
Abstract
Peptide toxins found in sea anemones venom have diverse properties that make them important research subjects in the fields of pharmacology, neuroscience and biotechnology. This study used high-throughput sequencing technology to systematically analyze the venom components of the tentacles, column, and mesenterial filaments of sea anemone Heteractis crispa, revealing the diversity and complexity of sea anemone toxins in different tissues. A total of 1049 transcripts were identified and categorized into 60 families, of which 91.0% were proteins and 9.0% were peptides. Of those 1049 transcripts, 416, 291, and 307 putative proteins and peptide precursors were identified from tentacles, column, and mesenterial filaments respectively, while 428 were identified when the datasets were combined. Of these putative toxin sequences, 42 were detected in all three tissues, including 33 proteins and 9 peptides, with the majority of peptides being ShKT domain, β-defensin, and Kunitz-type. In addition, this study applied bioinformatics approaches to predict the family classification, 3D structures, and functional annotation of these representative peptides, as well as the evolutionary relationships between peptides, laying the foundation for the next step of peptide pharmacological activity research.
Similar content being viewed by others

Introduction
Sea anemones (Cnidaria: Anthozoa: Hexacorallia: Actiniaria) are an order of marine animals found in deep oceans and shallow coastal regions around the world, including two suborders: Anenthemonae and Enthemonae1. Cnidarians are one of the oldest venomous lineages in existence. Molecular and fossil evidence suggesting that they appeared more than 750 million years ago, before the Ediacaran period2,3,4. Like other cnidarians, sea anemones store venom in specialized cells known as nematocysts, which have venom-filled capsules and inverted tubules5,6. Contact with prey causes an explosive eversion of the tubule, piercing the target organism and releasing venom for predation, defense, or competitive deterrence7. The endodermal and ectodermal gland tissue of sea anemones contains venom, revealing an alternative venom-delivery mechanism in sea anemones8,9.
Sea anemone venom contains complex proteinaceous (peptides and proteins) and non-proteinaceous components (e.g., quaternary ammonium compounds, purines, and biogenic amines)10. Sea anemone toxins disrupt many targets, including voltage-gated sodium (Nav) and potassium (Kv) channels, acid-sensing ion channels (ASIC), transient receptor potential vanilloid 1 (TRPV1), and transient receptor potential ankyrin 1 (TRPA1)11,12,13,14,15. Many peptide toxins in sea anemones have been studied for their potential as pharmaceutical tools or treatments. The sea anemone venom protein ShK of Stichodactyla helianthus inhibited Kv1.3 with an IC50 in the low picomolar range16,17. The efficacy of ShK and its homologs in the treatment of human autoimmune disorders, including rheumatoid arthritis, multiple sclerosis, and type I diabetes, have been demonstrated in animal models18,19,20. Moreover, the phase I trials of Dalazatide (formerly ShK-186) in the treatment of psoriasis have been successfully concluded21.
Sea anemone Heteractis crispa (H. crispa), also known as the leathery sea anemone, long tentacle anemone or purple tip anemone, belongs to Heteractidae family and is native to the Indo-Pacific region22,23,24. In 1994, Mebs mentioned the sea anemone Heteractis crispa25, which has the valid name Radianthus crispa on the WoRMS website. In 2010, Fedorov et al. published an article in which they mentioned that Radianthus macrodactylus is equivalent to Heteractis crispa26. Some toxins have been detected in H. crispa, mainly actinoporin, Kunitz-type protease inhibitors, Nav channel toxins, and Kv channel toxins26,27,28,29, and the venom assembly in the tentacles, mesentery filaments, and columns of three species of sea anemones (Anemonia sulcata, H. crispa, and Megalactis griffithsi) has been investigated, and the number of toxin-like genes varies significantly between tissues and species22,30,31. The high-throughput sequencing (HTS) technology has been increasingly applied in studying sea anemone venom components, such as Stichodactyla haddoni and Anthopleura dowii32,33,34. In this study, we applied HTS technology to identify the protein and peptide components in H. crispa and compared the distribution of peptide toxins in different tissues, this lays the foundation for in-depth research on H. crispa peptide toxins and provides new potentials for marine drug development.
Results
Transcriptome sequence assembly
We applied the BUSCO suite tools to assess the completeness of transcriptomes35. We found that overall, the BUSCO match values were within the expected range for both a complete single copy and duplicated copies of BUSCO (S/D). Whether in the Tentacles, Column, Mesenterial filaments, or their combination (Combine) dataset, there were a large number of Complete BUSCOs (C) accounting for more than 90% of the Total BUSCO groups, while the proportion of Fragmented BUSCOs (F) and Missing BUSCOs (M) in the Total BUSCO groups was extremely low (Table S1).
The sequence and assembly of H. crispa transcriptome were generated and then submitted to the National Center for Biotechnology Information (NCBI) (BioProject: PRJNA893400, and SRA accession: SRX17999840, SRX17999841, and SRX17999842). The total Reads of the Tentacles (83,799,076), Column (69,427,990), and Mesenterial filaments (67,486,092) were merged into one combined dataset (Combine). After filtering out low-quality reads, about 81.9 million (81,961,116), 67.8 million (67,828,874), 66.2 million (66,235,446), and 120 million reads were obtained from the Tentacles, Column, Mesenterial filaments, and the Combine dataset, respectively. The HTS data was assembled into transcript sequences by using Inchworm, Chrysalis, and Butterfly assembly tools36,37 which generated 288,563 contigs with a mean length of 1,001 bp and an N50 length of 1,829 bp. Meanwhile, 183,198 non-redundant unigenes with a mean length of 769 bp and an N50 length of 1,199 bp were obtained by splicing and removing redundant sequences (Table S2).
Annotation was performed based on five databases to examine unigene functions: Nr (NCBI non-redundant protein sequence), KOG (Eukaryotic Orthologous Groups), Uniprot (Universal Protein), GO (Gene Ontology), and KEGG (Kyoto Encyclopedia of Genes and Genomes). A total of 183,198 unigenes were grouped into these databases: Nr (37,932 unigenes), Uniprot (40,920 unigenes), GO (29,531 unigenes), KEGG (16,167 unigenes), and KOG (20,259 unigenes) respectively, while there were 139,585 unigenes lacking annotation in these databases (Figure S1). Moreover, 19,414 unigenes were enriched into 33 KEGG pathways and assigned into five primary categories: processing environmental information (2,799, 14.42%), cellular processes (2,801, 14.42%), genetic information processing (2,539, 13.08%), metabolism (6,221, 32.04%), and organismal systems (5,054, 26.03%). Most of these unigenes were assigned to metabolism, and the global and overview maps of metabolism contained the most annotated unigenes (Figure S2). 18,016 unigenes were annotated in KOG and categorized into 25 different molecular families (Figure S3)."General function cluster prediction only" (3,831 unigenes, 21.26%) was the largest of these KOG categories, followed by "Signal transduction pathways" (2,419 unigenes, 13.43%), "Posttranslational modification, protein turnover, chaperones" (1,893 unigenes, 10.51%), and "Nuclear structure" (10 unigenes, 0.06%). GO analysis demonstrated that 110,279 unigenes were categorized into three categories: Cellular Component (27,969, 25.36%), Biological Process (46,361, 42.13%), and Molecular Function (35,849, 32.51%), which were further classified into 49 functional groups. The top four enriched functional groups were binding (14,521 unigenes), cellular process (14,127 unigenes), catalytic activity (13,471 unigenes), and metabolic function (13,024 unigenes) (Figure S4).
Hierarchical clustering analysis demonstrated that the Tentacles, Column, and Mesenterial filaments in H. crispa were well distinguished, and all gene sequences were divided into three categories (Fig. 1a). GO analysis of gene expression indicated (Fig. 1b–d) that the number of genes expressed in Tentacles was greater than the number of genes expressed in the Column and Mesenterial filaments. In Tentacles, ribosomes, structural constitutions of ribosomes, translation, and internal anatomical structure have the largest number of genes. Meanwhile, the extracellular region and proteolysis have the largest gene numbers in the Column and Mesenterial filaments.

Cluster heatmap and genes annotated by GO analysis in three different tissues. (a) Cluster heatmap of three different tissues. (b) Bar chart of the number of genes in tentacles. (c) Bar chart of the number of genes in the column. (d) Bar chart of the number of genes in the mesenterial filaments.
Family classification of proteins and peptide toxins in the H. crispa transcriptome
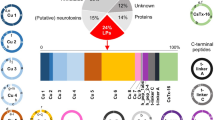
A total of 1049 transcripts were assessed and categorized into 60 families regarding predicted functions, which were assessed according to their amino acid sequence identity, of which 91.0% were proteins and 9.0% were peptides (amino acids ≤ 80) (Fig. 2, Table S3). Most protein components matched Peptidase S1, metalloprotease, G-protein coupled receptor, and Factor 5/8 C-domain. The important and well-known actinoporins family was included in the above statement (Figures S5,6). Moreover, the peptide components were related to the ShKT domain, β-defensin, and Kunitz-type.

Families of putative protein and peptide toxins in H. crispa transcriptome. Based on their amino acid sequences and cysteine scaffolds, the 956 protein sequences and 93 peptide sequences with significant BLAST hits to manually curated lists of animal toxins in UniProt (www.uniprot.org/program/Toxins) were assigned to distinct toxin families.
