
Abstract
In these studies, we designed and investigated the potential anticancer activity of five iron(II) cyclopentadienyl complexes bearing different phosphine and phosphite ligands. All complexes were characterized with spectroscopic analysis viz. NMR, FT–IR, ESI–MS, UV–Vis, fluorescence, XRD (for four complexes) and elemental analyses. For biological studies, we used three types of cells—normal peripheral blood mononuclear (PBM) cells, leukemic HL-60 cells and non-small-cell lung cancer A549 cells. We evaluated cell viability and DNA damage after cell incubation with these complexes. We observed that all iron(II) complexes were more cytotoxic for HL-60 cells than for A549 cells. The complex CpFe(CO)(P(OPh)3)(η1-N-maleimidato) 3b was the most cytotoxic with IC50 = 9.09 µM in HL-60 cells, IC50 = 19.16 µM in A549 and IC50 = 5.80 µM in PBM cells. The complex CpFe(CO)(P(Fu)3)(η1-N-maleimidato) 2b was cytotoxic only for both cancer cell lines, with IC50 = 10.03 µM in HL-60 cells and IC50 = 73.54 µM in A549 cells. We also found the genotoxic potential of the complex 2b in both types of cancer cells. However, the complex CpFe(CO)2(η1-N-maleimidato) 1 which we studied previously, was much more genotoxic than complex 2b, especially for A549 cells. The plasmid relaxation assay showed that iron(II) complexes do not induce strand breaks in fully paired ds-DNA. The DNA titration experiment showed no intercalation of complex 2b into DNA. Molecular docking revealed however that complexes CpFe(CO)(PPh3) (η1-N-maleimidato) 2a, 2b, 3b and CpFe(CO)(P(OiPr)3)(η1-N-maleimidato) 3c have the greatest potential to bind to mismatched DNA. Our studies demonstrated that the iron(II) complex 1 and 2b are the most interesting compounds in terms of selective cytotoxic action against cancer cells. However, the cellular mechanism of their anticancer activity requires further research.
Similar content being viewed by others
Introduction
The discovery of a simple coordination compound cis-PtCl2(NH3)2, known as cisplatin and CDDP, with therapeutic potential against a broad spectrum of solid tumors in 1969 was the landmark for the design and development of organometallic compounds as anticancer drug candidates1. Further research led to the development of second- and third generation of platinum FDA-approved drugs, namely, carboplatin and oxaliplatin. Moreover, three other cisplatin derivatives (nedaplatin, lobaplatin and heptaplatin) have been approved regionally in China, Japan and South Korea, respectively. The widespread use of platinum drugs is estimated to reach half of all cancer patients who require chemotherapy2. Despite the prevalence of platinum drugs in chemotherapeutic regimens, their usage is limited due to intrinsic or acquired drug resistance and serious systemic toxicity including hepatotoxicity, nephrotoxicity, neurotoxicity, and ototoxicity3. To overcome the abovementioned drawbacks associated with Pt-based drugs, several strategies have been exploited in the design of small-molecule anticancer metallocompounds. A common approach is the exploration of complexes based on alternative transition metals. In particular, the complexes with metals of group 8, 9 and 10 have been investigated leading to an emergence of a library of non-platinum metal complexes designed as putative anticancer agents.
DNA is the main target of many anticancer drugs. These drugs often induce various damage mechanisms to DNA which inhibit replication and transcription in rapidly dividing cancer cells, leading to their death. For example, cisplatin mainly forms intrastrand DNA adducts, and has a well-documented selectivity for adjacent GG dinucleotide sequences (60–65%) over AG sequences (20–25%). Due to their redox potential and multiple oxidation states, metal complexes (including Pt drugs) can generate reactive oxygen and nitrogen radicals which can also cause DNA damage via base modifications and DNA strand breaks4.
Among the transition metal complexes, ruthenium complexes have attracted considerable attention as non-platinum anticancer drug candidates5. The most prominent representatives of Ru-based complexes include NAMI-A6, KP10197, NKP1339 (IT-139; BOLD-100)8 and TLD14439 which have entered clinical trials. Although NAMI-A has passed phase I clinical trials, the phase II was prematurely terminated due to its limited efficacy6. The KP10197 and NKP13398 successfully completed phase I trials and TLD-1433 is currently under phase II clinical trial (NCT03945162).
Apart from ruthenium complexes, an increasing interest in anticancer properties of iron complexes has recently been observed10,11,12. The development of iron-based drugs is an expected trend since iron naturally occurs in human organism and thus has lower intrinsic toxicity. The discovery of cytotoxic properties of ferrocene and its oxidized form ferrocenium in 1984, was a groundwork for the development of Fe-based putative anticancer agents13,14. The seminal work by Köpf, Köpf-Maier and Neuse laid the foundation for the subsequent research on iron-containing cytotoxic agents which was mainly focused on ferrocene derivatives. One example is ferrocifen which contains ferrocene moiety bound to hydroxytamoxifen—a selective oestrogen receptor modulator used in chemotherapy regiments for breast cancer15. The main obstacle hindering entrance of ferrocene derivatives into clinical trial is their poor bioavailability13. The half-sandwich cyclopentadienyl-iron complexes overcome this drawback since the structure allows for the attachment of three ligands influencing the final properties of a compound.
The encouraging results obtained by our group16,17 and others 18,19,20,21,22,23 with Ru(η5-cyclopentadienyl) piano stool complexes led us to the design of five Fe(η5-cyclopentadienyl)-compounds as anticancer drug candidates. The complexes have been intentionally functionalized with ligands enhancing anticancer properties including maleimidato, carbonyl, phosphine and phosphite moieties (Fig. 1). All these complexes (2a, 2b, 3a, 3b and 3c) were derivatized from complex 1 in visible light induced CO ligand exchange process. Triphenylphosphine, tri(2-furyl)phosphine, triethyl phosphite, triphenyl phosphite and triisopropyl phosphite have been chosen for the study. The NMR (1H, 31P, 13C), FT-IR, ESI–MS, UV–Vis, fluorescence, XRD and elemental studies were executed for the characterization and structural analysis of these complexes.

The structures of the half-sandwich complexes 1, 2a,b and 3a–c.
The cytotoxic potential of these complexes was analyzed in peripheral blood mononuclear (PBM) cells as normal cells, and two human cancer cell lines—leukemic HL-60 cells and non-small cell lung cancer A549 cells. To study anticancer potential of iron(II) complexes, we selected those complexes that were the most cytotoxic for cancer cells and were less cytotoxic for normal PBM cells, comparing IC50 doses. We evaluated the ability of selected iron(II) complexes to induce DNA damage. We also used the plasmid relaxation assay, DFT studies and docking studies to determine the potential of iron(II) complexes to directly damage DNA.
Results and discussion
Synthesis of complexes 1, 2a,b and 3a–c
The CpFe(CO)2(η1-N-maleimidato) 1 was obtained following previously described photochemical reaction of CpFe(CO)2I with maleimide in presence of diisopropylamine24.
To synthesize complexes 2a,b and 3a–c, we modified photochemical ligand exchange reaction of CO by phosphine/phosphite of complex 1 (Scheme 1). A similar synthetic procedure for 2a has been previously reported by our group where 1 was irradiated with triphenylphosphine by visible light to produce CpFe(CO)(PPh3)(η1-N-maleimidato) in benzene25. Here, we replace benzene with toluene to obtain 2a,b and 3a–c.

Synthesis of complexes 2a,b and 3a–c.
Complex 1 was irradiated with four 150W tungsten lamps in the presence of preferred phosphines (triphenylphosphine, tri(2-furyl)phosphine) or phosphites (triethyl phosphite, triphenyl phosphite and triisopropyl phosphite) under argon to obtain complexes 2a,b and 3a–c respectively (Scheme 1).
The crude products were purified by column chromatography and were subsequently characterised by different spectroscopic methods (Figs. S3–S22). Single crystals of 2a, 2b, 3b and 3c were analysed using X-ray diffraction method.
