulk-sorting to further enrich GFP+ cells. Cells were then grown for 14 days, then GFP+ cells were single-cell sorted into flat-bottom 96-well plates in pre-conditioned growth medium using a MoFlo Astrios EQ cell sorter (Beckman Coulter). Single-cell sorts were allowed to grow in 96-well plates in YTS pre-conditioned medium for 14 days, followed by clonal screening for MGAT1 expression by western blot, L-PHA and 3G8 staining by flow cytometry and genotyping analysis.
Immunoblotting
A total of 3 × 106 cells were collected and lysed by continuous pipetting in 120 μL of SDS-PAGE buffer A (50 mM TRIS, 1% SDS, pH 6.8, Halt protease inhibitor), vortexed briefly, then 1 mL of cold acetone was added, followed by an additional vortex step, then incubated on ice for 10 min. Samples were centrifuged at 4400g for 15 min at 4 °C. The protein pellet was resuspended in 50 mM NaOH, and protein concentration was determined using Pierce BCA Protein Kit. Samples of 25 μg total lysate were resolved on a 10% SDS–polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane using the one-step electroblotting system (Pierce Power Station; ThermoFisher). The membrane was blocked with 5% dry milk in TBS Tween 20 (TBST) buffer for 1 h at RT. Following blocking, the membrane was stained for 18 h at 4 °C with anti-MGAT1 (rabbit, 1:1000, Abcam #ab180578) in 5% milk in TBST. The blot was washed 3 × 10 min with TBST and stained with secondary anti-rabbit AF800 (1:10,000, Invitrogen #A32735) in 5% milk in TBST for 1 h at room temperature. Then, blot was washed 3 × 15 min and blocked once more with 5% milk in TBST for 1 h at room temperature. The membrane was then stained for 1 h at room temperature with anti-hGAPDH (goat, 1:10,000, R&D Systems #AF57185P), followed by 3 × 10 min washes with TBST. The blot was then incubated for 1 h at RT with the anti-goat AF680 (1:10,000, Invitrogen #AF32860) secondary antibody in 5% milk TBST, and washed 3 × 15 min with TBST. The dual-labeled blot was imaged on an Odyssey CLx Image system (LICOR Biosciences). Densitometry was performed using ImageJ software.
MGAT1 genotyping and indel analysis
Unmodified wild-type YTS-CD16a cells as well as cells with reduced MGAT1 levels (3 × 106 cells) were pelleted and genomic DNA was isolated using QIAamp Blood Mini Kit per the manufacturer’s protocol (Qiagen). A PCR reaction with published primers encapsulating the chosen MGAT1 gRNA were used: 5ʹ-CCAGGATGCTGAAGAAGCAGTCT-3ʹ (forward), 5ʹ-TGGCTAACGATGATGGGGAAG-3ʹ (reverse)25. DNA was amplified using GoTaq Green MasterMix (Promega), and PCR products were purified by 1% agarose gel using SV Wizard PCR and Gel Clean-Up Kit (Promega). Purified DNA was sent for Next-Generation-Sequencing analysis by Amplicon-EZ (Azenta Life Sciences).
Clonal screening by flow cytometry
A total of 500,000 effector cells were stained for viability using Zombie Violet (1:1000, BioLegend #423114) for 30 min at RT. Cells were washed 3 × 5 min with PBS+ azide, followed by 10 min blocking with 10% human serum at 4 °C. Cells were then stained with anti-hCD16 (3G8-APC, BioLegend #302011) or an isotype control (Iso-APC, BioLegend #400120) for 30 min at 4 °C. Cells were washed 3 × 5 min with PBS+ azide, and fixed with 1% PFA for 20 min at RT, followed by 3 × 5 min washes with PBS+ azide. Cells were then incubated with lectin L-PHA-AF594 (5 μg/mL, Invitrogen #L32456) for 20 min at 37 °C, and washed 3 × 5 min with PBS azide. A collection of 5 × 104 events per sample were acquired on a CytoFLEX instrument (Beckman Coulter), and analysis performed using FlowJo 10 (BD Biosciences).