Comparative analysis of protein and peptide toxins in different tissues of H. crispa
A total of 416, 291, 307, and 428 putative proteins and peptide toxins precursors were detected from the tentacles, column, mesenterial filaments, and their combined dataset, respectively. Figure 3a depicts the comparative distributions of protein and peptide toxins. Of these putative proteins and toxic precursors, 42 were common in these four datasets, of which 33 were proteins and 9 were peptides. A total of 81 were shared by tentacles and column, 74 were shared by tentacles and mesenterial filaments, and 74 were common in both column and mesenterial filaments. These 42 common toxins precursors across four databases were classified into 14 families: β-defensin, metalloproteinase, Kunitz-type, and ShKT domain (Fig. 3b). For each protein and peptide toxin, transcripts Per Kilobase of exon model per Million mapped reads (TPM) values were calculated representing transcription levels. The top ten protein and peptide toxins (with the highest TPM values) in each dataset were assigned. The metalloprotease and ShKT domains derived from the tentacles were expressed at high levels, while the ShKT domain, metalloprotease, and β-defensin derived from the column were expressed at high levels too. However, various proteins and peptide toxins derived from mesenterial filaments were downregulated, among which metalloprotease and ShKT domains were still the highest (Fig. 3c). Therefore, protein and peptide toxins in the ShKT domain and metalloprotease were highly expressed in three H. crispa tissues. Additionally, the ShKT domain included protein and peptide toxins with the highest expressions in the column. Surprisingly, only β-defensin-like peptides were highly expressed in the column but not in the other two tissues, which deserves further studies.

Transcripts of protein and peptide toxins from several H. crispa tissues are compared. (a) Correlation between datasets of putative protein and neurotoxic peptide detected from H. crispa combine, tentacles, column, and mesenterial filaments. (b) 42 putative protein and peptide transcripts from various H. crispa tissues. (c) The ten most greatly expressed protein and peptide transcripts from different H. crispa tissues.
Cysteine pattern analysis of sea anemone peptide toxins
The nomenclature and classification of cysteine patterns in sea anemone neurotoxic peptides have been reported by Kozlov38 and Gao et al39. In this study, a total of 93 peptide toxins were obtained from the tentacles, column, mesenterial filaments, and combined datasets and named Hc-01~Hc-93 in order (Table S4). Many cysteines exist in sea anemone peptide toxins, and cysteine structural scaffolds are diverse. According to our previously proposed classification method39,40, cysteine patterns of these 93 peptides were split into eight broad categories and several subcategories (Fig. 4). The most common peptide structures have six cysteines and three disulfide bond patterns (VI), accounting for 47.31% followed by those having four cysteines producing two disulfide bond patterns (IV), accounting for 25.81%. Furthermore, although most peptides possess an even number of cysteines, there is a small proportion of peptides characterized by an odd number of cysteine residues. The peptide toxins of IV-type and VI-type in sea anemones may be engaged in capturing prey, defending against predators, or repulsing competitors, indicating that these peptide toxins have rich targeting activities41,42. These peptide toxins have potential biotechnological applications and provide rich resources for the development of new drugs.

Classification of peptide toxic cysteine patterns from H. crispa.
Sequence analysis of typical sea anemone peptide toxins
Sea anemone venom has a high concentration of peptide toxins, serving a crucial function in prey and defense7, and they operate on various ion channels, such as Nav channels, Kv channels, ASIC, TRPV1, and TRPA129,43,44,45. The three-dimensional (3D) structure or cysteine pattern or both of nine representative anemone peptide toxins have been determined, including ShKT domain, epidermal growth factor-like (EGF-like), β-defensin-like, Kunitz-type, Anemonia sulcata toxin III (ATX-III), inhibitor cystine-knot (ICK), small cysteine-rich peptides (SCRiPs), proline-hinged asymmetric β-hairpin (PHAB), and boundless β-hairpin (BBH)41. Herein, we described sea anemone peptide toxins with typical and unique homologs, including ShKT domain (11 homologs), β-defensin-like (11 homologs), Kunitz-type (7 homologs), and EGF-like (3 homologs). The peptide toxin sequences from these representative families were analysed to assess their similarity. The distribution of these sequences in these four datasets was examined. Additionally, the 3D structures of select significant peptide toxins were predicted (Figs 5, 6, 7 and 8).

Sea anemone ShKT domain mature peptide sequences. (a–b) The conserved cysteine residues are highlighted with green text on yellow background. T, C, F, and M respectively represent tentacles, column, mesenterial filaments, and combine, highlighted in blue, orange, green, and yellow. (c) Homology modeling and ShK prediction of sea anemone mature peptides HC-36, HC-37, and HC-43 (PDB 4LFQ).

β-defensin-like sea anemone mature peptide sequences. (a–b) The conserved cysteine residues are highlighted with green text on yellow background. T, C, F, and M respectively represent tentacles, column, mesenterial filaments, and combine, highlighted in blue, orange, green, and yellow. (c) Homology modeling prediction of several mature peptides from sea anemones with CgNa (PDB 2H9X), BDS I (PDB 1BDS), and APETx2 (PDB 2MUB).

Representative mature peptide sequences from sea anemones containing Kunitz-type peptides. (a) The conserved cysteine residues are highlighted with green text on yellow background. T, C, F, and M respectively represent tentacles, column, mesenterial filaments, and combine, highlighted in blue, orange, green, and yellow. (b) Homology modeling prediction of mature peptides sea anemone HC-50 and HC-47 with SHPI-1 (PDB 3M7Q).

Representative Sea anemone mature peptide sequences in other families. (a) The conserved cysteine residues are highlighted with green text on yellow background. T, C, F, and M respectively represent tentacles, column, mesenterial filaments, and combine, highlighted in blue, orange, green, and yellow. (b) Homology modeling prediction of several representative sea anemone mature peptides with human EGF (PDB 7SZ1), PI-actitoxin-Avd5a (PDB 1Y1B).
The ShK toxin, isolated from the sea anemon Stichodactyla helianthus46, is termed the ShKT domain inhibits Kv channels16,17,47,48,49,50. A total of 11 homologous sequences with ShK that have not been reported previously were assessed, their cysteine pattern was C-C-C-CX3CX2C, and it predicted that the connection mode of disulfide bonds was C1-C6, C2-C4, C3-C5 (Fig. 5a,b). The sequence alignment demonstrated that HC-39 had the highest similarity with the previously identified sequence ID (GenBank No. XP_031556600.1), and the sequence identity was 85.71%. Additionally, the sequence similarity of HC-36/37/43 with ShK was 30.77%, 52.94%, and 30.30%, respectively. The homology modeling prediction indicated that HC-36/37/43 and ShK have 3D structure similarities. This suggests they may simultaneously act on Kv channels (Fig. 5c). These peptide sequences are all analogues of ShK and deserve further study.
β-defensins are ubiquitous in vertebrate antimicrobial peptides and are part of the main components of the innate immune system51,52,53. However, β-defensin-like peptides in sea anemone venom including CgNa, Rc I, Am II, BDS I, APETx1, APETx2, and Magnificamide are potential toxins that may disrupt voltage- and ligand-gated ion channels as Nav types 1/2/4, Kv type 3, ASIC, and ASIC354,55,56,57,58,59,60. CgNa can be purified from the sea anemone Condylactis gigantea and inhibit Nav types 1/261. Rc I is a peptide toxin in H. crispa, which can inhibit Nav channels62. Am II is a neurotoxin from Antheopsis maculata with toxin-paralyzing activity against crabs63. BDS I is a peptide toxin with an anti-angiogenic activity from the sea anemone Anemonia viridis64. APETx1 and APETx2 are peptide toxins with antibacterial and neurotoxic activity from Anthopleura Elegantissima. These toxins act on ERG Kv and Nav channels and ASIC356,65,66,67,68. Magnificamide, a peptide inhibitor of mammalian α-amylases, isolated from the venom of sea anemone Heteractis magnifica, can be used to control postprandial hyperglycemia in diabetes mellitus69. Therefore, its functionally active recombinant analogue is a promising agent that awaits further investigation as a potential drug candidate for the treatment of type 2 diabetes mellitus70.
In this study, we identified 11 homologous sequences to sea anemone toxin β-defensin-like peptides with a cysteine pattern of CXC-C-C-CC (Fig. 6a,b). This compact core β-defensin structure relies on disulfide connections formed between cysteines C1-C5, C2-C4, and C3-C6. All these 11 novel homologous sequences were not previously reported. Sequence similarity analysis indicated that the sequence identity of HC-71 and Rc I (GenBank No. P0C5G5.1), HC-66/70 and Am II (GenBank No. P69930.1) was 97.87%, 97.83%, and 95.65%, respectively. Furthermore, HC-64/73 showed high similarities with previously reported sequences (GenBank No. ALL34531.1 and GenBank No. ALL34540.1), 93.33% and 80.00%, respectively. Taking CgNa (PDB 2H9X), BDS I (PDB 1BDS), and APETx2 (PDB 2MUB) as the templates, homology modeling prediction showed that HC-71 and Rc I, HC-64/68, and BDS I, HC-65/69 and APETx2 have similar 3D structures, respectively (Fig. 6c). Therefore, these peptide toxins may block multiple ion channels, including Nav, Kv, and ASIC.