In our previous studies it was evident from the 31P NMR analysis that the phosphorus atoms of phosphine and phosphite ligands are being deshielded upon reaction with (η5-cyclopentadienyl) ruthenium(II) dicarbonyl maleimidato complex16. For this reason, peaks for the 31P were found to be shifted downfield in complexes compared to the corresponding ligands. Similar phenomenon has also been observed in iron(II) complexes 2a,b and 3a–c where the degree of downfield shift of 31P signal is higher than that of the Ru(II) complex with same ligands as shown in Table 1.
The 1H and 13C NMR spectra of complexes 2a,b and 3a–c corroborate to the proposed formulas. The Cp protons of 2a,b and 3a–c were shifted upfield compared to the dicarbonyl complex 1. The most shifted was the Cp signal of complex 3b bearing the triphenyl phosphite ligand (4.61 ppm for 3b and 5.05 ppm for 1). The presence of phosphine or phosphite ligand at the complex has also affected the position of the olefinic protons of maleimide ligand, which was shifted upfield in complexes 2a,b and 3a–c. The olefinic protons of 2a are shifted upfield by 0.45 ppm as compared to those of 1, which is highest for the corresponding signal among all other complexes.
The 13C NMR spectra show the expected signals in the appropriate regions. The olefinic carbons (C=C) were noted around 137 ppm and the imide carbon (C=O) around 185 ppm. The CC≡O resonating at 221–218 ppm like in similar half-sandwich carbonyl compounds we had studied previously. This signal has been shifted downfield by 6–9 ppm compared to that of 1 in all the complexes, probably due to the deshielding effect by phosphorus atom. The CC≡O signal is splitted (d) as a result of the coupling with the phosphorus atom.
No significant change in the signals from the phosphines and phosphites have been observed in the complexes corresponding to the ligands in both 1H and 13C NMR spectra. Hence, it can be said that the coordination with Fe(II) atom does not affect much the structure of the ligand part in the complexes 2a,b and 3a–c.
The absorbance and emission spectra of all complexes were recorded in chloroform. For example, compound 2a and 2b shows emission peaks at 345 nm and 347 nm, with the excitation at 300 nm and 280 nm respectively. It is observed that the characteristic emission peaks of most of the phosphine/phosphite ligands have been suppressed after formation of the complex with iron (Figs. S24 and S25).
Crystal structure description
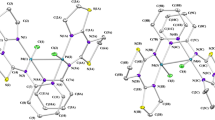
The results of crystal structure determination and molecular structures with corresponding atom labelling schemes are presented in Table 2 and Fig. 2. As seen, in all the cases iron Fe1 atom is bonded to cyclopentadienyl ring (C11–C12–C13–C14–C15), carbonyl ligand (C10–O10), nitrogen atom N1 of maleimidato ligand and phosphorus P1 of phosphine or phosphite ligand.

Molecular structures of the investigated compounds with atom labelling scheme: 2a, 2b, 3b and 3c. Displacement ellipsoid are drawn with 40% probability level, hydrogen atoms and minor disorder component of 3b are omitted for clarity.
All the investigated compounds can be classified as half-sandwich complexes with cyclopentadienyl moiety arranged on the opposite side of central Fe1 atom in respect to the three other ligands. Among the coordination bonds the longest are Fe1-P1 (from 2.144(4)Å to 2.221(2)Å) and a little shorter, of length about 1.77 Å, Fe1-N1 to maleimidato ligands (compare Table S1). Bond distances between iron and carbon atoms of carbonyl ligand (Fe1-C10) are changing from 1.754(2)Å to 1.773(5)Å. In turn, the shortest bonds are observed to cyclopentadienyl moiety with Fe1-Cg1 distance of about 1.72 Å, excluding data for the disordered structure 3b (Cg1 corresponds to the centre of gravity of cyclopentadienyl ring).
The valence angles around Fe1 and between N1 (maleimidato), P1 (phosphine/phosphite) and C10 (carbonyl) atoms are very close to 90°. It may be stated, that Fe1-N1, Fe1-P1 and Fe1-O10 bonds are perpendicular and cross each other in the position of the central iron atom. In turn, the valence angles to the cyclopentadienyl ring (Cg1) are all above 120°. All these values of geometric parameters are in agreement with the known for half-sandwich compounds “piano-stool” conformation17.
Assuming that cyclopentadienyl is treated as a single ligand, we may state that coordination number for iron is equal to four and call the coordination sphere in molecules of all compounds a strongly distorted tetrahedron. For such tetrahedral four-coordinated Fe1 atom, similarly like for asymmetric sp3 carbon atom, there can be indicated as a formal configuration. Using the Cahn-Ingold-Prelog26 rules, the coordinative ligands can be listed in the following order: I—phosphine/phosphite, II—maleimidato, III—carbonyl, IV—cyclopentadienyl, and hence two opposite conformations (corresponding to R and S stereoisomers) can be indicated.
Compounds 2a, 3b and 3c crystallize in centrosymmetric space groups of triclinic or monoclinic system. In such cases, because of the presence of inversion centre, there exist two opposite conformers in the crystalline state. Interestingly, compound 2b crystallizes in the non-centrosymmetric orthorhombic Pna21 space group, nevertheless, there are also observed R and S stereoisomers due to the presence of glide mirror planes (nx and ay) among space group symmetry elements. This is in contrast to analogical ruthenium complex which due to spontaneous resolution during crystallization shows P212121 space group symmetry and the reported crystal forms only one R stereoisomer16.
Figure 3 presents overlay and comparison of molecular conformations including both currently investigated iron complexes and previously published results of their ruthenium analogues.

Comparison of molecular conformations observed in crystal structures: (a) 2a (purple), 2b (blue), 3b (lilac) and 3c (cyan); analogic complexes of ruthenium (cyan) and iron (purple)—2a (b) and 2b (c).
For comparison of the overall molecular geometry the same isomers have been considered in overlapping procedure (Table S1—compare torsion angles). As seen, there are no significant differences between iron–ruthenium pairs of compounds (see Fig. 3b,c). Sligh conformational changes result from rotation about P1-C21/C31/C41 bonds of aromatic rings. In turn, larger differences are seen in a group of iron complexes, in particular with phosphite ligands. This is due to the higher flexibility of sp3 oxygen O20/O30/O40 atoms (compare Figs. 2 and 3a).
Even though, molecular conformations in the investigated structures are in general rather similar, their final crystal packings vary significantly because of different crystal systems and space groups. This is mainly the result of various size, shape and electron donor–acceptor properties of phosphoroorganic ligands. However, the one thing is common to all structures—they belong to the class of molecular crystals of structure stabilized by non-covalent interactions. In the observed relatively weak C–H…O hydrogen bonds oxygen atoms from carbonyl groups are taking part as proton acceptors. Some molecular complexes linked by these bonds are shown in the Fig. S23 and the corresponding interaction geometric parameters are gathered in the Table S2.
DFT studies
In order to gain insight into the reactivity of the investigated compound, a frontier orbital analysis was conducted using DFT calculations. For this purpose, a full optimization was performed on isolated molecules, utilizing the wB97XD27 functional in conjunction with the def2TZVP basis set28, implemented in the Gaussian16 suite29. The molecular structure obtained from X-ray data served as the starting point for geometry optimization. The HOMO/LUMO energies were calculated for the optimized geometries and are presented in Table 3, optimized geometries of studied compounds are presented in Fig. 4.

Optimized geometries of model system chosen for computational analysis; 2a (a), 2b, (b), 3b, (c), and 3c, (d).
Based on the DFT calculations, the HOMO/LUMO gap for all four derivatives is approximately 0.3 hartrees. The smallest gap was observed for the 3c molecule with similar value for 2a, differing by only about 1 kcal/mol. In the case of the other two compounds, the HOMO/LUMO gap is relatively higher, with the highest value found for 3b being above 7 kcal/mol. Therefore, based on the thermodynamic properties of the isolated molecular models, 3c and 2a are expected to be the most active, while the other two compounds appear to have lower activity.