Analysis of CD16a on NK cells using flow cytometry
A total of 500,000 YTS-CD16a cells containing various percentages (from 0 to 100%) of untreated and kifunensine (20 μM) treated cells were stained for viability using Zombie Green (1:1000, BioLegend #423111) for 30 min at RT. Cells were washed 3 × 5 min with PBS+ azide, followed by 10 min blocking with 10% human serum at 4 °C. Cells were then stained with anti-hCD16 antibodies (2 µg/mL 3G8-APC, BioLegend #302011; 2 µg/mL B73.1-BV421, BioLegend #360724) or their isotype controls (Iso-APC, BioLegend #400120; Iso-BV421, BioLegend #400158) for 30 min at 4 °C. Antibody concentrations for CD16 staining were chosen to be in great excess of the receptor concentration. For example, previous reports indicate ~ 25,000 CD16a/NK cell thus for 5 × 105 cells/mL = 1.3 × 1010 CD16a/mL = 2.1 × 10–11 M. 3G8 (2 µg/mL) = 1.3 × 10–8 M, and the KD = 3–6 × 10–9 M, thus we expect the vast majority of receptors to be bound with antibody. Cells were washed 3 × 5 min with PBS+ azide, and fixed with 1% PFA for 20 min at RT, followed by 3 × 5 min washes with PBS+ azide. A collection of 5 × 104 events per sample were acquired on a CytoFLEX instrument (Beckman Coulter), and analysis performed using FlowJo 10 (BD Biosciences). The same hCD16a staining was done for isolated donor primary NK cells (NK122, NK123, NK124, NK138, NK139, NK140) with L-PHA staining as mentioned previously.
PBMC data analysis for B73.1 and 3G8
Raw data from PBMCs isolated from healthy children and adults was acquired from a previously published study29. Demographic information on these donors is reported in Table S2. Dual staining analysis for CD16a based on B73.1 and 3G8 epitopes was performed using FlowJo 10 (BD Biosciences).
CD107a quantitation
Briefly, effector cells were labeled with CellTrace™ Yellow (CTY) in PBS using the CellTrace™ Yellow Cell Proliferation Kit (Invitrogen) for 7 min at 37 °C as previously described38,39, then 10 mL of assay medium (RPMI 1640 medium, 10% FBS, 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1 mM non-essential amino acids, 50 U/mL penicillin/streptomycin) were added and cells were incubated for an additional 5 min. The cells were then washed 5 × 10 mL with assay medium, and resuspended at 5 × 104 cells/mL in assay medium. Target Raji cells (50,000 cells, 100 μL) were incubated for 15 min with 0–20 μg/mL antibody (rituximab; Selleckchem) in a V-bottom 96-well plate, followed by addition of effector cells at 1:1 target-to-effector (T:E) ratio, as well as anti-hCD107a (CD107a-FITC, BioLegend #328606) or the isotype control (Iso-FTIC, BioLegend #400108) and further incubated for a total of 1 h at 37 °C, 5% CO2 in triplicates. Effector cells were previously treated with kifunensine (20 μM) for 3 days. For positive controls, effector cells were activated with 1 μg/mL phorbol-myristate-acetate (PMA) and 1 μg/mL ionomycin. Following the first hour of incubation, 6 μg/mL GolgiStop and 10 μg/mL GolgiPlug were added to the sample wells and further incubated for 3 h at 37 °C, 5% CO2. Cells were then washed with PBS+ azide and stained for viability using Zombie Violet (1:1000, BioLegend #423114) for 30 min at RT. Cells were washed 3 × 5 min with PBS+ azide, and fixed with 1% PFA for 20 min at RT, followed by 3 × 5 min washes with PBS+ azide. A collection of 2500 events gated for CTY+ cells per sample were acquired on a CytoFLEX instrument (Beckman Coulter), and analysis performed using FlowJo 10 (BD Biosciences). CTY+ cells were further gated for viability, and viable cells were assessed for CD107a upregulation by FITC geometric mean fluorescence intensity analysis. Controls in each experiment monitored viability upon labeling of target cells with CellTrace reagent, as well as viability upon incubation with antibody as well.
hIFNγ quantitation
Serum human IFNγ levels of isolated NK cell donors were examined by collecting supernatant from the ADCC assays and using IFNγ Human ProQuantum Immunoassay Kit (A35576, Invitrogen) using 20 μL volume reactions. Data was acquired on a StepOnePlus Real-Time PCR system (Applied Biosystems) and analyzed per manufacturer’s instructions by the ProQuantum cloud-based software.