Kunitz-type peptides are ubiquitous in marine organism venom71,72. Kunitz-type peptides block ion channels and are anti-inflammatory73. Kunitz-type peptides in sea anemones affect TRPV and type II Kv channels74,75,76. The ShPI-1 belongs to the Kunitz-type peptide, which is derived from the sea anemone Stichodactyla helianthus77,78,79. The ShPI-1 is toxin-suppressing Kv channels and proteases78. SHPI-1 is active against serine proteases, cysteine proteases, and aspartic proteases80,81. Seven homologous sequences with ShPI-1 were identified, and except for HC-52, the cysteine patterns of other sequences were C-C-C-C-CX3C, comprising three disulfide bridges with C1-C6, C3-C5, C2-C4 connectivity (Fig. 7a). Of the seven newly discovered sequences in Kunitz-type, HC-49 has the highest similarity of 84.75% to the reported sequence (XP_031567285.1). Furthermore, HC-47/50 and SHPI-1 have similar 3D structures, with a similarity of 56.36% and 54.72%, respectively (Fig. 7b). Therefore, HC-47 and HC-50 may have similar biological activities to SHPI-1.
EGF has a crucial role in the growth, proliferation, and differentiation of various cells of vertebrates82,83. The tight association between EGF-like peptides and the pathogenesis of human cancer is indicated by their diverse functionality in several cancer cell types, including bladder and liver cancer, as observed in mammalian EGF and its family members84,85,86,87. In invertebrates, particularly among hazardous marine creatures, the associated proteins have garnered considerable recognition due to their transformation into defensive and predatory toxins88. L-EGF is a growth factor released by the gastropod mollusk Lymnaea stagnalis88,89. The peptide toxin Gigantoxin I (ω-stichotoxin-Sgt1a) from Stichodactyla gigantea, acting on the Nav channel, can paralyze crabs89,90. Gigantoxin I is the first peptide toxin of the EGF family and is representative88. Here, we detected three homologous sequences with Gigantoxin I, and their cysteine patterns were C-C-C-CXC-C (Fig. 8a). The sequence identity of Gigantoxin I, HC-77, and Human EGF are 35.71% and 36.36%, respectively. Using Human EGF (BDP 7SZ1) as a model revealed similar 3D structures of Gigantoxin I and HC-77 (Fig. 8b). Therefore, invertebrate EGF family members, including three identified homologous sequences, may have similar biological activities to mammalian EGF.
Kazal-like belongs to serine protease inhibitor family and plays crucial roles in host physiological blood coagulation91,92, development regulation, and immunological functions93, in which protease activity is modulated by protease inhibitors94. PI-actitoxin-Avd5a is an elastase inhibitor from Anemonia sulcata, a 'non-classical' Kazal-type protein, and PI-actitoxin-Avd5a reveals strong inhibition against Streptomyces griseus protease B (SGPB)95,96. Taking PI-actitoxin-Avd5a (PDB 1Y1B) as a template for homology modeling, PI-actitoxin-Avd5a and HC-56 revealed similar 3D structures. Accordingly, HC-56 could strongly inhibit SGPB as PI-actitoxin-Avd5a.
ICK is a family of structural peptides that exerts its effects by targeting ion channels and serving as a defense mechanism against pathogens97. ICK is found abundantly in various species, and ICK toxins are also prevalent in animal venom that contribute to predation and defense97,98. BcsTx3, an ICK representative, is a Kv channel blocker from Bunodosoma caissarum. BcsTx3 mainly inhibits Kv channels, including but unlimited to Kv1.1, rKv1.2, hKv1.3, and rKv1.6. It also paralyzes swimming crabs when injected at the junction between the body and the walking leg99. Using blast alignment, the similarity between HC-87 and BcsTx3 (GenBank No.C0HJC4.1) sequences is as high as 72%. BcsTx3 and HC-87 have the same cysteine pattern (C-C-CC-C-C-C-C) (Fig. 8a). Therefore, it can be deduced that HC-87 may have one or all activities of BcsTx3.
MS 9.1, a positive modulator of mammalian TRPA1, is a typical representative of the BBH family41. TRPA1 is a non-selective cation channel involved in various physiological processes and exhibits significant anti-inflammatory and analgesic activities15,100,101,102. The homologous alignment results showed that HC-18 was the same as the sequence (GenBank No. BAS68532.1) from sea anemone Heteractis aurora. The similarity of HC-18/19 was 87.5%, and they share the same cysteine pattern. The HC-18/19 compounds that have been identified belong to the BBH family. It is hypothesized that the target of these compounds is TRPA1, providing a basis for developing drug screening assays aimed at identifying potential anti-inflammatory and analgesic medications.
Acrorhagin Ic obtained from red waratah sea anemone Actinia tenebrosa in New Zealand and Australian, is a member of the Acrorhagin family. HC-85/86 sequence is highly similar to the previously reported Acrorhagin Ic sequence (GenBank No.ATY39990.1), and the sequence identity is 61.90% and 59.52%, respectively. Although HC-85/86 and Acrorhagin Ic have similar 3D structures, Acrorhagin Ic is a toxin lethal to crabs inactively against any ion channels with no bacteriostatic activity, suggesting this peptide could have various biological functions103,104. Furthermore, HC-89 is also highly similar to the Supwaprin-a sequence (GenBank No.XP_048584021.1), a peptide from Nematostella vectensis (starlet sea anemone), and its sequence identity is 60.78%.
Phylogenetic analysis of H. crispa sea anemone peptides
A total of 106 peptide sequences, including the mature regions of 93 peptide sequences from sea anemone H. crispa and 13 peptides of known families, were clustered using MEGA 7.0.14 software105,106. The results of phylogenetic analysis indicated that all peptides were divided into five major categories, some of which were consistent with the family classification based on the cysteine pattern, and there were also large differences in the family classification of a large part of peptides (Fig. 9). Among them, these β-defensin-like peptides (except for HC-72), Acrorhagin family, EGF (except for HC-78), and Kunitz-type family peptides can be clustered into one branch, respectively, showing good evolutionary and affinity relationship, consistent with the classification results based on cysteine pattern and structural characteristics. ShKT domain, Kazal-like, and BBH family peptides are clustered into two or more branches. This indicates their low similarity amino acid sequences, suggesting that there may be more diversity in target and activity. HC-05/06 were unknown families but were embedded in the ShKT domain. It is speculated that they are highly similar to the ShKT domain sequences with similar activities. A total of 52 unknown family peptides are distributed in nine major categories in the phylogenetic tree, indicating that family classification relies on amino acid similarity which provides more basis for the family classification of sea anemone peptide toxins.

Phylogenetic tree from sequenced 93 peptide sequences and reported sequences in the Blast database. The tree was established by the NJ approach. Sequences with the same background color indicate peptides from the same family.
Discussion
HTS technology has been applied to multiple venom components of sea anemones39, including Exaiptasia diaphana, Nematostella vectensis, Oulactis sp., Anemonia sulcata, Megalactis griffithsi, Epiactis japonicus, Exaiptasia pallida, Anthopleura dowii, and Stichodactyla haddoni30,32,33,40,107,108,109,110,111,112,113. The HTS and bioinformatics techniques have been used to determine venom assemblage in tentacles, column, and mesenterial filaments for three species of sea anemone: H. crispa, Anemonia sulcata, and Megalactis griffithsi. A significant diversity was reported in the abundance of toxin-like genes across tissues and species30. However, specific sea anemone H. crispa crude venom components are unclear. Integrating HTS and bioinformatics technology can explore the peptide components in crude venom of sea anemone H. crispa, predict its family classification, and 3D structural and functional annotation.
Three sea anemone H. crispa tissues were sequenced and analyzed to explore peptide toxin distribution. Our HTS analysis detected 1049 transcripts, including 416, 291, and 307 putative protein and peptide toxin transcripts, respectively, from the tentacles, column, and mesenterial filaments. Whether these putative protein and peptide toxins are present in the venom needs to be further verified through technologies such as proteomics. Using the combined proteomic and transcriptomic techniques, a holistic overview of the venom arsenal of the well-studied sea anemone was obtained32. Macrander et al30, analyzed three sea anemone H. crispa tissues, including the tentacles, column, and filament, obtaining 840 protein and peptide toxins, and the toxin expression levels in the tentacles were significantly higher than those in the column and filament. However, we found that the protein and peptide families of the top 10 TPM sequences in the three tissues are similar, with the overall expression levels of all toxins being highest in the column of sea anemone H. crispa, followed by the tentacles and mesenterial filaments. Through comparative analysis, we found that there were significant differences between individuals even within the same species of sea anemone, and determining organizational boundaries may affect the data results. Among the 1049 protein and peptide sequences identified in this study, 88 sequences had a similarity of over 80% with the 840 sequences identified by Jason Macrander30. Among these 88 sequences, the main families of protein toxins are metalloproteinase and protein inhibitor, and the family of peptides is β-defensins and Kunitz-type (Figure S7a). However, a comparative analysis of 93 peptide sequences with previous study data revealed that only 22 sequences shared a similarity of over 80% (Figure S7b). Therefore, using our peptide screening principle, only 208 peptides were screened from 840 sequences identified by Jason Macrander30. Comparative analysis of cysteine in peptides showed that in previous studies, 142 peptides had cysteine residues less than 4, and only 66 peptides had cysteine residues greater than or equal to 4. These 93 peptide sequences all contain 4 or more cysteine residues, with the sequences containing 6 cysteine residues being the most abundant ones (Figure S7c). As is well known, most active sea anemone peptides such as β-defensins, ShK, Kunitz-type and other family toxins all contain 6 cysteine residues. In addition, comparative analysis was conducted with the traditional isolation of 48 protein and peptide sequences from sea anemone H. crispa and H. magnifica, and it was found that HC-71 and Rc I from H. crispa only differ by one amino acid, but there is no similar sequence from H. magnifica, indicating the diversity of sea anemone peptide toxins. Therefore, there are significant differences in toxin levels between individuals of the same species, especially anemones from different sea areas.