Results of the cytotoxicity study
We examined the viability of cells after 2 h and 24 h incubation with iron(II) complexes using the resazurin reduction assay (Tables S3 and S4, respectively). We observed no change in the viability of PBM cells during short, 2 h, incubation with complexes 1, 3a and 3b. At high concentrations, the complex 2b presented cytotoxic properties while the complex 3c increased PBM cells viability which could be explained by the increased activity of the cells after 2 h treatment with the compound. The complex 2a slightly decreased PBM cell viability at low concentrations (Table S3). In the case of 24 h incubation, we observed a decrease in the viability of PBM cells with the increasing concentrations of all complexes with the exception of complex 3a (Table S4).
We observed a decrease in the viability of HL-60 cells with increasing concentrations of complexes 1, 2b and 3a after 2 h incubation. The complex 2a caused a slight increase in the viability of these cells. We observed a sharp increase (up to approximately 140%) in the HL-60 cell viability for complexes 3b and 3c (Table S3). However, the highest concentration (250 μM) of these compounds led to a sharp decrease in HL-60 cell viability. The viability of HL-60 cells was decreased for all complexes with increasing concentrations after 24 h incubation. Among the studied compounds, the complex 3b was the most cytotoxic (Table S4).
In the case of A549 cells was observed a steady decrease in cell viability with increasing concentration of complexes 1, 2a and 3a after a short, 2 h incubation (Table S3). We also observed an increased viability (123%) of HL-60 cells after short treatment with complex 2b at the concentration of 50 μM which later dropped sharply to 40% at 250 μM. Incubation with 3c resulted in steady rise in the HL-60 cell viability to the point of 134% at 250 μM, and in the case of 3b no change in the viability was observed. Regarding 24 h incubation, an increase in the viability of A549 cells was observed with increasing concentrations of complexes 2a and 3a to 120% and 153%, respectively (Table S4). The complexes 1, 2b and 3b sharply decreased A549 cell viability at higher concentrations, while complex 3c decreased viability to approximately 69% at concentration of 5 μM and increased viability at higher concentrations.
Then, we determined IC50 doses for all iron(II) complexes against the three tested cell types after a 24 h incubation (Table 4). HL-60 cells showed a decrease in the viability with increasing concentrations for all complexes. Complexes 1, 2b, 3a and 3c were cytotoxic for HL-60 cells while maintaining low cytotoxicity for normal PBM cells. Among studied compounds the most promising against HL-60 cells were complexes 2b and 3a (IC50 = 10.03 μM and 15.93 μM, respectively) without detected cytotoxicity against PBM cells (IC50 > 250 μM). Our studies also indicated that the complex 2b bearing maleimide and phosphine ligands was selectively cytotoxic for both cancer cell lines, HL-60 cells (IC50 = 10.03 µM) and A549 cells (IC50 = 73.54 µM) and not for the normal PBM cells (IC50 > 250). The selective targeting of cancer cells by complex 2b is promising and fits into the development of new generation therapeutic agents oriented toward protection of normal cells.
Interestingly, the iron(II) complex 2b was more active to HL-60 cells than the analogous ruthenium compound 2b CpRu(CO)(PPh3) (η1-N-maleimidato) that we studied previously15 (IC50 = 10.03 µM for Fe and IC50 = 35.64 µM for Ru). It should be noted that the iron complex 2b is not active to PBM cells (IC50 > 250 µM), but the ruthenium complex 2b was cytotoxic to PBM cells (IC50 = 8.48 µM). The iron(II) complex 3a showed a very similar IC50 for HL-60 cells (IC50 = 15.93 µM) to the analogous ruthenium(II) complex 3a (CpRu(CO)(P(OEt)3)(η1-N-maleimidato)) (IC50 = 12.25 µM). Both complexes were also non-cytotoxic to normal PBM cells (IC50 > 250 µM).
Based on the cytotoxicity results, iron(II) complexes 1, 2b, 3a and 3c were selected for genotoxic studies involving HL-60 cells. In the case of A549 cells, among iron(II) complexes with limited cytotoxicity against PBM cells (IC50 > 250 μM) complex 2b was the most cytotoxic (IC50 = 73.54 μM). The complexes 2a, 3a and 3c did not show cytotoxicity against A549 cells. Although, the complex 1 demonstrated the highest cytotoxic properties among studied compounds against A549 cells (IC50 = 54.2 μM), it was also cytotoxic for PBM cells at the concentration twice as high (IC50 = 105.63 μM). Thus, complexes 1 and 2b were selected for further studies on A549 cell line.
Results of the genotoxicity study
We performed genotoxic assays to investigate the mechanism of cytotoxic properties of iron(II) complexes. The ability to induce DNA damage was examined by both the comet assay and the plasmid relaxation assay. The comet assay allows for the analysis of DNA damage in single cell whereas the plasmid relaxation assay discriminate the genotoxic potential at a molecular level.
We performed alkaline version of the comet assay, which allows for the detection of DNA double and single strand breaks and alkali labile sites, after 2 h incubation of cancer cells with selected iron(II) complexes. We deliberately chosen short incubation period to eliminate the possibility of DNA damage induction as secondary effect e.g., as a result of apoptosis. Additionally, we performed the cell viability assay after 2 h incubation with cells to further exclude this effect.
We observed that the complex 1 and 2b induced DNA damage in HL-60 cells in the concentration range 5–50 μM (p < 0.001) (Fig. 5a). The complex 1 was more genotoxic than complex 2b, inducing 25% DNA in tail vs. 6% DNA in tail at concentration 50 μM. In the case of complex 3a, we observed DNA damage at the concentrations of 10, 25 and 50 μM (p < 0.001), however, the percentage of DNA in tail has not increased in a concentration-dependent manner. The DNA damage for complex 3c was not detected in these conditions.

DNA damage in (a) HL-60 cells and (b) A549 cells incubated for 2 h at 37 °C with selected iron(II) complexes analysed by the alkaline comet assay. The figure shows mean results ± SEM, n = 100; ***p < 0.001.
We did not observe any increase in the level of DNA damage in A549 cells in the range of tested concentrations for complex 2b with the exception at the concentration of 50 μM (p < 0.001) (Fig. 5b). The percentage of DNA damage in tail was equal to about 2%.
Previously, we studied iron(II) complex 1 (η5-C5H5)Fe(CO)2(η1-N-maleimidato) in HL-60 cells. We have shown that this complex damages DNA and causes a significant increase in the expression of HO-1 gene, in contrast to the complex of iron(II) with succinimide ligand (η5-C5H5)Fe(CO)2(η1-N-succinimidato). Furthermore, DNA damage induced by complex 1 was not effectively repaired in HL-60 cells30.
In recent years, many iron complexes have been synthesized and their biological activity was described. For example, a series of diiron cyclopentadienyl complexes containing bridging vinyliminium ligands against cisplatin sensitive and resistant human ovarian carcinoma (A2780 and A2780cisR) cell lines were investigated31. Notable selectivity towards these cancerous cell lines was observed as compared to the non-cancerous 293 T cell line. The anticancer activity of these complexes was associated with the induction of reactive oxygen species (ROS). Moreover, cyclopentadienyl iron complexes with the general formula [CpFe(CO)(PPh3)(NCR)]+ (NCR = benzonitriles) were tested against breast MDA-MB-231 and colorectal SW480 cancer cells with IC50 at low micromolar range. These compounds caused apoptosis, inhibited colony formation and affected cell cytoskeleton organization32. Another study evaluated the antiproliferative activity of iron(II) cyclopentadienyl complexes bearing n-heterocyclic carbene ligands in human colorectal (HCT116) and ovarian (A2780) carcinoma cells and in vivo. The complex ([Cp(IMes)Fe(CO)2]I) (IMes = 1,3-bis(2,4,6-trimethyl-phenyl)imidazol-2-ylidene) displayed higher than cisplatin cytotoxic activity both in HCT116 and A2789 cells with IC50 values in the low micromolar range. Interestingly, this complex decreased the proliferation of colorectal HCT116 cancer cell in vivo while demonstrated low in vivo toxicity as analyzed in zebrafish (Danio rerio) xenograft32.