Mass spectrometry of permethylated N-glycans
NK cells were collected and boiled for 10 min at 100 °C, cooled down, and trypsin (Promega) was added prior to overnight incubation at 37 °C. Samples were lyophilized and resuspended in GlycoBuffer2 and PNGase F (New England Biolabs) was added and samples incubated overnight at 37 °C. Samples were lyophilized, then resuspended in 5% acetic acid (in water), and released N-glycans were eluted from C18 cartridges according to manufacturer’s instructions (Sep-Pak, 100 mg; Waters). Samples were lyophilized and permethylated as previously described40. The samples were analyzed on an Orbitrap Fusion Tribrid (ThermoFisher) connected in-line to a Thermo Ultimate RSLCnano chromatography system (ThermoFisher). A commercial LC-column (Acclaim PepMap 100 C18 (3 μm, 100 Å, 75 μm × 15 cm) was used for separation. A gradient including mobile phase A consisting of 98% water, 2% acetonitrile, and 1 mM sodium acetate and mobile phase B consisting of 20% water and 80% acetonitrile were used for a linear gradient from low to high percentage of mobile phase B within a 72-min gradient. The data dependent precursor ion scan was collected from m/z 600–2000 m/z at 120,000 resolution inOrbitrap mode and precursors at a time frame of 3 s were selected for subsequent MS/MS fragmentation (CID; 40%) at 15,000 resolution. Precursors with either an unknown charge state or +1 charge state were excluded, and dynamic exclusion was enabled for a 30 s duration. MS2 fragmentation patterns were evaluated using GlycoWorkBench software41. Mass spectrometry by MALDI was also performed on the samples and collected on a RapifleX Tissuetyper (Bruker) in linear positive mode with 2,5-dihydroxybenzoic acid used as a matrix. MS1 (LC-NSI-MS; MALDI) spectra for identified N-glycans can be found in Supplemental Data (Figs. S2, S7).
Surface plasmon resonance
GFP-CD16a (1 µg/mL) expressed from HEK293F cells using Freestyle medium (ThermoFisher) was coupled to the surface of a Series S CM5 sensor chip on an Akta T200 instrument (Cytiva) in 10 mM sodium acetate pH 5.0. Following coupling, the sensor surface was equilibrated with 1 × phosphate buffered saline, 0.05% P20, pH 7.4; this buffer was used as the “running buffer” for all following experiments. Rituximab was likewise expressed from HEK293F cells. Affinities were estimated using a single cycle kinetics method where the sensor surface was washed (± 100 nM B73.1) for 450 s, followed by an additional 150 s of washing with running buffer. Five injection cycles of increasing rituximab concentrations in running buffer were applied to the surface as follows (150 s injection, 70 s wash with running buffer). The sensor surface was regenerated between experiments by applying 100 mM glycine, pH 3.0 for 30 s followed by equilibration with running buffer for 500 s. Affinities were determined by fitting the signal once an equilibrium had been established during the application of rituximab. Affinities were fitted with a 1:1 Langmuir binding isotherm.
Statistical analyses
All statistical analyses were performed with Excel (Microsoft), Prism 6.09 (GraphPad software), or R Studio (Version 2023.12.0+369, Posit Software, PBC).
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files). Additional information is available to the corresponding author on reasonable request.
Abbreviations
- NK:
-
Natural killer
- E:T:
-
Effector-to-target
- MFI:
-
Mean fluorescence intensity
- MGAT-1:
-
Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase
- TR-F:
-
Time-resolved fluorescence
- MS:
-
Mass spectrometry
- LC:
-
Liquid chromatography
- NSI:
-
Nanoelectrospray ionization
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- mAb:
-
Monoclonal antibody
- L-PHA :
-
Phaseolus vulgaris hemagglutinin
- IFNγ:
-
Interferon-gamma
References
Caligiuri, M. A. Human natural killer cells. Blood 112, 461–469 (2008).
Xu, X. et al. Association of germline variants in natural killer cells with tumor immune microenvironment subtypes, tumor-infiltrating lymphocytes, immunotherapy response, clinical outcomes, and cancer risk. JAMA Netw. Open 2, e199292. https://doi.org/10.1001/jamanetworkopen.2019.9292 (2019).
Imai, K., Matsuyama, S., Miyake, S., Suga, K. & Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 356, 1795–1799. https://doi.org/10.1016/S0140-6736(00)03231-1 (2000).
Tartter, P. I., Steinberg, B., Barron, D. M. & Martinelli, G. The prognostic significance of natural killer cytotoxicity in patients with colorectal cancer. Arh. Surg. 122, 1264–1268 (1987).
Schantz, S. P., Savage, H. E., Racz, T., Taylor, D. L. & Sacks, P. G. Natural killer cells and metastases from pharyngeal carcinoma. Am. J. Surg. 158, 361–366 (1989).
