
Abstract
With growing public concern about water quality particular focus should be placed on organic micropollutants, which are harmful to the environment and people. Hence, the objective of this research is to enhance the security and resilience of water resources by developing an efficient system for reclaiming industrial/military wastewater and protecting recipients from the toxic and cancerogenic explosive compound—2,4,6-trinitrotoluene (TNT), which has been widely distributed in the environment. This research used an anodic oxidation (AO) process on a boron-doped diamond (BDD) electrode for the TNT removal from artificial and real-life matrices: marine water and treated wastewater. During experiments, TNT concentrations were significantly decreased, reaching the anodic degradation efficiency of above 92% within two hours and > 99.9% after six hours of environmental sample treatment. The presented results show the great potential of AO performed on BDD anodes for full-scale application in the industry and military sectors for TNT removal.
Similar content being viewed by others

Introduction
Commencing in the nineteenth century, chiefly driven by armed conflicts like the First and Second World Wars, there was a substantial increase in the mass production of nitro-organic explosive compounds1. Among them, 2,4,6-trinitrotoluene (TNT) was the most widely used due to its cost-effective production and low sensitivity to shock and friction, which reduces the possibility of an unexpected detonation2,3. However, the widespread use of TNT increases the risks of environment contamination. TNT is categorised as a class C human carcinogen4, and the United States Environmental Protection Agency (US EPA) has classified TNT as a contaminant of emerging concern (CEC), which has resulted in 2,4,6-trinitrotoluene being routinely monitored, and being found at low levels in many water sources. Even at these low levels it may cause may cause ecological or human health impacts5. Additionally, there is evidence showing that explosive materials and their by-products are being accumulated in the marine food chain, which in turn affects human food chains6,7.
2,4,6-trinitrotoluene is still being produced and used for military and commercial applications. In the course of manufacturing and processing, the generation of wastewater occurs, wherein the concentration of TNT may reach up to 160 mg L−18,9,10. Moreover, a single 2,4,6-trinitrotoluene ammunition factory can produce 1,900,000 dm3 of explosive-contaminated effluent per day3. Historically, “pinkwater” (or “redwater”), the effluent containing mainly TNT, was often released directly into lagoons10. Presently, numerous countries are tightening regulations concerning the quality of wastewater released into the environment. Commonly used wastewater treatment methods, such as biological and physicochemical approaches, have various drawbacks. Firstly, there is a limitation in the microbiological applicability, especially in the presence of other xenobiotics, which may result in inadequate treatment. Secondly, the physical processes such as filtration, reverse osmosis, and adsorption separate contaminants rather than degrade them, thereby generating another waste contaminated with explosive compounds (at a higher concentration) that still requires treatment8,9,11.
Despite the hazards associated with the presence of TNT in the environment, in the newest Directive (EU) 2020/2184 on the quality of water intended for human consumption12, there is no information regarding TNT acceptable levels. Similarly, established “Watch List” regulations (Watch List no. 313, Directive 2013/39/EU14) requiring countries to monitor selected contaminants in water do not include explosive materials. So far, only the US EPA has assessed the risks associated with the presence of TNT in drinking water. Suggesting a limit of 0.1 mg L−1, for which, an estimated lifetime cancer risk is 1 in 10,00015. Nevertheless, we believe efforts should focus on sustaining TNT concentrations below the 2 μg L−1 recognized as a safe dose15. Thus, the authors propose this level as a recommended drinking water limit for EU countries. To achieve this concentration level, a cost effective and scalable wastewater treatment method is required.
Anodic oxidation (AO) is a method that exhibits potential in effectively eliminating persistent organic compounds. This technique encompasses the indirect oxidation of organic pollutants through oxidizing agents and/or direct oxidation at the anode surface16,17. Among the anodic materials used in AO processes, boron-doped diamond (BDD) is distinguished its excellent resistance to fouling, and ability to withstand extreme conditions (high potentials, high temperature/pressure, corrosive environments)18,19,20. Furthermore, using BDD in AO enables to direct production of a strong oxidising agent, the hydroxyl radical (·OH), via reaction (1)21. Compared to other anode materials, BDD exhibits a higher kinetic overpotential of O2 generation, which reduces the competition from oxygen evolution reaction, and increases the efficiency of ·OH production. The hydroxyl radical is known for its ability to mineralize a wide variety of organic species, including persistent organic pollutants21,22,23. Furthermore, BDD is weakly interacting with generated ·OH radicals so that most of the generated oxidant can react with the pollutants available in the solution instead of being chemisorbed at the electrode21. In addition to the hydroxyl radical, other oxidising compounds can also be formed according to the Eqs. (1–6), which may improve the oxidation process24,25.