The latest studies indicate that iron complexes can inhibit the activity of the multidrug resistance protein ABCB133. The research was carried out using doxorubicin-sensitive cells (Colo205) and doxorubicin-resistant (Colo320) human colon adenocarcinoma cell lines. Compound [CpFe(CO)(PPh3) (1-benzylimidazole)](CF3SO3) was the most active in both cell lines with IC50 values of 1.26 ± 0.11 and 2.21 ± 0.26 μM, respectively, being also slightly selective towards cancer cells vs. MRC5 human embryonic fibroblast cell lines. This compound, together with [CpFe(CO)(PPh3)(1H-1,3-benzodiazole)](CF3SO3), was found to display very potent ABCB1 inhibitor activity. [CpFe(CO)(PPh3)(1-benzylimidazole)](CF3SO3) also showed the ability to induce apoptosis. Iron cellular accumulation studies by ICP-MS and ICP-OES methods revealed that cytotoxicity of these complexes was not related to the extent of iron accumulation. On the other hand, [CpFe(CO)(PPh3)(1-benzylimidazole)][CF3SO3] was the only one where iron accumulation was greater in the resistant cell line than in the sensitive one, validating the possible role of ABCB1 inhibition33.
We also investigated the induction of DNA damage by the iron(II) complexes using the plasmid relaxation assay. We isolated the pUC19 plasmid from the DH5α E. coli cells in its supercoiled form (CCC) (Fig. 6, line 2). We treated the pUC19 plasmid with restrictase PstI overnight at 37 °C, obtaining linear (L) form of the plasmid (Fig. 6, line 3). Next, we incubated native plasmid (CCC) with iron(II) complexes at concentrations 5 μM and 50 μM. We observed no degradation of the plasmid after 2 h and 24 h incubation (Fig. 6a,b, respectively). Neither open circular form of pUC19 plasmid (OC) nor the L form of the plasmid was visible. These results suggest that the iron(II) complexes do not cause DNA damage at a molecular level.

Plasmid relaxation assay. Plasmid pUC19 was incubated for 2 h (a) and 24 h (b) in 37 °C with iron(II) complexes at concentrations 5 μM and 50 μM, and then was resolved on a 1% agarose gel, stained with ethidium bromide and visualized in UV light. Lines 1 and 16—DNA ladder; line 2—pUC19 plasmid in the supercoiled (CCC) form; line 3—pUC19 plasmid incubated with restrictase Pstl in the linear (L) form; line 4 and 5—pUC19 plasmid incubated with complex 1 at 5 μM and 50 μM, respectively; line 6 and 7—pUC19 plasmid incubated with complex 2a at 5 μM and 50 μM, respectively; line 8 and 9—pUC19 plasmid incubated with complex 2b at 5 μM and 50 μM, respectively; line 10 and 11—pUC19 plasmid incubated with complex 3a at 5 μM and 50 μM, respectively; line 12 and 13—pUC19 plasmid incubated with complex 3b at 5 μM and 50 μM, respectively; line 14 and 15—pUC19 plasmid incubated with complex 3c at 5 μM and 50 μM, respectively.
Similarly, new dinuclear iron(II) complexes containing iminopyridine ligands based on a methanodibenzodioxocine (DBDOC) backbone did not induce DNA breaks in the plasmid relaxation assay34. However, in the presence of H2O2 these complexes promoted DNA double strand breaks. The Cu2+/Zn2+/Fe2+/Fe3+ complexes containing different ligand scaffolds such as phenanthroline, bipyridine, terpyridine, cyclen, hydrazones, triazole, imidazole and protected phenol have been reported to concurrently bind and cleave DNA without any external redox agent(s). In most cases, ROS such as superoxide ions (O2⋅−), hydroxyl radicals (⋅OH), singlet oxygen (1O2)/singlet oxygen like species or H2O2 played active roles in DNA cleavage. In some cases, transient metal bound species were also created and were responsible for DNA cleavage. Metal complexes with nucleolytic activity that do not require the participation of additional activating factors are of great interest to researchers because such complexes could be used in anticancer therapy35. Unlike the iron(II) complexes studied here, the analogous ruthenium(II) complexes, studied previously, induced DNA breaks in vitro16.
Cancer cells accumulate and use more iron than normal cells, due to the higher proliferation and DNA synthesis. Furthermore, iron plays a crucial role in the regulation of cell cycle by affecting both the formation and the activity of cyclin proteins (cyclin A, B, D, and E) and cyclin-dependent kinase (CDKs) complexes. Cancer cells overexpress genes involved in the iron metabolism and the iron-sulfur (Fe-S) cluster biogenesis. The initial Fe–S cluster synthesis occurs within the mitochondria; however, the maturation of Fe–S clusters culminating in their ultimate insertion into appropriate cytosolic/nuclear proteins is coordinated by a late-acting cytosolic iron-sulfur assembly (CIA) complex in the cytosol. Several nuclear proteins involved in DNA replication and repair interact with the CIA complex and contain Fe–S clusters necessary for proper enzymatic activity. Moreover, it is currently hypothesized that the late-acting CIA complex regulates the maintenance of genome integrity and is an integral feature of DNA metabolism36. Iron metabolism disorders including Fe–S clusters leading to iron accumulation in the cell and lipid peroxidation can cause cell death by ferroptosis37.
Results of the Docking studies
The binding energy analysis suggests the highest DNA binding potential for 2a, 2b and 3b (Table 5 and Fig. S26). This may be linked with the fact that all these compounds are aromatic derivatives. Complex 3b is toxic to all cell lines, including normal PBM cells (Table 4). Complex 2a is toxic towards cancerous HL-60 cells, but also towards normal cell line and both compounds (2a and 3b) are phenyl derivatives. Interestingly, 2b analog is an aromatic derivative, but with furan not phenyl substituents; this seems to be most promising compound expressing toxicity towards cancerous cells (HL-60 and A549), however without detectable cytotoxicity towards normal human blood derived PBM cells (Table 4). What is interesting all tested compounds (2a,b and 3b,c) have relatively low binding energy with fully paired DNA fragment (Table 5). However, tested iron(II) compounds bind 3 times stronger with mismatched DNA, where all tested complexes appear to be located at the level of T-T mismatch (Fig. S27a,b). This suggests that when DNA is damaged it can recruit Ru(II)16, or for that matter also Fe(II) complexes, which might cause additional DNA degradation and/or alter the DNA repair mechanisms inside cells. These results corroborate with cellular experiments and might explain higher cytotoxicity; however, additional tests are necessary to prove this mechanism.
Results of the DNA titration study
We performed UV–Vis spectroscopic analysis to assess whether these iron(II) complexes can impose structural changes in DNA structure. For this purpose, we analyzed UV–Vis absorption spectra (240–300 nm) for DNA, complex 2b, and DNA incubated with complex 2b at 0.5, 5, and 50 µM (Fig. 7). We deliberately chose complex 2b for this study as the most potent agent with anticancer properties.

UV–Vis absorption spectra (240–300 nm) of DNA and complex 2b at 50 µM (A), 5 µM (B), and 0.5 µM (C).
In this study, we analyzed UV–Vis absorption spectra (240–300 nm) of DNA, complex 2b alone and after incubation with DNA. First of all, a significant absorbance for complex 2b at 260 nm was observed restricting the analysis (Fig. 7A). We observed an increase in absorbance at 260 nm when DNA was incubated with complex 2b (Fig. 7), however, we attribute this change to the additive effect of absorbance for complex 2b and DNA, measured separately. Since we have not observed hyperchromic, hypochromic, hypsochromic or bathochromic effect we exclude the possibility of inducing DNA structural changes by complex formation with 2b. The spectroscopic titration is a sensitive method for assessing changes in DNA structure especially by intercalators, which insert a planar aromatic ring between the stacked base pairs of double-stranded DNA38. Thus, we can conclude that complex 2b does not interact in an intercalative manner with DNA.