Liu, S. et al. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 14, 7. https://doi.org/10.1186/s13045-020-01014-w (2021).
Snyder, K. M. et al. Expression of a recombinant high affinity IgG Fc receptor by engineered NK cells as a docking platform for therapeutic mAbs to target cancer cells. Front. Immunol. 9, 2873. https://doi.org/10.3389/fimmu.2018.02873 (2018).
Jochems, C. et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 7, 86359–86373 (2016).
Hayes, J. M. et al. Identification of Fc gamma receptor glycoforms that produce differential binding kinetics for rituximab. Mol. Cell. Proteom. 16, 1770–1788. https://doi.org/10.1074/mcp.M117.066944 (2017).
Musolino, A. et al. Role of Fcgamma receptors in HER2-targeted breast cancer therapy. J. Immunother. Cancer 10, 25. https://doi.org/10.1136/jitc-2021-003171 (2022).
Dekkers, G. et al. Decoding the human immunoglobulin G-glycan repertoire reveals a spectrum of Fc-receptor- and complement-mediated-effector activities. Front. Immunol. 8, 877. https://doi.org/10.3389/fimmu.2017.00877 (2017).
Hatjiharissi, E. et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood 110, 2561–2564 (2007).
Binyamin, L. et al. Blocking NK cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. J. Immunol. 180, 6392–6401. https://doi.org/10.4049/jimmunol.180.9.6392 (2008).
Mössner, E. et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood 115, 4393–4402 (2010).
Patel, K. R., Roberts, J. T., Subedi, G. P. & Barb, A. W. Restricted processing of CD16a/Fc gamma receptor IIIa N-glycans from primary human NK cells impacts structure and function. J. Biol. Chem. 293, 3477–3489. https://doi.org/10.1074/jbc.RA117.001207 (2018).
Subedi, G. P. & Barb, A. W. CD16a with oligomannose-type N-glycans is the only “low-affinity” Fc gamma receptor that binds the IgG crystallizable fragment with high affinity in vitro. J. Biol. Chem. 293, 16842–16850. https://doi.org/10.1074/jbc.RA118.004998 (2018).
Moremen, K. W., Tiemeyer, M. & Nairn, A. V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell. Biol. 13, 448–462. https://doi.org/10.1038/nrm3383 (2012).
Patel, K. R., Nott, J. D. & Barb, A. W. Primary human natural killer cells retain proinflammatory IgG1 at the cell surface and express CD16a glycoforms with donor-dependent variability. Mol. Cell. Proteom. 18, 2178–2190 (2019).
Rodriguez Benavente, M. C., Hughes, H. B., Kremer, P. G., Subedi, G. P. & Barb, A. W. Inhibiting N-glycan processing increases the antibody binding affinity and effector function of human natural killer cells. Immunology https://doi.org/10.1111/imm.13662 (2023).
Yodoi, J. et al. TCGF (IL 2)-receptor inducing factor(s). I. Regulation of IL 2 receptor on a natural killer-like cell line (YT cells). J. Immunol. 134, 1623–1630 (1985).
Yoneda, N. et al. Detection of Epstein-Barr virus genome in natural-killer-like cell line, YT. Leukemia 6, 136–141 (1992).
Wang, W., Erbe, A. K., Hank, J. A., Morris, Z. S. & Sondel, P. M. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front. Immunol. 6, 368. https://doi.org/10.3389/fimmu.2015.00368 (2015).
Stanley, P., Narasimhan, S., Siminovitch, L. & Schachter, H. Chinese hamster ovary cells selected for resistance to the cytotoxicity of phytohemagglutinin are deficient in a UDP-N-acetylglucosamine-glycoprotein N-acetylglucosaminyltransferase activity. Proc. Nat. Acad. Sci. USA 72, 3323–3327 (1975).
Kumar, R., Ynag, J., Larsen, R. D. & Stanley, P. Cloning and expression of N-acetylglucosaminyltransferase I, the medial Golgi transferase that initiates complex N-linked carbohydrate formation. Proc. Nat. Acad. Sci. USA 87, 25 (1990).
Narimatsu, Y. et al. A validated gRNA library for CRISPR/Cas9 targeting of the human glycosyltransferase genome. Glycobiology 28, 295–305. https://doi.org/10.1093/glycob/cwx101 (2018).