Herein, the proportion of identified proteins and peptide toxins is significantly different from previous studies. For example, the proportion of peptide sequences (9%) in sea anemone Stichodactyla haddoni32 and Exaiptasia diaphana40, was significantly lower than the proportion of protein sequences (91%) in our study. The relatively low proportion of peptides in the entire transcriptome of sea anemones can be attributed to the high concentration of protein in the column and mesenterial filaments. Most protein components corresponded to Peptidase S1, metalloprotease, G-protein coupled receptor, Factor 5/8 C-domain, and actinoporins. The actinoporins family, which holds significant recognition, has been identified for its possession of hemolytic action28. The sea anemone H. crispa actinoporin has in vitro anticancer activity, and it is expected to become an anticancer drug with high anti-migration potential26,114. In addition, the peptide constituents were shown to align with the ShKT domain, β-defensin, and Kunitz-type. In general, comparable findings were noted in transcriptome data obtained from various sea anemones32,33,40. The protein component metalloproteinases were highly expressed in the tentacles and the mesenterial filaments, with lower expression levels in the column than in ShKT domain. Metalloproteinases significantly participate in wound-healing processes in cnidarians, including tentacles regeneration and transdifferentiating115, they also have potential dual involvement in food digestion development116. For peptide toxin components, the ShKT domain was highest expression in the column tissue of sea anemone H. crispa, followed by expression in the tentacles and a small amount in the mesenterial filaments. This also verifies that sea anemones do not have a centralized venom system, and the toxin peptide is expressed throughout the sea anemone, not just in the tentacles30,117. Surprisingly, β-defensin-like peptides are only highly expressed in the column, not in the other two tissues. β-defensin-like peptides, as antimicrobial peptides, can accelerate wound healing, are widely present in vertebrates, and are one of the main components of the innate immune system51,52,53,118. β-defensin peptides showed paralytic activity in crustaceans, indicating that it had evolved into a weapon to capture prey119. Here, β-defensin-like peptides are not highly expressed in the tentacles and may have other biological functions.
The most common and pharmacologically valuable peptides in sea anemones are the ShKT domain, β-defensin-like, Kunitz-type, and EGF-like peptides, and they influence various ion channels, including Nav channels, Kv channels, ASIC, TRPV1, and TRPA129,43,44,45. The ShKT domain is one of the families with the highest presence in the transcriptome data of sea anemone H. crispa, suggesting that this type of peptide toxin may play a crucial role in its predation, defense, and competition17,47,48. ShK inhibits Kv channels, blocking Kv1.1/1.2/1.3/1.6/3.2 and Kca3.1 channels, especially in Kv1.1/KCNA1 and Kv1.3/KCNA3 channels16,49,50. Kv1.3 is involved in various autoimmune diseases and many cancers by contributing to cell proliferation, malignant angiogenesis, and metastasis18,20,120,121,122,123,124,125. ShK is a Kv1.3 channel blocker analogue with significant roles in T and B lymphocyte subsets related to autoimmune conditions. Therefore, ShK is a potential immune modulator for autoimmune disease therapy46. Of these, ShK-186, also known as Dalazatide, was the first representative of the ShKT domain to be detected and characterized and the first drug to complete Phase I trials17,46,126. ShK and its analogues, including 11 homologs in the ShKT domain found in this study, may act on Kv1.3, suggesting that they may have significant involvements in treating human autoimmune disorders127,128,129.
β-defensin-like peptides block ligated-gated and voltage-gated ion channels, as Nav types 1/2/4, Kv type 3, and ASIC54,55,56,57,130. Eleven identified β-defensin-like homologous sequences may act on Nav types 1/2/4 channels related to acute and chronic pain, and it can potentially treat pain131,132. Additionally, these peptides acting on Nav channels, considered insecticidal lead compounds, have insecticidal effects133. Kunitz-type peptides block ion channels and are anti-inflammatory73. HCRG1/2 are the first Kunitz-type peptides to block Kv1.3 found in sea anemones27,134. The first Kunitz-type representative bovine pancreatic trypsin inhibitor (BPTI) is a serine protease inhibitor resisting inflammatory responses135,136. In sea anemones, Kunitz-type peptides act on TRPV1 and Kv channels29,74,75, indirect TRPV1 activation contributes to EGF receptor/PLA2/arachidonic acid/lipoxygenase pathway, resulting in Kunitz-type peptides regulating TRPV1 channel activity41,137. APHC1-3 is earlier shown to possess a unique property of inhibiting of the pain vanilloid receptor TRPV1 in vitro and providing the analgesic effects in vivo in addition to their trypsin inhibitory activity76. The activated ion channel TRPV1 produces pain, so TRPV1 is the most important therapeutic target for pain and inflammatory stimulation14,29,75,138. Besides blocking TRPV1 channels, various anemone Kv channel toxins inhibit serine protease activity, participating in various functions, like blood clotting, tumor immunity, fibrinolysis, inflammatory modulation, and resistance against bacterial and fungal infections73,139,140. Seven homologous Kunitz-type peptide sequences were identified, contributing to the anti-inflammatory responses by inhibiting serine protease activity and Kv channels.
The phylogenetic tree of typical family sea anemone peptides exhibited a pattern in which the majority of the sequences were clustered based on families, while a subset of individual sequences remained dispersed among alternative family groupings. The peptide sequence families of the transcriptomes in this study were based on changes in cysteine patterns and 3D structures, resulting in some sequences not being clustered together. Therefore, 3D structural alignment is a very powerful tool for inferring the evolutionary relationship between two low homology peptides.
Conclusions
The transcriptome analysis of H. crispa sea anemone venom from the tentacles, column, and mesenterial filaments was performed using HTS technology. A total of 1049 putative protein toxins were obtained, including 956 (91.0%) protein sequences and 93 (9.0%) peptide toxin sequences, which were divided into 60 known families. ShKT domain in peptide toxins was predominantly expressed in the tentacles, column, and mesenterial filaments and contributes to prey capture, defines, and intraspecific competition. Our study demonstrated that the venom assemblages within these different sea anemone H. crispa tissues are complex and diverse. Combining HTS and bioinformatics technologies new peptides can be systematically identified in addition to predicting their family categorization, 3D structures, and functional annotations. These advances lay the foundation for enhanced understanding and development of sea anemone venom as potential marine pharmaceuticals.
Materials and methods
Specimens and RNA extraction
The sea anemone was collected from Paracel Islands located at [Lat 15°46' N, Lon 111°11' E] in the Southern Sea of China and maintained in the lab in aquariums containing artificial seawater. The sea anemone sample was identified by the mitochondrial genome as H. crispa. A total of three H. crispa were collected, and over a week, different tissues were removed from H. crispa using tweezers and a scalpel, starting with the tentacles, column, and mesenterial filaments including the pharynx and gonads. Three different H.crispa tissue samples were mixed separately, and then the total RNA from these three tissues were extracted after liquid nitrogen flash evaporation (TIANGEN biotech Co., Ltd., China), and their RNA integrity number values were measured using an Agilent 2100 Bioanalyzer (Agilent Tech., Palo Alto, CA, USA). Then, BGI-Tech (Shenzhen, Guangdong, China) was used to build three Illumina cDNA libraries from qualifying RNAs and sequenced them using an Illumina HiSeq4000 platform (San Diego, CA, USA).
Sequence analyses and assembly
This study evaluated the assembly integrity of four assembled transcripts using BUSCO v5.2.2 software and databases: etazoan_ Odb10 (Creation date: 2021-02-17, genomes: 65, number of BUSCOs: 954). BUSCO was run in mode: transcriptome. Illumina HTS, raw image data, was converted into raw reads after base calling by Illumina CASAVA software (v1.8.4). High-quality clean reads were obtained by removing the adapter and reads with > 10% of non-sequenced bases or > 50% of low-quality bases (≤ 10 was the base quality score). We compared the transcriptomes after assembly to evaluate the impact of the cleanup step on overall completeness and also conducted a reciprocal BLAST (Basic Local Alignment Search Tool) search of known sea anemone venom genes to determine whether the alternative cleanup strategies would result in a different number of candidate toxin genes. The transcriptome sequence assembly strategy was used to assemble HTS data into transcript sequences through three steps of Inchworm, Chrysalis, and Butterfly36,37. (A) Inchworm: Use Kmer-based assembly strategy to assemble reads into contigs, (B) Chrysalis: Cluster contigs sequences, define components, and align reads back to components to verify correctness, (C) Butterfly: De Bruijn graph-based assembly strategy to assemble components into possible transcripts. This study generated four transcriptome reference sequences for Tentacles, Column, Mesenterial filaments, and their combination (Combine).