Conclusions
We designed and investigated the potential anticancer activity of five iron(II) cyclopentadienyl complexes bearing different phosphine and phosphite ligands. All complexes have been characterized by 1H NMR, 13C NMR, 31P NMR, FT–IR, ESI–MS, UV–Vis, elemental analyses and single crystal X-ray diffraction analysis (for 2a, 2b and 3b, 3c). Our biological studies indicated that the complex 2b bearing tri(2-furyl)phosphine was cytotoxic for both cancer cell lines, HL-60 and A549 cells and not for the normal PBM cells. Having considered the results of the present work with our previous findings regarding Ru complexes16,39 it can be postulated that both ruthenium and iron-based (η5-cyclopentadienyl) piano stool complexes bearing maleimide and carbonyl ligands exert anticancer activity against HL-60 cells. It is worth noting that complex 3a and its ruthenium analogue 3a-Ru were potent against HL-60 cells without cytotoxicity toward PBM cells, indicating that the triethyl phosphite ligand can effect selectivity toward HL-60 cancer cells16.
The replacement of the central atom in both complexes containing phosphines, 2a and 2b, modulated the cytotoxicity toward PBM cells and HL-60 cells. On one hand, iron-bearing complex 2b displayed no cytotoxicity toward PBM cells compared to its ruthenium counterpart 2b-Ru which is cytotoxic. On the other hand, the complex 2a showed cytotoxicity toward PBM cells compared to its non-toxic ruthenium analogue 2a-Ru16. This observation led us to the conclusion that the substitution of central atom in metal containing complexes can influence their selectivity which is crucial for bioinorganic chemical activity.
Interestingly, comparing the activity of complex 1 to complex 3c bearing triisopropyl phosphite we did not observe differences in the cytotoxicity toward HL-60 cells and PBM cells, but in case of A549 cells the 3c was no cytotoxic when 1 showed significant cytotoxicity.
The DFT studies showed that the complexes 2a and 3c should be the most active; however, we demonstrated their high cytotoxic activity mainly towards HL-60 leukemic cells. These complexes were not cytotoxic to A549 cells (IC50 > 250 µM). We also found that the complex 2b induced DNA damage in both types of cancer cells. It is noteworthy that all the studied compounds were less toxic than complex 1 that is lacking phosphine and phosphite ligand. Molecular docking revealed that complexes 2a, 2b and 3b, 3c have potential to bind to mismatched DNA. However, the plasmid relaxation assay showed that these iron(II) complexes did not induce the DNA breaks. Furthermore, the DNA titration experiment showed no intercalation of complex 2b into DNA. Our studies demonstrated that the iron(II) complex 1 and 2b are the most interesting compounds in terms of selective cytotoxic action against cancer cells. Therefore, we suggest that DNA damage probably occurs as a result of impaired cellular metabolic processes under the influence of iron(II) complexes rather than direct interaction with DNA.
Materials and methods
Chemicals
Bis(cyclopentadienylirondicarbonyl) dimer, maleimide, diisopropylamine, and all the phosphines and phosphites were purchased from Sigma-Aldrich (Merck). Solvents were purchased from POCH (Polish Chemical Reagents) and used without further purification. All syntheses were carried out under argon. Chromatographic purification of the crude products were performed on silica gel 60 (230–400 mesh) purchased from Merck. FTIR spectra were recorded in KBr on a Fourier Transform InfraRed (FTIR) NEXUS (Thermo Nicolet) spectrometer. NMR spectra were recorded on Bruker Avance III BBFO (600 MHz) and Bruker AvanceNeo Cryoprobe Prodigy spectrometer (600 MHz). NMR data were collected in CDCl3 (Merck) solution. The chemical shifts were calculated in part per million (ppm) unit. Coupling constants were calculated in Hertz (Hz). Electrospray ionization mass spectrometry (ESI–MS) spectra were recorded on the Varian 500-MS LC ion trap spectrometer. Elemental analyses were obtained with a Vario EL III (Elementar Analysensysteme GmbH) instrument. Photochemical syntheses were carried out using UV lampTQ 150 Z3. PerkinElmer Lambda 45 UV/Vis spectrometer and PerkinElmer LS55 Fluorimeter have been used to measure the absorbance and emission of all compounds, respectively. Relevant guidelines and regulations were followed in each consecutive step. Dulbecco's Modified Eagle Medium (DMEM), IMDM medium and fetal bovine serum (FBS) were obtained from Biowest (Cytogen, Zgierz, Poland). Dimethyl sulfoxide (DMSO), hydrogen peroxide (H2O2), low-melting-point (LMP), normal-melting-point (NMP) agarose, phosphate buffered saline (PBS), and 4ʹ,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (USA). The pUC19 plasmid isolation kit (Plazmid Mini AX Kit) was obtained from A&A Biotechnology and restrictase PstI from New English Biolabs. All other reagents were obtained at the highest commercially available grades. A stock solutions of iron complexes (10 mM) was dissolved in DMSO.
Synthetic procedures
Complex 1 was synthesized according to previously published method, by photochemical reaction of CpFe(CO)2I with maleimide in the presence of diisopropyl amine in toluene24.
General procedure for synthesis of 2a,b and 3a–c
A stirred, water–ice cooled, and argon-saturated solution of 1 (70 mg, 0.25 mmol) and phosphine or phosphite (0.7 equiv.) was illuminated under visible light (4 × 150W lamps) for 2 h in toluene (10 mL). The progress of the reaction was continuously monitored with TLC. After completion of the reaction, solvent was evaporated in vacuum.
Synthesis of 2a The crude product was purified by column chromatography using CHCl3-EtOAc (3:1) as eluent to afford a brown–red solid (2a). The product was recrystallized from chloroform/heptane. Yield 32 mg (55%). 1H NMR (δ, ppm, 600 MHz, CDCl3): 7.38 (dd, J = 7.2 Hz, 6.6 Hz, 3H, p-Ph), 7.32 (m, 12H, o,m-Ph), 6.17 (s, 2H, olefinic), 4.60 (s, 5H, Cp). 13C NMR (δ, ppm, 151 MHz, CDCl3): 221.79 (d, J = 30.6 Hz, C≡O); 185.23 (s, imide C=O); 137.41 (s, olefinic C=C); 133.98 (d, J = 42.3 Hz, phenyl C–P); 133.69 (d, J = 10 Hz, o-C of Ph); 130.07 (d, J = 1.8 Hz, p–C of Ph); 128.35 (d, J = 9.7 Hz, m-C of Ph); 82.65 (s, Cp). 31P NMR (δ, ppm, 242.99 MHz, CDCl3): 75.27. FTIR (cm−1): 1951 (C≡O); 1639 (C=O, imide); 1327, 695 (P-Ph). ESI–MS: m/z calcd for C28H22FeNO3P (M + H)+, 508.07; found, 508.13. Anal. calcd for C28H22FeNO3P (507.0687): C 66.29; H 4.37; N 2.76; found C 66.17; H 4.35; N 2.57.
Synthesis of 2b The crude product was purified by column chromatography using CHCl3-EtOAc (3:2) as eluent to afford a red solid (2b). The product was recrystallized from chloroform/heptane. Yield: 56 mg (68%). 1H NMR (δ, ppm, 600 MHz, CDCl3): 7.64 (s, 3H, furyl H), 6.71 (d, J = 3.6 Hz, 3H, furyl H), 6.44 (ddd, J = 1.2 Hz, 3 Hz, 3H, furyl H), 6.41 (s, 2H, olefinic), 4.80 (d, J = 1.2 Hz, 5H, Cp). 13C NMR (δ, ppm, 151 MHz, CDCl3): 219.25 (d, J = 31.6 Hz, C≡O); 185.13 (s, imide C=O); 147.89 (d, J = 5.2 Hz, furyl C–O); 146.86 (d, J = 70.8 Hz, furyl C–P); 137.67 (s, olefinic C=C); 121.44 (d, J = 15.7 Hz, furyl C=C); 111.35 (d, J = 6.7 Hz, furyl C=C); 82.48 (s, Cp). 31P NMR (δ, ppm, 242.99 MHz, CDCl3): 29.25. FTIR (cm−1): 1966 (C≡O); 1645 (C=O, imide); 1019, 758 (P-Fu). ESI–MS: m/z calcd. for C22H16FeNO6P (M + H)+, 478.0098; found, 478.17. Anal. calcd for C22H16FeNO6P (477.0065): C 55.37; H 3.38; N 2.94; found C 55.33; H 3.15; N 2.75.