In this work, we present for the first time the effective anodic oxidation of TNT using BDD electrodes, together with an analysis of the by-products of this process. The research involved testing the efficiency of TNT removal depending on solution matrix. Studies were carried out in both laboratory-grade purity matrices and environmental samples, which were selected to investigate removal efficiency in matrices where higher concentrations of TNT can be expected: seawater and samples from wastewater treatment plants. The TNT removal process was controlled by applying two techniques in parallel: (i) liquid chromatography with a photodiode array detector and (ii) electrochemical sensing. An analysis of the by-products generated during the process was also carried out using gas chromatography-tandem mass spectrometry. An analysis of energy consumption was also conducted, which could support the possibility of introducing this method into industry. The presented work is a first step towards the application of the AO method on BDD in explosives removal from aquatic environments.
Results and discussion
Anodic oxidation of TNT-spiked samples
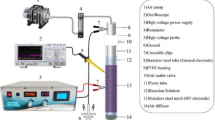
The AO experiments were performed using a BDD electrode as an anode and stainless-steel mesh as a cathode. The electrodes were placed in a 500-mL undivided electrolytic cell, which was put in a specially designed experimental set-up consisting of a cooling bath and magnetic stirrer (Fig. 1). The anode geometric surface area was 10.5 cm2. The distance between them was kept constant at about 2.5 cm. All AO tests were performed under galvanostatic conditions, with a current density of 50 mA cm−2, which was adjusted to the treated wastewater sample conductivity, which was the lowest among studied matrices.

Advanced oxidation setup (batch reactor) scheme using BDD anode and stainless-steel cathode in the undivided electrolytic cell.
The ability to remove aromatic compounds was first tested in an uncomplicated, laboratory-grade purity matrix of 0.1 M phosphate buffer solution (0.1M_PBS). Next, TNT degradation was investigated in real-life samples—treated wastewater (TWW) and Baltic Sea marine water (MW) (full characterisation of TWW and MW samples can be found in SI 2). Considering the elevated concentration of chlorides in the real-life samples, the decision was made to broaden the scope of the research to examine the impact of chlorides on the effectiveness of the treatment process. For this purpose, NaCl was dissolved in the PBS solution to obtain a solution with a concentration of 100 mg Cl− L−1 (0.1M_PBS_100mgCl) and 200 mg Cl− L−1 (0.1M_PBS_200mgCl). Each AO process was carried out for 8 h at a temperature of 20 ± 3 °C, and the solutions were spiked with TNT stock solution resulting in the same initial concentration of 50 mg L−1 (50 ppm).
Anodic oxidation of TNT-polluted samples was first conducted in the PBS solution. Phosphate buffer of pH 7.4 was prepared to mimic the conditions of wastewater treatment plant effluents26. Chloride-free electrolytes as phosphate buffer support electrogeneration of reactive oxygen species including ·OH, H2O2, O3, or O2−27, and do not have a supportive effect on the anodic oxidation, as it would be expected in other electrolytes containing chloride or sulphate ions28. The efficiency of TNT removal by AO is presented as the concentration (determined by the HPLC–PDA technique) as a function of time (Fig. 2a). As can be seen, with increasing time the concentration successively decreases reaching the value of normalised concentration Ct/C0: 0.56, 0.08, and 0.006 after 1 h, 4 h, and 6 h, respectively. Furthermore, after 8 h of AO, 99.8% of initial TNT was degraded reaching the value of 93 µg L−1.

Trend with a time of oxidising TNT, normalised concerning the initial concentration (C0 = 50 mg L−1) based on HPLC–PDA results in (a) 0.1 M PBS, (b) saline artificial samples: 0.1M_PBS_100mgCl and 0.1M_PBS_200mgCl, (c) environmental samples: TWW and MW (0.1M_PBS sample is left as reference), (d) kinetic order description and kinetic equation of TNT removal rate (A—reaction rate constant [h−1], t—time [h]).
As TNT degradation products cannot be identified using the HPLC–PDA method, select samples have been analysed using a GC–MS/MS technique. The gas chromatography results confirmed a significant decrease in the concentration of TNT in the spiked samples (see Table 1). After 8 h treatment, a concentration of 86 µg L−1 was achieved, which agrees with previous HPLC–PDA results. The concentration is anticipated to continue decreasing; however, further investigation is needed to access the total TNT mineralisation. This significant reduction in the concentration of TNT indicates a high efficiency of removing this compound by anodic oxidation on the BDD electrode.