Cummings, R. D. & Kornfeld, S. Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectins. J. Biol. Chem. 257, 11230–11234. https://doi.org/10.1016/s0021-9258(18)33746-3 (1982).
Mortales, C. L., Lee, S. U. & Demetriou, M. N-Glycan branching is required for development of mature B cells. J. Immunol. 205, 630–636. https://doi.org/10.4049/jimmunol.2000101 (2020).
Mortales, C. L., Lee, S. U., Manousadjian, A., Hayama, K. L. & Demetriou, M. N-Glycan branching decouples B cell innate and adaptive immunity to control inflammatory demyelination. iScience 23, 101380. https://doi.org/10.1016/j.isci.2020.101380 (2020).
Mahapatra, S. et al. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PLoS One 12, e0181134. https://doi.org/10.1371/journal.pone.0181134 (2017).
Kristic, J. et al. Glycans are a novel biomarker of chronological and biological ages. J. Gerontol. A. Biol. Sci. Med. Sci. 69, 779–789. https://doi.org/10.1093/gerona/glt190 (2014).
Kristic, J., Lauc, G. & Pezer, M. Immunoglobulin G glycans—biomarkers and molecular effectors of aging. Clin. Chim. Acta 535, 30–45. https://doi.org/10.1016/j.cca.2022.08.006 (2022).
Franzka, P. et al. Altered glycosylation in the aging heart. Front. Mol. Biosci. 8, 673044. https://doi.org/10.3389/fmolb.2021.673044 (2021).
Wu, J. et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J. Clin. Invest. 100, 1059–1070 (1997).
Koene, H. R. et al. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood 90, 1109–1114 (1997).
de Haas, M. et al. A triallelic Fc gamma receptor type IIIA polymorphism influences the binding of human IgG by NK cell Fc gamma RIIIa. J. Immunol. 156, 2948–2955 (1996).
DiazdeLeon, J. A., Aguilar, I. & Barb, A. W. Macrophage N-glycan processing inhibits antibody-dependent cellular phagocytosis. Glycobiology https://doi.org/10.1093/glycob/cwad078 (2023).
Blomberg, K. et al. Time-resolved fluorometric assay for natural killer activity using target cells labelled with a fluorescence enhancing ligand. J. Immunol. Methods 193, 199–206 (1996).
Miller, M. L. & Finn, O. J. Flow cytometry-based assessment of direct-targeting anti-cancer antibody immune effector functions. Methods Enzymol. 632, 431–456. https://doi.org/10.1016/bs.mie.2019.07.026 (2020).
Alter, G., Malenfant, J. M. & Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 294, 15–22. https://doi.org/10.1016/j.jim.2004.08.008 (2004).
Feasley, C. L., Hykollari, A., Paschinger, K., Wilson, I. B. & West, C. M. N-Glycomic and N-glycoproteomic studies in the social amoebae. Methods Mol. Biol. 983, 205–229. https://doi.org/10.1007/978-1-62703-302-2_11 (2013).
Ceroni, A. et al. GlycoWorkbench: A tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res. 7, 1650–1659 (2008).
Acknowledgements
We thank the donors and staff at the Shepeard Community Blood Center (Augusta, GA) for their generous donations and assistance, Julie Nelson for assistance in single-cell sorting cells (CTEGD Flow Cytometry Core, UGA), Robert Sabatini (UGA) for use of the nucleofection device, Christopher West (UGA) for use of the Li-Cor imager, Jordan S. Orange (Columbia University College of Physicians and Surgeons) for providing the unprocessed NK cell flow cytometry data for the donors listed in Table S2, Harrison B. Hughes for BSL-2 assistance (UGA), Elisabet Gas Pascual and Hanke van der Wel (UGA) for their guidance in permethylation.
Funding
Funding to A.W.B. and E.M.M. by the National Institutes of Health under Award No. U01 AI148114 (NIAID), and R24GM137782 to P.A., and from the Biochemistry and Molecular Biology Department at the University of Georgia, Athens. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.C.R.B., A.R.D., Z.H. and N.B.M. carried out experiments. E.M.M. provided data. M.C.R.B. performed statistical analysis. A.W.B., P.A. and E.M.M. are responsible for funding acquisition. M.C.R.B. and A.W.B. designed the study, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benavente, M.C.R., Hakeem, Z.A., Davis, A.R. et al. Distinct CD16a features on human NK cells observed by flow cytometry correlate with increased ADCC. Sci Rep 14, 7938 (2024). https://doi.org/10.1038/s41598-024-58541-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58541-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