Synthesis of 3a The crude product was purified by column chromatography using EtOAc–Petroleum Ether (1:1) as eluent to afford a brown–red liquid product (3a). The product was recrystallized from chloroform/heptane. Yield 38 mg (79%). 1H NMR (δ, ppm, 600 MHz, CDCl3): 6.53 (s, 2H, olefinic), 4.70 (s, 5H, Cp), 3.92–3.86 (m, 6H, –CH2), 1.22 (t, J = 7.06 Hz, 9H, –CH3). 13C NMR (δ, ppm, 151 Hz, CDCl3): 220.11 (d, J = 45 Hz, C≡O); 185.33 (s, imide C=O); 137.86 (s, olefinic C=C); 82.57 (s, Cp); 61.47 (s, -CH2); 16.32 (s, –CH3). 31P NMR (δ, ppm, 242.99 MHz, CDCl3): 175.37. FTIR (cm−1): 1972 (C≡O); 1649 (C=O, imide); 1032, 947 (P-OEt). ESI–MS: m/z calcd. for C12H22FeNO6P (M + H)+, 412.05; found, 412.11. Anal. calcd for C16H22FeNO6P (411.0534): C 46.74; H 5.39; N 3.41; found C 46.53; H 5.34; N 3.24.
Synthesis of 3b The crude product was purified by column chromatography using CHCl3-EtOAc (3:1) as eluent to afford an orange solid (3b). The product was recrystallized from chloroform/heptane. Yield 50 mg (70%). 1H NMR (δ, ppm, 600 MHz, CDCl3): 7.31–7.28 (m, 6H, meta-H of phenyl), 7.14 (dd, J = 7.9 Hz, 13.8 Hz, 9H, ortho-, para-H of phenyl), 6.35 (s, 2H, olefinic), 4.60 (s, 5H, Cp). 13C NMR (δ, ppm, 151 Hz, CDCl3): 218.30 (d, J = 42 Hz, C≡O); 184.72 (s, imide C=O); 151.33 (d, J = 9 Hz, phenyl C–O); 137.60 (s, olefinic C=C); 129.78 (s, meta-C of Ph); 125.14 (s, ortho-C of phenyl); 121.26 (d, J = 4.5 Hz, para-C of phenyl); 82.85 (s, Cp). 31P NMR (δ, ppm, 242.99 MHz, CDCl3): 168.28. FTIR (cm−1): 1970 (C≡O); 1646 (C=O, imide); 1194, 921 (P-OPh). ESI–MS: m/z calcd for C28H22FeNO6P (M + H)+, 556.05; found, 556.21. Anal. calcd for C28H22FeNO6P (555.0534): C 60.56; H 3.99; N 2.52, found C 60.66; H 3.96; N 2.4.
Synthesis of 3c The crude product was purified by column chromatography using CHCl3-EtOAc (3:1) as eluent to afford brown–red crystals (3c). The product was recrystallized from chloroform/heptane. Yield 38 mg (38%). 1H NMR (δ, ppm, 600 MHz, CDCl3): 6.54 (s, 2H, olefinic), 4.65 (s, 5H, Cp), 4.45–4.39 (m, 3H of iPr), 1.22 (dd, J = 9 Hz, 6 Hz, 18H of iPr). 13C NMR (δ, ppm, 151 Hz, CDCl3): 220.45 (d, J = 48 Hz, C≡O); 185.47 (s, imide C=O); 137.92 (s, olefinic C=C); 82.87 (s, Cp); 69.85 (d, J = 6 Hz, CH of iPr); 24.20 (t, J = 4.5 Hz, CH3 of iPr). 31P NMR (δ, ppm, 242.99 MHz, CDCl3): 170.93. FTIR (cm−1): 1964 (C≡O); 1647 (C=O, imide); 1168, 1006, 696 (P-OiPr). ESI–MS: m/z calcd. for C19H28FeNO6P (M + H)+, 454.10; found, 454.36. Anal. calcd for C19H28FeNO6P (453.1004): C 50.35; H 6.23; N 3.09; found C 50.32; H 6.04; N 2.96.
X-ray structure determination
X-ray diffraction data for 2a, 2b, 3b and 3c compounds were measured on a four-circle Oxford Diffraction Supernova Dual diffractometer using a two-dimensional area CCD detector and a low-temperature device Oxford Cryosystem cooler. Integration of the intensities, corrections for Lorentz effects, polarization effects and analytical absorption were performed with CrysAlis PRO40. The crystal structures were solved by direct methods and refined on F2 using a full-matrix least-squares procedure (SHELXL-2014)41. During anisotropic refinement there were observed some evidence of crystal disorder (elongated displacement ellipsoids) in case of 3b. Finally the crystal structure was refined as a disordered one with two positions of cyclopentadienyl ring of occupancies ratio 0.56:0.44. Moreover, in the refinement additional restrains were applied to displacement parameters of carbon atoms (C11A/B, C12A/B, C13A/B, C14A/B, C15A/B) including SADI instruction in Shelx.
In all the investigated crystal structures the positions of the hydrogen were introduced in the calculated positions with an idealized geometry and constrained using a rigid body model with isotropic displacement parameters equal to 1.2 of equivalent displacement parameters of their parent atoms. The molecular geometry was calculated by Platon42 and WinGX programs43. The relevant crystallographic data are given in Table 2. Atomic coordinates, displacement parameters, an structure factors of the analysed crystal structures are deposited with Cambridge Crystallographic Data Centre CCDC44. Deposit numbers are submitted in the Table 2.
Cell culture
The A549 (human non-small cell lung cancer) cell line was obtained from the American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 2 mM l-Glutamine, 25 mM HEPES and penicillin/streptomycin solution (100 U/ml and 100 µg/ml, respectively). The HL-60 (human promyelocytic leukemia) cell line was also obtained from the American Type Culture Collection (ATCC) and cultured in Iscove’s Modified Dulbecco's Medium (IMDM) with 15% fetal bovine serum (FBS), 2 mM l-Glutamine, 25 mM HEPES and penicillin/streptomycin solution (100 U/ml and 100 µg/ml, respectively). Both cell lines were cultured in flasks at 37 °C in 5% CO2 and sub-cultured every 2–3 days to maintain exponential growth.
Peripheral blood mononuclear (PBM) cells were isolated from a leukocyte-buffy coat collected from the blood of healthy, non-smoking donors at the Blood Bank in Lodz, Poland. The study protocol received approval from the Committee for Research on Human Subjects of the University of Lodz (17/KBBN-UŁ/III/2019). The initial step of isolation process involved mixing buffy coats with PBS in a 1:1 ratio. Subsequently, the mixture was centrifuged using a density gradient of Lymphosep (Cytogen, Zgierz, Poland) at 2200 RPM for 20 min, employing the lowest values for acceleration and deceleration. PBM cells were collected and washed three times by centrifugation with 1 × PBS. Following isolation, the cells were suspended in RPMI 1640 medium.