The chromatography results indicated the formation of TNT oxidation intermediates such as 1,3,5-trinitrobenzene (TNB) and 1,3-dinitrobenzene (1,3-DNB). The appearance of the 21 µg L-1 TNB in the 0.1M_PBS sample after 4 h of the AO process and its absence after 8 h agrees with the mechanism of TNT decomposition shown in Fig. 3a. The proposed oxidation process is a chain of H-abstraction reactions. First, a methyl group is oxidised to an aldehyde moiety –CHO, which in turn is transformed into a carboxyl –COOH group. Next is the decarboxylation process that leads to the TNB compound. Then, with time, the full demineralization of the TNB to water, carbon dioxide and nitrate ions could be is expected29,30. With the increasing duration of the AO process, there is a noticeable decrease in the pH of the reaction mixture from pH 7.77 to 7.06 and from 7.4 to 7.18 (Table S4, SI 3) in the samples with low Cl- influence: 0.1 M PBS and TWW, respectively. The decrease in pH could be explained by the increasing concentration of nitrate ions in the presence of week-base cations and/or short-chain carboxylic acids evolution during TNT degradation31.

Utilizing identified intermediates, proposed pathways for (a) electrooxidative decomposition of TNT, and (b) 2,4-DNT; (c) 2,4-DNT and 4-NT formation through TNT-Meisenheimer complex formation; (d) creation of (d) 2-amino-4,6-dinitrotoluene, (e) 4-amino-2,6-dinitrotoluene via electroreduction.
1,3-DNB is in turn formed from the oxidation of 2,4-dinitrotoluene (2,4-DNT) (Fig. 3b). The presence of dinitrotoluenes (2,4-DNT and 2,6-dinitrotoluene—2,6-DNT) and 4-nitrotoluene (4-NT) may be explained by the formation of hydride Meisenheimer complexes32,33,34 (Fig. 3c). The electronegative nitro groups extract the π electrons from the aromatic ring of TNT, thereby rendering the aromatic nucleus electrophilic and enabling nucleophilic attack. In this way, it is possible to form a negatively charged nonaromatic complex, which can be rearomatized after -NO2 release forming dinitrotoluenes34.
Apart from the electrooxidation products, Table 1 also displays products obtained through the TNT reduction process. The presence of 4-amino-2,6-dinitrotoluene (4-A-2,6-DNT) and 2-amino-4,6-dinitrotoluene (2-A-4,6-DNT) can be explained by the cathodic reaction taking place at the stainless-steel counter electrode (Fig. 3d,e). It should be noted that although only the diamond anode was polarised, current must also be passed through the cathode to complete the cell. While 4-A-2,6-DNT and 2-A-4,6-DNT exhibit lower toxicity compared to TNT35, their presence is still linked to health risks. However, it should be emphasized, that identified TNT degradation by-product concentrations are almost 100 times lower (< 0.2 mg L−1) than the initial concentration of TNT (50 mg L−1). To avoid the formation of electroreduction products, (1) the anode and cathode could be separated by a membrane or (2) different cathode material can be evaluated, and (3) different advanced oxidation processes can be merged.
To explore the impact of chloride anions, present in environmental samples on the anodic oxidation process, the analyses were also performed in chloride-spiked PBS (100 and 200 mg Cl− L−1). The results of the HPLC–PDA investigation of the saline artificial samples are presented in Fig. 2b. During the first hour of treatment Ct=1/C0 decreases to 0.33 and 0.26 for 0.1M_PBS_100mgCl and 0.1M_PBS_200mgCl samples, respectively. The rate of explosive material decomposition in 0.1M_PBS_200mgCl solution is more than double compared to PBS-only sample. After 3 h of treatment, Ct=3h/C0 was recorded below 0.1 for both salty solutions (Ct=3h/C0 = 0.066 for 0.1M_PBS_100mgCl, and Ct=3h/C0 = 0.052 for 0.1M_PBS_200mgCl), whereas in pristine 0.1M_PBS the Ct/C0 value reached almost 0.2 (Ct=3h/C0 = 0.195). Moreover, after 7 h of AO, the concentration of TNT recorded for 0.1M_PBS_200mgCl was below the limit of detection (LOD) value of the HPLC–PDA apparatus (LOD = 2.9 µg L−1).