Gene annotation
The Unigene gene's coding region was predicted using the translation approach, and possible coding protein sequences were predicted. The resulting protein sequences were cross-referenced against Uniprot and the non-redundant (NR) protein database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The protein coding model was determined by utilizing the coding mode of the alignment with the highest alignment score. Unigene gene annotation was conducted based on NR, Uniprot, KEGG141,142,143, and KOG (eukaryotic)/COG (prokaryotic) databases.
Cluster heatmap and GO analysis
Differentially expressed genes (DEGs) between two tissues were determined by a Fold Change (FC) of | LogFC |>2. The gene expression heatmap is plotted using the TPM value and the R language's photomap package (Pretty Heatmaps (Version 1.0.12) method. GO enrichment analysis of DEGs was conducted by using the cluster profiler program144. Fisher's exact test pvalue or Benjamin’s corrected pvalue less than 0.05 was set as the significant enrichment level.
Identification of protein and peptide toxins
Prediction of sea anemone protein and peptide toxins using four datasets, homolog searches, and an ab initio prediction method (tentacles, column, and mesenterial filaments, Combine). The BLAST database was queried for proteins and peptides for sequence similarity prediction. After assembly, the sequences were checked against a local database using BLASTX (with an E-value of 1e-5). The BLASTX-hit unigenes were used to generate amino acid sequences. According to the BLAST database's superfamily and family classifications, those four datasets were divided into different groups.
Prediction and comparison of 183,198 transcripts of H. crispa were completed by using SPM Predictor (length ≤ 200, hydrophobic ≥ 70%), Diamond ATDB database (with an E-value ≤ 1e-8) and Diamond NR database (with an E-value ≤ 1e-8). A Python script was developed to trim all of the sea anemone toxin-candidate transcripts to allow only the open-reading frame (ORF) identified by Transdecoder (http://transdecoder.github.io).
Classification of protein and peptide toxins superfamilies
Using the BLAST (default setting), predicted sea anemone peptide and protein transcripts were identified. Peptide and protein toxins with the highest resemblance to known superfamilies in the BLAST database were assigned based on cysteine structural scaffold. Those protein and peptide toxins with low similarities (< 75%) were classified into unknown groups.
Alignment and homology modelling
MEGA 7.0.14 software was used to create new protein sequence alignments and perform amino acid alignments on all peptide sequences, where the MUSCLE algorithm was chosen to intelligently align amino acids105,106. Genedoc software was used to export the sequence in FASTA format.
Protein 3D structure was predicted using homology computational structure prediction modeling from amino acid sequences145. The SWISS-MODEL, available through the Expasy web server or Deep View software (Swiss Pdb-Viewer) were applied. The homologous sequences with high sequence identity were assigned as templates, and then the cartoon mode was used to build the model.
Phylogenetic analyses
Representative peptide sequences from various families of sea anemones were obtained from UniProt and BLAST databases (www.uniprot.org/, https://blast.ncbi.nlm.nih.gov/), and they were comparable to those sequences obtained in this study. The mature regions of 93 peptide sequences were aligned using MEGA 7.0.14. A phylogenetic tree was established using the Neighbor-Joining approach (Bootstrap method 1000 and Pairwise deletion 50%).
Human and animal resources
The article does not involve human or animal experiments, and all sea anemones are collected according to the collection permit issued by the China Fisheries Administration.
Data availability
The datasets described are accessible through internet repositories. The repository(s) and accession number(s) can be accessed at the following URL: https://www.ncbi.nlm.nih.gov/sra/PRJNA893400 (accession: SRX17999840, SRX17999841, and SRX17999842).
References
Rodríguez, E. et al. Hidden among sea anemones: the first comprehensive phylogenetic reconstruction of the order Actiniaria (Cnidaria, Anthozoa, Hexacorallia) reveals a novel group of hexacorals. PLoS ONE 9, e96998. https://doi.org/10.1371/journal.pone.0096998 (2014).
Jouiaei, M. et al. 2015 Ancient venom systems: A review on cnidaria toxins. Toxins. 7(6), 2251–2271 (2015).
Erwin, D. H. et al. The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science 334, 1091–1097. https://doi.org/10.1126/science.1206375 (2011).
Park, E. et al. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Mol. Phylogenet. Evol. 62, 329–345. https://doi.org/10.1016/j.ympev.2011.10.008 (2012).
David, C. N. et al. Evolution of complex structures: Minicollagens shape the cnidarian nematocyst. Trends. Genet. 24, 431–438. https://doi.org/10.1016/j.tig.2008.07.001 (2008).
Beckmann, A. & Ozbek, S. The nematocyst: A molecular map of the cnidarian stinging organelle. Int. J. Dev. Biol. 56, 577–582. https://doi.org/10.1387/ijdb.113472ab (2012).
Lotan, A., Fishman, L., Loya, Y. & Zlotkin, E. Delivery of a nematocyst toxin. Nature 375, 456. https://doi.org/10.1038/375456a0 (1995).
Columbus-Shenkar, Y. Y. et al. Dynamics of venom composition across a complex life cycle. Elife 7, e35014. https://doi.org/10.7554/eLife.35014 (2018).
Moran, Y. et al. Neurotoxin localization to ectodermal gland cells uncovers an alternative mechanism of venom delivery in sea anemones. Proc. Biol. Sci. 279, 1351–1358. https://doi.org/10.1098/rspb.2011.1731 (2012).
Frazao, B., Vasconcelos, V. & Antunes, A. Sea anemone (Cnidaria, Anthozoa, Actiniaria) toxins: an overview. Mar. drugs 10, 1812–1851. https://doi.org/10.3390/md10081812 (2012).
Moran, Y., Gordon, D. & Gurevitz, M. Sea anemone toxins affecting voltage-gated sodium channels–molecular and evolutionary features. Toxicon 54, 1089–1101. https://doi.org/10.1016/j.toxicon.2009.02.028 (2009).
Diochot, S. & Lazdunski, M. Sea anemone toxins affecting potassium channels. Prog. Mol. Subcell. Biol. 46, 99–122. https://doi.org/10.1007/978-3-540-87895-7_4 (2009).
Rodríguez, A. A. et al. A novel sea anemone peptide that inhibits acid-sensing ion channels. Peptides 53, 3–12. https://doi.org/10.1016/j.peptides.2013.06.003 (2014).
Andreev, Y. A. et al. Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 283, 23914–23921. https://doi.org/10.1074/jbc.M800776200 (2008).
Logashina, Y. A. et al. Peptide from sea anemone metridium senile affects transient receptor potential ankyrin-repeat 1 (TRPA1) function and produces analgesic effect. J. Biol. Chem. 292, 2992–3004. https://doi.org/10.1074/jbc.M116.757369 (2017).
Kalman, K. et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 273, 32697–32707. https://doi.org/10.1074/jbc.273.49.32697 (1998).
Castaneda, O. et al. Characterization of a potassium channel toxin from the Caribbean Sea anemone Stichodactyla helianthus. Toxicon 33, 603–613. https://doi.org/10.1016/0041-0101(95)00013-c (1995).
Chandy, K. G. & Norton, R. S. Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Curr. Opin. Chem. Biol. 38, 97–107. https://doi.org/10.1016/j.cbpa.2017.02.015 (2017).
Pennington, M. W. et al. Development of highly selective Kv1.3-blocking peptides based on the sea anemone peptide ShK. Mar. Drugs 13, 529–542. https://doi.org/10.3390/md13010529 (2015).
Beeton, C. et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA. 103, 17414–17419. https://doi.org/10.1073/pnas.0605136103 (2006).
Tarcha, E. J. et al. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS One 12, e0180762. https://doi.org/10.1371/journal.pone.0180762 (2017).
Kashimoto, R. et al. Transcriptomes of giant sea anemones from okinawa as a tool for understanding their phylogeny and symbiotic relationships with anemonefish. Zool. Sci. 39, 374–387. https://doi.org/10.2108/zs210111 (2022).
Nguyen, H. T. T., Dang, B. T., Glenner, H. & Geffen, A. J. Cophylogenetic analysis of the relationship between anemonefish amphiprion (Perciformes: Pomacentridae) and their symbiotic host anemones (Anthozoa: Actiniaria). Mar. Biol. Res. 16, 117–133. https://doi.org/10.1080/17451000.2020.1711952 (2020).
Titus, B. M. et al. Phylogenetic relationships among the clownfish-hosting sea anemones. Mol. Phylogenet. Evol. 139, 106526. https://doi.org/10.1016/j.ympev.2019.106526 (2019).
Mebs, D. Anemonefish symbiosis: vulnerability and resistance of fish to the toxin of the sea anemone. Toxicon 32, 1059–1068. https://doi.org/10.1016/0041-0101(94)90390-5 (1994).
Fedorov, S. et al. The anticancer effects of actinoporin RTX-A from the sea anemone Heteractis crispa (=Radianthus macrodactylus). Toxicon 55, 811–817. https://doi.org/10.1016/j.toxicon.2009.11.016 (2010).
Gladkikh, I. et al. Kunitz-type peptides from the sea anemone Heteractis crispa demonstrate potassium channel blocking and anti-inflammatory activities. Biomedicines 8, 473. https://doi.org/10.3390/biomedicines8110473 (2020).
Kalina, R. S. et al. New sea anemone toxin RTX-VI selectively modulates voltage-gated sodium channels. Dokl. Biochem. Biophys. 495, 292–295. https://doi.org/10.1134/S1607672920060071 (2020).