Cell viability resazurin assay
Firstly, resazurin salt powder was dissolved in sterile 1 × PBS. Cells were then seeded on the 96-well plates in the count of 1 × 104 in the case of HL-60 cells and of 5 × 104 for PBM cells and of 5 × 103 for A549 cells per well. A549 cells were seeded 24 h prior to treatment with the complexes to allow cell adherence to 96-well plate. Iron(II) complexes were added to wells to obtain final concentrations of 0.5, 1, 2.5, 5, 10, 25, 50, 100 and 250 μM. Subsequently, plates were incubated at 37 °C in 5% CO2 for 2 h and 24 h. After that 10 μl of resazurin salt was added to each well and plates again were incubated in 37 °C in 5% CO2 for 2 h. Finally, fluorescence was measured with microplate reader Synergy HT (Bio-Tek Instruments, USA) using an excitation wavelength of 530/25 and an emission wavelength of 590/35 nm. The effects of iron(II) complexes were calculated as the percentage of control fluorescence. IC50 (half maximal inhibitory concentration) values were calculated using a website algorithm (https://www.aatbio.com/tools/ic50-calculator). All assays were performed in octuplicate.
DNA damage by the comet assay
Selected iron(II) complexes were added to the suspension of the cells to give final concentrations of 1, 5, 10, 25 and 50 μM. HL-60 and A549 cells were incubated with the complexes for 2 h at 37 °C. The comet assay was performed under alkaline conditions according to the procedure of Tokarz et al.45. After incubation, a freshly prepared cells suspension in 0.75% LMP agarose dissolved in PBS was layered onto microscope slides (Superior, Germany), which were pre-coated with 0.5% NMP agarose. Next, the cells were lysed for 1 h at 4 °C in a buffer containing 2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, 1% Triton X-100, pH 10. Following the lysis, the slides were placed in an electrophoresis unit. DNA was allowed to unwind for 20 min in the solution containing 300 mM NaOH and 1 mM EDTA, pH > 13. Electrophoretic separation was performed in the solution containing 30 mM NaOH and 1 mM EDTA, pH > 13 at ambient temperature of 4 °C (the temperature of the running buffer did not exceed 12 °C) for 20 min at an electric field strength of 0.73 V/cm (28 mA). After separation, the slides were washed in water, drained, stained with 2 μg/ml DAPI and covered with cover slips. As a prevention from additional DNA damage, the procedure described above was conducted under limited light or in the dark. Each experiment included the positive control—cells incubated with H2O2 at 20 µM for 15 min on ice (data not shown).
Comet analysis
The comets obtained after single cell gel electrophoresis were analysed in the following manner. The comets were observed at 200× magnification in an Eclipse fluorescence microscope (Nikon, Japan) attached to a ProgRes MF cool monochrome video camera (JENOPTIK, Jena, Germany) and connected to a personal computer-based image analysis system Lucia Comet Assay version 7.30 (Laboratory Imaging, Praha, Czech Republic). Hundred images of singular comets were randomly selected from each sample and the mean value of DNA in comet tail was taken as an index of DNA damage (expressed in percent).
Plasmid relaxation assay
The pUC19 plasmid was isolated from the DH5α E. coli cells with Plasmid Mini AX Kit (A&A Biotechnology) according to the manufacturer’s instruction. The isolated plasmid quantity and quality were determined by A260/A280 ratio and gel electrophoresis, respectively. The native form of pUC19 exists mainly in the supercoiled form (CCC) which is characterized by a relatively high electrophoretic mobility. The plasmid was digested with the restrictase PstI (New English Biolabs) to induce linear (L) form. Topological differences between CCC and L forms of the plasmid account for their different electrophoretic mobility. The plasmid at 50 ng/μl was incubated for 2 h and 24 h with iron(II) complexes at the concentration of 5 µM and 50 µM. Then the samples were subjected to 1% agarose gel electrophoresis with ethidium bromide staining, visualization under UV light (302 nm), scanning by a CCD camera, and analysis with the GeneTools by Syngene (Cambridge, UK) software. During electrophoresis, we also separated 4 μl of 1 kb DNA ladder (GeneRuler 1 kb DNA Ladder, Thermo Scientific, Waltham, MA, USA).
Docking studies
The molecular docking was performed using AutoDock46 with the grid box designed based on the DNA structure as a substrate with iron(II) complex as a ligand. Fully paired 12nt DNA fragment was used as an example of ds-DNA. For the mismatched DNA also 12nt fragment with T-T mismatch was selected and structure optimized as in previous study16 to mimic damaged DNA strand. To produce global structure of complexes, a grid-based docking was applied using the Lamarckian genetic algorithm. Appropriate parameters for iron(II) were added to the AutoDock parameters file. Every docking was analysed as 10 best possible conformations with 3 repeats. Metal complex ligand conformations of the lowest energy, was analysed and if necessary used for additional MD simulations.
DNA titration
Genomic DNA was isolated from A549 cell line using a commercially available kit (Extractme Genomic DNA Kit, Blirt, Gdansk, Poland) following the manufacturer’s protocol. The concentration and purity of DNA were measured by BioTek Synergy HT Microplate Reader (BioTek Instruments, Winooski, VT, USA). Genomic DNA in the amount of 20 µg was incubated with complex 2b for 15 min at room temp. at concentrations 0.5, 5, and 50 µM in a final volume of 2 ml. Next UV–Vis absorbance spectra (240–300 nm) was measured by UV–Vis JASCO V-630 spectrophotometer (JASCO, Japan).
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Rosenberg, B., Vancamp, L., Trosko, J. E. & Mansour, V. H. Platinum compounds: A new class of potent antitumour agents. Nature 222, 385–386. https://doi.org/10.1038/222385a0 (1969).
Johnstone, T. C., Park, G. Y. & Lippard, S. J. Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res. 34, 471–476 (2014).
Oun, R., Moussa, Y. E. & Wheate, N. J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 47, 6645–6653. https://doi.org/10.1039/c8dt00838h (2018).
Fong, C. W. Platinum anti-cancer drugs: Free radical mechanism of Pt-DNA adduct formation and anti-neoplastic effect. Free Radical Biol. Med. 95, 216–229. https://doi.org/10.1016/j.freeradbiomed.2016.03.006 (2016).
Lee, S. Y., Kim, C. Y. & Nam, T. G. Ruthenium complexes as anticancer agents: A brief history and perspectives. Drug Des. Devel. Ther. 14, 5375–5392. https://doi.org/10.2147/dddt.s275007 (2020).
Leijen, S. et al. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Invest New Drugs 33, 201–214. https://doi.org/10.1007/s10637-014-0179-1 (2015).
Hartinger, C. G. et al. KP1019, a new redox-active anticancer agent–preclinical development and results of a clinical phase I study in tumor patients. Chem. Biodivers. 5, 2140–2155. https://doi.org/10.1002/cbdv.200890195 (2008).
Trondl, R. et al. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 5, 2925–2932. https://doi.org/10.1039/C3SC53243G (2014).
Monro, S. et al. Transition metal complexes and photodynamic therapy from a tumor-centered approach: Challenges, opportunities, and highlights from the development of TLD1433. Chem. Rev. 119, 797–828. https://doi.org/10.1021/acs.chemrev.8b00211 (2019).
Wani, W. A. et al. Recent advances in iron complexes as potential anticancer agents. N. J. Chem. 40, 1063–1090. https://doi.org/10.1039/C5NJ01449B (2016).
Bouché, M., Hognon, C., Grandemange, S., Monari, A. & Gros, P. C. Recent advances in iron-complexes as drug candidates for cancer therapy: Reactivity, mechanism of action and metabolites. Dalton Trans. 49, 11451–11466. https://doi.org/10.1039/D0DT02135K (2020).
Basu, U., Roy, M. & Chakravarty, A. R. Recent advances in the chemistry of iron-based chemotherapeutic agents. Coordination Chem. Rev. 417, 213339. https://doi.org/10.1016/j.ccr.2020.213339 (2020).
Köpf-Maier, P., Köpf, H. & Neuse, E. W. Ferrocenium salts—The first antineoplastic iron compounds. Angewandte Chemie Intl. Edn. Eng. 23, 456–457. https://doi.org/10.1002/anie.198404561 (1984).