The results show that a saline environment enhances the rate of nitroaromatic TNT degradation over time. A similar effect was also observed by Kuo et al.36. The effect can be explained by the presence of active chlorine species originating from the chloride ions oxidation to active chlorine species like dissolved (Cl2,aq), hypochlorous acid (HClO) and hypochlorite ion (ClO−) via reactions (Eqs. 7–9)23. The acceleration of the degradation of aromatic compound pollution may be attributed to the presence of active chlorine species.
The electrooxidation process was then carried out in a complex, real-life environmental matrices. For this purpose, the measurements were made in samples collected from the Baltic Sea (MW) and a wastewater treatment plant (TWW). The results of the HPLC–PDA investigation of TWW and MW are presented in Fig. 2c. Normalized concentration Ct/C0 in TWW matrix was 0.4, 0.004 after 1 h and 4 h of treatment, respectively; and in Baltic Sea marine water matrix: 0.18, 0.006 after 1 h, and 4 h, respectively. After 6 h of AO, for both environmental solutions the value of Ct=6h/C0 was below LOD of HPLC–PDA which, when converted, gives > 99.9% removal of TNT. Compared to the 0.1M_PBS sample, environmental matrices have a positive effect on the nitroaromatic compounds' degradation rate. After 1 h of anodic oxidation, 42%, 61%, and 80% of the initial concentration of TNT were reduced for 0.1M_PBS, TWW and MW matrices, respectively. After only two hours of treatment, TNT removal efficiencies were 92% for TWW and 96% for MW, while electro-assisted advanced oxidation processes e.g., combined electrolytic and H2O2 system led to only 70% removal of TNT in the same timeframe37.
In all studied matrixes TNT removal represents 1st kinetic order (Fig. 2d), where the highest reaction rate constant, A, is noticed in the marine water matrix (A = 1.23 h−1) and under these conditions we are observing the fastest initial disappearance of the analyte. Hence, reaction rates analysis also confirms that saline environment enhances the TNT AO removal.
It is worth noting, that well-conductive matrices, with conductivity above 10 mS cm−1, showed reduced energy consumption in comparison to the less-conductive solution of treated wastewater (conductivity < 2 mS cm−1) (for details please see Table S4, SI 3). During 8 h of treatment using anodic oxidation on BDD energy consumption needed for the AO process varied between 68.0 and 84.8 kWh m-3 for most tested samples (only the TWW environment generates consumption of 281.6 kWh m−3). The energy consumption associated with the electrochemical oxidation of organic micropollutants is contingent upon various factors such as the type of anode, current density, reaction time, and the structure of the micropollutant. This complexity makes it challenging to directly compare the resulting energy consumption data with existing literature. However, referring to our previous work38, where BDD anode was also applied for the removal of forever chemicals (polyfluorinated alkyl compounds) from landfill leachates, energy consumption varied between 88 to 114 kW hm−3 after 8 h of AO (j = 75 mA cm−2). In comparison to our other study39, the energy consumption at the specific current 50 mA cm−2 after 8 h of treatment was proportional to present results regarding the saline environment. This confirms that electrochemical oxidation is the preferable technique for more complex matrices with higher conductivity.
The presented results show potential for the application of AO in the “pink water” treatment. It should be emphasized that the presented method can be operated without introducing additional species such as iron catalyzers or hydrogen peroxide/oxide streams often used in other purification methods (see Table 2). The technique is robust, easy to operate and flexible through electrode material modification, and current adjustment in case of fluctuating load of contaminants in the wastewater streams.
Nevertheless, in line to the circular economy, a holistic approach for efficient wastewater management is needed. The presented technology has a great potential for modular application as well for integrated treatment (together with other treatment processes). Moreover, in the full-scale application it may be used for any type of wastewater stream treatment (inc. main wastewater stream, any by-pass stream or concentrate obtained after reverse osmosis or nano-/ultra-filtration). The application as a fit-of-purpose manner is coherent with EU Green Deal ambitions44, (e.g., Towards Zero Pollution, Water-Smart Society) and UN Sustainable Development concepts45. Such an approach will help to close the water cycles at industrial production sites. This in fact would decouple industrial growth from resource consumption. Moreover, all the circular economy policy would significantly reduce the input of chemical compounds into the environment contributing to an improved water supply for human beings.