Monastyrnaya, M. et al. Kunitz-type peptide HCRG21 from the sea anemone Heteractis crispa is a full antagonist of the TRPV1 receptor. Mar. Drugs 14, 229. https://doi.org/10.3390/md14120229 (2016).
Macrander, J., Broe, M. & Daly, M. Tissue-specific venom composition and differential Gene expression in sea anemones. Genome Biol. Evol. 8, 2358–2375. https://doi.org/10.1093/gbe/evw155 (2016).
Delgado, A., Benedict, C., Macrander, J. & Daly, M. Never, Ever make an enemy out of an anemone: Transcriptomic comparison of clownfish hosting sea anemone venoms. Mar. Drugs 20, 730. https://doi.org/10.3390/md20120730 (2022).
Madio, B., Undheim, E. A. B. & King, G. F. Revisiting venom of the sea anemone Stichodactyla haddoni: Omics techniques reveal the complete toxin arsenal of a well-studied sea anemone genus. J. Proteomics 166, 83–92. https://doi.org/10.1016/j.jprot.2017.07.007 (2017).
Ramirez-Carreto, S. et al. Transcriptomic and proteomic analysis of the tentacles and mucus of Anthopleura dowii Verrill, 1869. Mar. Drugs 17, 436. https://doi.org/10.3390/md17080436 (2019).
Rodriguez, A. A. et al. Peptide fingerprinting of the neurotoxic fractions isolated from the secretions of sea anemones Stichodactyla helianthus and Bunodosoma granulifera. New members of the APETx-like family identified by a 454 pyrosequencing approach. Peptides 34, 26–38. https://doi.org/10.1016/j.peptides.2011.10.011 (2012).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245. https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. https://doi.org/10.1038/nbt.1883 (2011).
Li, B. & Dewey, C. N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. https://doi.org/10.1186/1471-2105-12-323 (2011).
Kozlov, S. & Grishin, E. Convenient nomenclature of cysteine-rich polypeptide toxins from sea anemones. Peptides 33, 240–244. https://doi.org/10.1016/j.peptides.2011.12.008 (2012).
Fu, J., Liao, Y., Jin, A. H. & Gao, B. Discovery of novel peptide neurotoxins from sea anemone species. Front. Biosci. Landmark. 26(11), 1256–1273. https://doi.org/10.52586/5022 (2021).
Fu, J. X. et al. Transcriptome sequencing of the pale anemones (Exaiptasia diaphana) revealed functional peptide gene resources of sea anemone. Front. Mar. Sci. 9, 856501. https://doi.org/10.3389/fmars.2022.856501 (2022).
Madio, B., King, G. F. & Undheim, E. A. B. Sea anemone toxins: a structural overview. Mar. Drugs 17, 325. https://doi.org/10.3390/md17060325 (2019).
Menezes, C. & Thakur, N. L. Sea anemone venom: Ecological interactions and bioactive potential. Toxicon 208, 31–46. https://doi.org/10.1016/j.toxicon.2022.01.004 (2022).
Bosmans, F. & Tytgat, J. Sea anemone venom as a source of insecticidal peptides acting on voltage-gated Na+ channels. Toxicon 49, 550–560. https://doi.org/10.1016/j.toxicon.2006.11.029 (2007).
Castañeda, O. & Harvey, A. L. Discovery and characterization of cnidarian peptide toxins that affect neuronal potassium ion channels. Toxicon 54, 1119–1124. https://doi.org/10.1016/j.toxicon.2009.02.032 (2009).
Cristofori-Armstrong, B. & Rash, L. D. Acid-sensing ion channel (ASIC) structure and function: Insights from spider, snake and sea anemone venoms. Neuropharmacology 127, 173–184. https://doi.org/10.1016/j.neuropharm.2017.04.042 (2017).
Pennington, M. W. et al. Chemical synthesis and characterization of ShK toxin: a potent potassium channel inhibitor from a sea anemone. Int. J. Pept. Protein Res. 46, 354–358. https://doi.org/10.1111/j.1399-3011.1995.tb01068.x (1995).
Shafee, T., Mitchell, M. L. & Norton, R. S. Mapping the chemical and sequence space of the ShKT superfamily. Toxicon 165, 95–102. https://doi.org/10.1016/j.toxicon.2019.04.008 (2019).
Krishnarjuna, B. et al. Synthesis, folding, structure and activity of a predicted peptide from the sea anemone Oulactis sp. with an ShKT fold. Toxicon 150, 50–59. https://doi.org/10.1016/j.toxicon.2018.05.006 (2018).
Rauer, H., Pennington, M., Cahalan, M. & Chandy, K. G. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J. Biol. Chem. 274, 21885–21892. https://doi.org/10.1074/jbc.274.31.21885 (1999).
Yan, L. et al. Stichodactyla helianthus peptide, a pharmacological tool for studying Kv3.2 channels. Mol. Pharmacol. 67, 1513–1521. https://doi.org/10.1124/mol.105.011064 (2005).
Oguiura, N., Correa, P. G., Rosmino, I. L., de Souza, A. O. & Pasqualoto, K. F. M. Antimicrobial activity of snake beta-defensins and derived peptides. Toxins Basel 14, 1. https://doi.org/10.3390/toxins14010001 (2021).
Shafee, T. M., Lay, F. T., Hulett, M. D. & Anderson, M. A. The defensins consist of two independent, convergent protein superfamilies. Mol. Biol. Evol. 33, 2345–2356. https://doi.org/10.1093/molbev/msw106 (2016).
Zhu, S. & Gao, B. Evolutionary origin of beta-defensins. Dev. Comp. Immunol. 39, 79–84. https://doi.org/10.1016/j.dci.2012.02.011 (2013).
Cariello, L. et al. Calitoxin, a neurotoxic peptide from the sea anemone Calliactis parasitica: amino acid sequence and electrophysiological properties. Biochemistry 28, 2484–2489. https://doi.org/10.1021/bi00432a020 (1989).
Ishida, M., Yokoyama, A., Shimakura, K., Nagashima, Y. & Shiomi, K. H. a polypeptide toxin from the sea anemone Halcurias sp., with a structural resemblance to type 1 and 2 toxins. Toxicon 35, 537–544. https://doi.org/10.1016/s0041-0101(96)00143-2 (1997).
Diochot, S. et al. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. Embo. j. 23, 1516–1525. https://doi.org/10.1038/sj.emboj.7600177 (2004).
Diochot, S., Schweitz, H., Béress, L. & Lazdunski, M. Sea anemone peptides with a specific blocking activity against the fast inactivating potassium channel Kv3.4. J. Biol. Chem. 273, 6744–6749. https://doi.org/10.1074/jbc.273.12.6744 (1998).
Kalina, R. S. et al. APETx-like peptides from the sea anemone Heteractis crispa, diverse in their effect on ASIC1a and ASIC3 ion channels. Toxins Basel. https://doi.org/10.3390/toxins12040266 (2020).
Blanchard, M. G., Rash, L. D. & Kellenberger, S. Inhibition of voltage-gated Na(+) currents in sensory neurones by the sea anemone toxin APETx2. Br. J. Pharmacol. 165, 2167–2177. https://doi.org/10.1111/j.1476-5381.2011.01674.x (2012).
Jensen, J. E. et al. Cyclisation increases the stability of the sea anemone peptide APETx2 but decreases its activity at acid-sensing ion channel 3. Mar. Drugs 10, 1511–1527. https://doi.org/10.3390/md10071511 (2012).
Salceda, E. et al. CgNa, a type I toxin from the giant Caribbean sea anemone Condylactis gigantea shows structural similarities to both type I and II toxins, as well as distinctive structural and functional properties(1). Biochem. J. 406, 67–76. https://doi.org/10.1042/bj20070130 (2007).
Shiomi, K., Lin, X. Y., Nagashima, Y. & Ishida, M. Isolation and amino acid sequence of a polypeptide toxin from the sea anemone Radianthus crispus. Fisheries Sci. 62, 629–633 (2011).
Honma, T. et al. Isolation and molecular cloning of novel peptide toxins from the sea anemone Antheopsis maculata. Toxicon 45, 33–41. https://doi.org/10.1016/j.toxicon.2004.09.013 (2005).
Loret, E. P. et al. A low molecular weight protein from the sea anemone Anemonia viridis with an anti-angiogenic activity. Mar. Drugs 16, 134. https://doi.org/10.3390/md16040134 (2018).
Diochot, S., Loret, E., Bruhn, T., Béress, L. & Lazdunski, M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 64, 59–69. https://doi.org/10.1124/mol.64.1.59 (2003).
Restano-Cassulini, R. et al. Species diversity and peptide toxins blocking selectivity of ether-a-go-go-related gene subfamily K+ channels in the central nervous system. Mol. Pharmacol. 69, 1673–1683. https://doi.org/10.1124/mol.105.019729 (2006).
Zhang, M., Liu, X. S., Diochot, S., Lazdunski, M. & Tseng, G. N. APETx1 from sea anemone Anthopleura elegantissima is a gating modifier peptide toxin of the human ether-a-go-go- related potassium channel. Mol. Pharmacol. 72, 259–268. https://doi.org/10.1124/mol.107.035840 (2007).
Peigneur, S. et al. A natural point mutation changes both target selectivity and mechanism of action of sea anemone toxins. Faseb. J. 26, 5141–5151. https://doi.org/10.1096/fj.12-218479 (2012).