Köpf-Maier, P., Köpf, H. & Neuse, E. W. Ferricenium complexes: A new type of water-soluble antitumor agent. J. Cancer Res. Clin. Oncol. 108, 336–340. https://doi.org/10.1007/bf00390468 (1984).
Jaouen, G., Vessières, A. & Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 44, 8802–8817. https://doi.org/10.1039/C5CS00486A (2015).
Juszczak, M. et al. Piano-stool ruthenium(ii) complexes with maleimide and phosphine or phosphite ligands: Synthesis and activity against normal and cancer cells. Dalton Trans. 52, 4237–4250. https://doi.org/10.1039/D2DT04083B (2023).
Juszczak, M. et al. Cytotoxicity of piano-stool ruthenium cyclopentadienyl complexes bearing different imidato ligands. Appl. Organometallic Chem. https://doi.org/10.1002/aoc.6595 (2022).
Morais, T. S., Valente, A., Tomaz, A. I., Marques, F. & Garcia, M. H. Tracking antitumor metallodrugs: Promising agents with the Ru(II)- and Fe(II)-cyclopentadienyl scaffolds. Future Med. Chem. 8, 527–544. https://doi.org/10.4155/fmc.16.7 (2016).
Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 39, 1673–1688. https://doi.org/10.1039/b916860p (2010).
Golbaghi, G. et al. Synthesis and biological assessment of a ruthenium(II) cyclopentadienyl complex in breast cancer cells and on the development of zebrafish embryos. Eur. J. Med. Chem. 188, 112030. https://doi.org/10.1016/j.ejmech.2019.112030 (2020).
Côrte-Real, L. et al. Ruthenium-cyclopentadienyl bipyridine–biotin based compounds: Synthesis and biological effect. Inorg. Chem. 58, 9135–9149. https://doi.org/10.1021/acs.inorgchem.9b00735 (2019).
Côrte-Real, L. et al. Methyl-cyclopentadienyl ruthenium compounds with 2,2′-bipyridine derivatives display strong anticancer activity and multidrug resistance potential. Inorg. Chem. 57, 4629–4639. https://doi.org/10.1021/acs.inorgchem.8b00358 (2018).
Teixeira-Guedes, C., Brás, A. R., Teixeira, R. G., Valente, A. & Preto, A. Ruthenium(II)-cyclopentadienyl-derived complexes as new emerging anti-colorectal cancer drugs. Pharmaceutics. https://doi.org/10.3390/pharmaceutics14061293 (2022).
Rudolf, B. & Zakrzewski, J. (ETA(5)-Cyclopentadienyl)FE(CO)(2)-complex of maleimide anion—An organometallic carbonyl probe for biomolecules containing HS groups. Tetrahedron Lett. 35, 9611–9612. https://doi.org/10.1016/0040-4039(94)88524-9 (1994).
Rudolf, B., Walendowska, J. & Zakrzewski, J. Photosubstitution of the halide ligands (X-) in (eta(5)-C5H5)Fe(CO)(PPh3)X. J. Organometallic Chem. 648, 293–296. https://doi.org/10.1016/s0022-328x(01)01447-4 (2002).
Cahn, R. S., Ingold, C. & Prelog, V. Specification of the molecular chirality. Angew. Chem. Int. Ed. Engl. 5, 385–415 (1966).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. https://doi.org/10.1039/B810189B (2008).
Schäfer, A., Huber, C. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835. https://doi.org/10.1063/1.467146 (1994).
Gaussian 16, Revision C.01 (Gaussian Inc., Wallin, 2016).
Wysokiński, D. et al. Photoactive CO-releasing complexes containing iron—Genotoxicity and ability in HO-1 gene induction in HL-60 cells. Toxicol. Res. (Camb.) 8, 544–551. https://doi.org/10.1039/c9tx00070d (2019).
Rocco, D. et al. Anticancer potential of diiron vinyliminium complexes. Chemistry 25, 14801–14816. https://doi.org/10.1002/chem.201902885 (2019).
Pilon, A. et al. A new family of iron(II)-cyclopentadienyl compounds shows strong activity against colorectal and triple negative breast cancer cells. Molecules (Basel, Switzerland) 25, 1592. https://doi.org/10.3390/molecules25071592 (2020).
Pilon, A. et al. First iron(II) organometallic compound acting as ABCB1 inhibitor. Eur. J. Med. Chem. 256, 115466. https://doi.org/10.1016/j.ejmech.2023.115466 (2023).
Castro, J. et al. Dinuclear iron complexes of iminopyridine-based ligands as selective cytotoxins for tumor cells and inhibitors of cancer cell migration. Pharmaceutics. https://doi.org/10.3390/pharmaceutics14122801 (2022).
Kumar, P., Tomar, S., Kumar, K. & Kumar, S. Transition metal complexes as self-activating chemical nucleases: Proficient DNA cleavage without any exogenous redox agents. Dalton Trans. 52, 6961–6977. https://doi.org/10.1039/D3DT00368J (2023).
Petronek, M. S. & Allen, B. G. Maintenance of genome integrity by the late-acting cytoplasmic iron-sulfur assembly (CIA) complex. Front. Genet. 14, 1152398. https://doi.org/10.3389/fgene.2023.1152398 (2023).
Lee, J. & Roh, J. L. Targeting iron-sulfur clusters in cancer: Opportunities and challenges for ferroptosis-based therapy. Cancers (Basel). https://doi.org/10.3390/cancers15102694 (2023).
Kashanian, S. et al. In vitro study of DNA interaction with clodinafop-propargyl herbicide. DNA Cell Biol. 27, 581–586. https://doi.org/10.1089/dna.2008.0740 (2008).
Juszczak, M., Kluska, M., Wysokiński, D. & Woźniak, K. DNA damage and antioxidant properties of CORM-2 in normal and cancer cells. Sci. Rep. 10, 12200. https://doi.org/10.1038/s41598-020-68948-6 (2020).
CrysAlisPRO software system (Oxford Diffraction/Agilent Technologies UK Ltd, Yarnton, England).
Sheldrick, M. G. Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8 (2015).
Spek, A. L. Structure validation in chemical crystallography. Acta Crystallographica Section D 65, 148–155. https://doi.org/10.1107/S090744490804362X (2009).
Farrugia, L. J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 45, 849–854. https://doi.org/10.1107/S0021889812029111 (2012).
Groom, R. C., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The Cambridge Structural Database. Acta Cryst. B72, 171–179 (2016).
Tokarz, P., Piastowska-Ciesielska, A. W., Kaarniranta, K. & Blasiak, J. All-trans retinoic acid modulates DNA damage response and the expression of the VEGF-A and MKI67 genes in ARPE-19 cells subjected to oxidative stress. Int. J. Mol. Sci. https://doi.org/10.3390/ijms17060898 (2016).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. https://doi.org/10.1002/jcc.21256 (2009).
Acknowledgements
This work was financially supported by the University of Lodz, Poland, Faculty of Chemistry and Faculty of Biology and Environmental Protection. The research described in the article was carried out as part of the Interdisciplinary Research Grant of the University of Lodz (IDUB B2311013000182.07).
Author information
Authors and Affiliations
Contributions
S.D. Organic synthesis, investigation, A.K. Organic synthesis, investigation, M.S. Biological part, investigation, M.J. Biological part, investigation, P.T. Biological part, investigation, P.W. Biological part, plasmid relaxation assay, investigation, M.P. DFT studies, investigation, A.R-P. Crystallography, investigation, K.W-R. Crystallography, investigation, S.V. Docking studies, investigation, A.Ch. Docking studies, supervision, manuscript writing, K.W. Conceptualization, supervision, manuscript writing, B.R. Conceptualization, supervision, manuscript writing. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Das, S., Strachanowska, M., Wadowski, P. et al. Synthesis, anticancer activity, and molecular docking of half-sandwich iron(II) cyclopentadienyl complexes with maleimide and phosphine or phosphite ligands. Sci Rep 14, 5634 (2024). https://doi.org/10.1038/s41598-024-56339-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-56339-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