Popkova, D. et al. Bioprospecting of sea anemones (Cnidaria, Anthozoa, Actiniaria) for β-defensin-like α-amylase inhibitors. Biomedicines 11, 2682. https://doi.org/10.3390/biomedicines11102682 (2023).
Sintsova, O. et al. Magnificamide, a β-defensin-like peptide from the mucus of the sea anemone Heteractis magnifica, is a strong inhibitor of mammalian α-amylases. Mar. Drugs 17, 542. https://doi.org/10.3390/md17100542 (2019).
Kvetkina, A. et al. A new multigene HCIQ subfamily from the sea anemone Heteractis crispa encodes Kunitz-peptides exhibiting neuroprotective activity against 6-hydroxydopamine. Sci. Rep. 10(1), 4205 (2020).
Mourao, C. B. F. & Schwartz, E. F. Protease inhibitors from marine venomous animals and their counterparts in terrestrial venomous animals. Mar. Drugs 11, 2069–2112. https://doi.org/10.3390/md11062069 (2013).
Mishra, M. Evolutionary aspects of the structural convergence and functional diversification of Kunitz-domain inhibitors. J. Mol. Evol. 88, 537–548. https://doi.org/10.1007/s00239-020-09959-9 (2020).
Hwang, S. M. et al. Venom peptide toxins targeting the outer pore region of transient receptor potential vanilloid 1 in pain: implications for analgesic drug development. Int. J. Mol. Sci. 23, 5772. https://doi.org/10.3390/ijms23105772 (2022).
Sintsova, O. V. et al. Peptide blocker of ion channel TRPV1 exhibits a long analgesic effect in the heat stimulation model. Dokl. Biochem. Biophy. 493, 215–217. https://doi.org/10.1134/S1607672920030096 (2020).
Zelepuga, E. A. et al. Interaction of sea amemone Heteractis crispa Kunitz type polypeptides with pain vanilloid receptor TRPV1: in silico investigation. Bioorg. Khim. 38, 185–198. https://doi.org/10.1134/s106816201202015x (2012).
Schweitz, H. et al. Kalicludines and kaliseptine. Two different classes of sea anemone toxins for voltage sensitive K+ channels. J. Biol. Chem. 270, 25121–25126. https://doi.org/10.1074/jbc.270.42.25121 (1995).
Garcia-Fernandez, R. et al. The Kunitz-type protein ShPI-1 inhibits serine proteases and voltage-gated potassium channels. Toxins Basel 8, 110. https://doi.org/10.3390/toxins8040110 (2016).
García-Fernández, R. et al. Two variants of the major serine protease inhibitor from the sea anemone Stichodactyla helianthus, expressed in Pichia pastoris. Protein. Expr. Purif. 123, 42–50. https://doi.org/10.1016/j.pep.2016.03.003 (2016).
García-Fernández, R. et al. Structure of the recombinant BPTI/Kunitz-type inhibitor rShPI-1A from the marine invertebrate Stichodactyla helianthus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1289–1293. https://doi.org/10.1107/s1744309112039085 (2012).
Garcia-Fernandez, R. et al. Structural insights into serine protease inhibition by a marine invertebrate BPTI Kunitz-type inhibitor. J. Struct. Biol. 180, 271–279. https://doi.org/10.1016/j.jsb.2012.08.009 (2012).
Tokunaga, A. et al. Clinical significance of epidermal growth factor (EGF), EGF receptor, and c-erbB-2 in human gastric cancer. Cancer 75, 1418–1425. https://doi.org/10.1002/1097-0142(19950315)75:6+%3c1418::aid-cncr2820751505%3e3.0.co;2-y (1995).
Maruo, T., Matsuo, H., Otani, T. & Mochizuki, M. Role of epidermal growth factor (EGF) and its receptor in the development of the human placenta. Reprod. Fertil. Dev. 7, 1465–1470. https://doi.org/10.1071/rd9951465 (1995).
Normanno, N., Bianco, C., De Luca, A. & Salomon, D. S. The role of EGF-related peptides in tumor growth. Front. Biosci. 6, D685-707. https://doi.org/10.2741/normano (2001).
De Luca, A. et al. EGF-related peptides are involved in the proliferation and survival of MDA-MB-468 human breast carcinoma cells. Int. J. Cancer 80, 589–594. https://doi.org/10.1002/(sici)1097-0215(19990209)80:4%3c589::aid-ijc17%3e3.0.co;2-d (1999).
Masilamani, A. P. et al. Epidermal growth factor based targeted toxin for the treatment of bladder cancer. Anticancer Res. 41(8), 3741–3746. https://doi.org/10.21873/anticanres.15165 (2021).
Tokay, E. Epidermal growth factor mediates up-regulation of URGCP oncogene in human hepatoma cancer cells. Mol. Biol. Mosk 55, 676–682. https://doi.org/10.31857/S0026898421040133 (2021).
Hermann, P. M., Schein, C. H., Nagle, G. T. & Wildering, W. C. Lymnaea EGF and gigantoxin I, novel invertebrate members of the epidermal growth factor family. Curr. Pharm. Des. 10, 3885–3892. https://doi.org/10.2174/1381612043382503 (2004).
Shiomi, K. et al. An epidermal growth factor-like toxin and two sodium channel toxins from the sea anemone Stichodactyla gigantea. Toxicon 41, 229–236. https://doi.org/10.1016/s0041-0101(02)00281-7 (2003).
Honma, T. & Shiomi, K. Peptide toxins in sea anemones: Structural and functional aspects. Mar. Biotechnol. NY 8, 1–10. https://doi.org/10.1007/s10126-005-5093-2 (2006).
Meiser, C. K. et al. Kazal-type inhibitors in the stomach of Panstrongylus megistus (Triatominae, Reduviidae). Insect. Biochem. Mol. Biol. 40, 345–353. https://doi.org/10.1016/j.ibmb.2010.02.011 (2010).
Lovato, D. V., Nicolau de Campos, I. T., Amino, R. & Tanaka, A. S. The full-length cDNA of anticoagulant protein infestin revealed a novel releasable Kazal domain, a neutrophil elastase inhibitor lacking anticoagulant activity. Biochimie 88, 673–681. https://doi.org/10.1016/j.biochi.2005.11.011 (2006).
Soto, A. et al. Contribution of kazal-like domains of the serine protease inhibitor-1 from toxoplasma gondii in asthma therapeutic vaccination effectiveness. Int. Arch. Allergy Immunol. 183, 471–478. https://doi.org/10.1159/000520796 (2022).
van Hoef, V. et al. Phylogenetic distribution of protease inhibitors of the Kazal-family within the Arthropoda. Peptides 41, 59–65. https://doi.org/10.1016/j.peptides.2012.10.015 (2013).
Hemmi, H. et al. Structural and functional study of an Anemonia elastase inhibitor, a “nonclassical” Kazal-type inhibitor from Anemonia sulcata. Biochemistry 44, 9626–9636. https://doi.org/10.1021/bi0472806 (2005).
Zheng, Q. L., Sheng, Q. & Zhang, Y. Z. Progresses in the structure and function of Kazal-type proteinase inhibitors. Sheng Wu Gong Cheng Xue Bao 22, 695–700 (2006).
Meissner, G. O. et al. Molecular cloning and in silico characterization of knottin peptide, U2-SCRTX-Lit2, from brown spider (Loxosceles intermedia) venom glands. J. Mol. Model. 22, 196. https://doi.org/10.1007/s00894-016-3067-0 (2016).
Matsubara, F. H. et al. A novel ICK peptide from the Loxosceles intermedia (brown spider) venom gland: cloning, heterologous expression and immunological cross-reactivity approaches. Toxicon 71, 147–158. https://doi.org/10.1016/j.toxicon.2013.05.014 (2013).
Orts, D. J. et al. BcsTx3 is a founder of a novel sea anemone toxin family of potassium channel blocker. FEBS J. 280, 4839–4852. https://doi.org/10.1111/febs.12456 (2013).
Logashina, Y. A. et al. Analysis of structural determinants of peptide MS 9a–1 essential for potentiating of TRPA1 channel. Mar. Drugs 20, 465. https://doi.org/10.3390/md20070465 (2022).
Beskhmelnitsyna, E. A., Pokrovskii, M. V., Kulikov, A. L., Peresypkina, A. A. & Varavin, E. I. Study of anti-inflammatory activity of a new non-opioid analgesic on the basis of a selective inhibitor of TRPA1 ion channels. Antiinflamm. Antiallergy Agents Med. Chem. 18, 110–125. https://doi.org/10.2174/1871523018666190208123700 (2019).
Logashina, Y. A. et al. Anti-inflammatory and analgesic effects of TRPV1 polypeptide modulator APHC3 in models of osteo- and Rheumatoid Arthritis. Mar. Drugs https://doi.org/10.3390/md19010039 (2021).
Honma, T. et al. Novel peptide toxins from acrorhagi, aggressive organs of the sea anemone Actinia equina. Toxicon 46, 768–774. https://doi.org/10.1016/j.toxicon.2005.08.003 (2005).
Krishnarjuna, B. et al. A disulfide-stabilised helical hairpin fold in acrorhagin I: An emerging structural motif in peptide toxins. J. Struct. Biol. 213, 107692. https://doi.org/10.1016/j.jsb.2020.107692 (2021).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Hall, B. G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 30, 1229–1235. https://doi.org/10.1093/molbev/mst012 (2013).
Warner, J. F. & Röttinger, E. Transcriptomic analysis in the sea anemone nematostella vectensis. Methods Mol. Biol. 2219, 231–240. https://doi.org/10.1007/978-1-0716-0974-3_14 (2021).
Mitchell, M. L. et al. Identification, synthesis, conformation and activity of an insulin-like peptide from a sea anemone. Biomolecules 11, 1785. https://doi.org/10.3390/biom11121785 (2021).
Mitchell, M. L. et al. Tentacle transcriptomes of the speckled anemone (Actiniaria: Actiniidae: Oulactis sp.): Venom-related components and their domain structure. Mar. Biotechnol. NY 22, 207–219. https://doi.org/10.1007/s10126-020-09945-8 (2020).
Krishnarjuna, B. et al. Structure, folding and stability of a minimal homologue from Anemonia sulcata of the sea anemone potassium channel blocker ShK. Peptides 99, 169–178. https://doi.org/10.1016/j.peptides.2017.10.001 (2018).
Macrander, J., Brugler, M. R. & Daly, M. A RNA-seq approach to identify putative toxins from acrorhagi in aggressive and non-aggressive Anthopleura elegantissima polyps. BMC Genomics 16, 221. https://doi.org/10.1186/s12864-015-1417-4 (2015).
Davey, P. A., Rodrigues, M., Clarke, J. L. & Aldred, N. Transcriptional characterisation of the Exaiptasia pallida pedal disc. BMC Genomics 20, 581. https://doi.org/10.1186/s12864-019-5917-5 (2019).
Grafskaia, E. N. et al. Discovery of novel antimicrobial peptides: A transcriptomic study of the sea anemone Cnidopus japonicus. J. Bioinform. Comput. Biol. 16, 1840006. https://doi.org/10.1142/s0219720018400061 (2018).
Kvetkina, A. et al. Sea anemone Heteractis crispa actinoporin demonstrates in vitro anticancer activities and prevents HT-29 colorectal cancer cell migration. Molecules 25, 5979. https://doi.org/10.3390/molecules25245979 (2020).
Yan, L., Leontovich, A., Fei, K. & Sarras, M. P. Jr. Hydra metalloproteinase 1: A secreted astacin metalloproteinase whose apical axis expression is differentially regulated during head regeneration. Dev. Biol. 219, 115–128. https://doi.org/10.1006/dbio.1999.9568 (2000).
Pan, T., Gröger, H., Schmid, V. & Spring, J. A toxin homology domain in an astacin-like metalloproteinase of the jellyfish Podocoryne carnea with a dual role in digestion and development. Dev. Genes. Evol. 208, 259–266. https://doi.org/10.1007/s004270050180 (1998).
Surm, J. M. et al. A process of convergent amplification and tissue-specific expression dominates the evolution of toxin and toxin-like genes in sea anemones. Mol. Ecol. 28, 2272–2289. https://doi.org/10.1111/mec.15084 (2019).
Stewart, Z. K., Pavasovic, A., Hock, D. H. & Prentis, P. J. Transcriptomic investigation of wound healing and regeneration in the cnidarian Calliactis polypus. Sci. Rep. 7, 41458. https://doi.org/10.1038/srep41458 (2017).
Kim, C. H. et al. Defensin-neurotoxin dyad in a basally branching metazoan sea anemone. FEBS J. 284, 3320–3338. https://doi.org/10.1111/febs.14194 (2017).
Gubic, S. et al. Discovery of K(V)13 ion channel inhibitors: Medicinal chemistry approaches and challenges. Med. Res. Rev. 41, 2423–2473. https://doi.org/10.1002/med.21800 (2021).
Chandy, K. G. et al. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 25, 280–289. https://doi.org/10.1016/j.tips.2004.03.010 (2004).
Feske, S., Skolnik, E. Y. & Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 12, 532–547. https://doi.org/10.1038/nri3233 (2012).
Pardo, L. A. & Stühmer, W. The roles of K(+) channels in cancer. Nat. Rev. Cancer 14, 39–48. https://doi.org/10.1038/nrc3635 (2014).
Chandy, K. G. & Norton, R. S. Immunology: Channelling potassium to fight cancer. Nature 537, 497–499. https://doi.org/10.1038/nature19467 (2016).
Teisseyre, A., Palko-Labuz, A., Sroda-Pomianek, K. & Michalak, K. Voltage-gated potassium channel Kv1.3 as a target in therapy of cancer. Front. Oncol. 9, 933. https://doi.org/10.3389/fonc.2019.00933 (2019).
Prentis, P. J., Pavasovic, A. & Norton, R. S. Sea anemones: Quiet achievers in the field of peptide toxins. Toxins Basel https://doi.org/10.3390/toxins10010036 (2018).
Norton, R. S., Pennington, M. W. & Wulff, H. Potassium channel blockade by the sea anemone toxin ShK for the treatment of multiple sclerosis and other autoimmune diseases. Curr. Med. Chem. 11, 3041–3052. https://doi.org/10.2174/0929867043363947 (2004).
Chi, V. et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 59, 529–546. https://doi.org/10.1016/j.toxicon.2011.07.016 (2012).
Beeton, C., Pennington, M. W. & Norton, R. S. Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Inflamm. Allergy Drug Targets 10, 313–321. https://doi.org/10.2174/187152811797200641 (2011).
Matsumura, K. et al. Mechanism of hERG inhibition by gating-modifier toxin, APETx1, deduced by functional characterization. BMC Mol. Cell. Biol. 22, 3. https://doi.org/10.1186/s12860-020-00337-3 (2021).
Bagal, S. K., Marron, B. E., Owen, R. M., Storer, R. I. & Swain, N. A. Voltage gated sodium channels as drug discovery targets. Channels 9, 360–366. https://doi.org/10.1080/19336950.2015.1079674 (2015).
Cardoso, F. C. & Lewis, R. J. Sodium channels and pain: From toxins to therapies. Brit. J. Pharmacol. 175, 2138–2157. https://doi.org/10.1111/bph.13962 (2018).
Bosmans, F. & Tytgat, J. Voltage-gated sodium channel modulation by scorpion alpha-toxins. Toxicon 49, 142–158. https://doi.org/10.1016/j.toxicon.2006.09.023 (2007).
Gladkikh, I. et al. New Kunitz-type HCRG polypeptides from the sea anemone Heteractis crispa. Mar. Drugs 13, 6038–6063. https://doi.org/10.3390/md13106038 (2015).
Kunitz, M. & Northrop, J. H. Isolation from beef pancreas of crystalline trypsinogen, Trypsin, a trypsin inhibitor, and an inhibitor-trypsin compound. J. Gen. Physiol. 19, 991–1007. https://doi.org/10.1085/jgp.19.6.991 (1936).
Ascenzi, P. et al. The bovine basic pancreatic trypsin inhibitor (Kunitz inhibitor): a milestone protein. Curr. Protein Pept. Sci. 4, 231–251. https://doi.org/10.2174/1389203033487180 (2003).
Cuypers, E., Peigneur, S., Debaveye, S., Shiomi, K. & Tytgat, J. TRPV1 channel as new target for marine toxins: Example of gigantoxin I, a sea anemone toxin acting via modulation of the PLA2 pathway. Acta Chim. Slov. 58, 735–741 (2011).
Hu, B. et al. Purification and characterization of gigantoxin-4, a new actinoporin from the sea anemone Stichodactyla gigantea. Int. J. Biol. Sci. 7, 729–739. https://doi.org/10.7150/ijbs.7.729 (2011).
Liu, Y., Jiang, S., Li, Q. & Kong, Y. Advances of Kunitz-type serine protease inhibitors. Sheng Wu Gong Cheng Xue Bao 37, 3988–4000. https://doi.org/10.13345/j.cjb.200802 (2021).
Honma, T. et al. Novel peptide toxins from the sea anemone Stichodactyla haddoni. Peptides 29, 536–544. https://doi.org/10.1016/j.peptides.2007.12.010 (2008).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res. 51, D587-d592. https://doi.org/10.1093/nar/gkac963 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Yu, G. C., Wang, L. G., Han, Y. Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics. 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Muhammed, M. T. & Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 93, 12–20. https://doi.org/10.1111/cbdd.13388 (2019).
Funding
This work was funded by the Chinese National Natural Science Foundation (no. 82060686), Hainan Province Health Industry Research Project (22A200358), special scientific research project of Hainan academician innovation platform (YSPTZX202132), Hainan Provincial Key Point Research and Invention Program (ZDYF2022SHFZ309), Hainan Provincial Natural Science Foundation (no. 820RC636) and Hainan Medical University graduate innovation and entrepreneurship training program (no. HYYS2021A24).
Author information
Authors and Affiliations
Contributions
Bingmiao Gao and Junqing Zhang were responsible for the conception and design of the project. Qiqi Guo, Jinxing Fu, Yanling Liao, and Lin Yuan performed data analysis. The first draft of the manuscript was written by Qiqi Guo and Bingmiao Gao. Ming Li, Xinzhong Li, and Bo Yi revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, Q., Fu, J., Yuan, L. et al. Diversity analysis of sea anemone peptide toxins in different tissues of Heteractis crispa based on transcriptomics. Sci Rep 14, 7684 (2024). https://doi.org/10.1038/s41598-024-58402-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58402-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