Electrochemical detection of TNT and its by-products before and after AO
TNT removal control was also carried out by electrochemical sensing. While most analyses of effluent compounds in the aqueous environment are performed using chromatographic methods, electrochemical sensors provide a low-cost substitute, as well as enabling on-line control of the process. For this purpose, samples before and after electro-oxidation were tested using electrodes with a high sp2 carbon content, the presence of which increases the sensitivity of the sensor (TNT easily adsorbs onto π-donor graphene surfaces, unlike BDD)46,47. Ability of 2,4,6-trinitrotoluene detection on boron-doped diamond/few-layer graphene nanowall electrodes has previously been published in our previous work46. Limit of detection for TNT is 73 ppb, whereas the sensitivity is 36.25 µA cm−2 ppm−1.
Electrochemical detection was performed by the differential pulse technique (Fig. 4). The measurements were first provided in blank solutions (0.1M_PBS, MW, TWW), and then the sample with spiked 50 mg L−1 TNT were tested. The last step was an electrochemical investigation of the solutions after AO.

Differential pulse voltammetry made before and after electrooxidation process recorded on BDGNW electrode immersed in (a) 0.1 M PBS, (b) marine water, (c) treated wastewater.
It was found that TNT is presented in all the samples before the AO process provide three well-separated peaks observed at negative potentials: − 0.31 V (peak “a”), − 0.47 V (peak “b”) and − 0.61 V (peak “c”) in 0.1M_PBS, − 0.35, − 0.50 and − 0.63 V in marine water, and − 0.44, − 0.62 and − 0.76 V in treated wastewater vs. Ag/AgCl/3 M KCl. The reduction signals are observed in the potential range typical for the reduction of nitro groups46,48,49. The reduction of TNT is a multistep process as the molecule contains three electroactive nitro (–NO2) moieties (oxygen and nitrogen atoms building –NO2 group are highly electronegative, which causes strong polarization of –N–O bond, making nitro moiety easily reducible1). Hence, each of the peaks corresponds visible in Fig. 4 corresponds to the individual reduction of –NO2 group to amine (–NH2) moiety46. The peaks in environmental samples are shifted towards more negative potentials, which may be caused by their lower pH value compared to PBS (the reduction process heavily depends on pH46). Then the samples after electrooxidation were tested. It should be noted that after AO there is no evidence of the explosive materials in all the investigated samples (the values are below LOD).
Conclusions
The efficacy of anodic oxidation in breaking down TNT has been demonstrated at the laboratory scale in aquatic environments employing BDD electrodes. Our results show that the AO process using diamond electrodes is very effective towards TNT degradation (> 92% removal within 2 h and above 99.9% within 6 h in environmental matrices). In each tested matrix, the anodic oxidation caused a significant decrease of explosive material reaching concentration below 0.1 mg L−1.
The research presented here is a first step towards the application of the AO method using BDD to remove explosives from aquatic environments. Our results show that electrochemical oxidation is the preferable technique for more complex matrices with higher conductivity. This technique creates a route for water reuse (water circular economy approach) in the industrial and military sectors. Employing AO process for wastewater treatment proves particularly advantageous for factories engaged in explosives production, mitigating the significant environmental pollution caused by these compounds. Furthermore, suitable, and sustainable technology for the backwash salt water (potentially generated during the chemical weapon removal in the Baltic Sea shore), is suggested to purify saline water contaminated with explosive compounds.
The anodic oxidation process on the BDD anode is promising for direct treatment of wastewater from the TNT. Given high removal efficiency AO process on BDD shows great potential for full-scale application, hence process optimisation, scalability, operation, and maintenance cost need to be studied further for its actual field application.
Methods
Fabrication and characterisation of BDD electrodes
Following the previously reported synthesis procedure50, all the BDD electrodes were fabricated by microwave plasma-assisted chemical vapour deposition system (SEKI Technotron AX5400S, Japan) on an Nb substrate. Before deposition, the substrates were subjected to surface roughening by sandblasting using a compressed air sandblasting gun and corundum sand, cleaned in analytical-grade acetone and isopropanol in an ultrasonic bath, and seeded in a water-based nanodiamond slurry. The growth conditions were: substrate temperature of 700 °C, microwave power of 1300 W, and process duration of 12 h. The boron doping level expressed as the [B]/[C] ratio in the gas phase, was 2000 ppm. No post-treatment cleaning procedure was performed on the electrodes. Electrodes preparation and characterisation (scanning electron microscopy, Raman spectroscopy, electrochemical behaviour) details are presented in SI 1.
Chemicals and solutions
TNT was obtained by the chemical plant “NITRO-CHEM S.A.” and purified for testing purposes by the Military Institute of Armament Technology. The purification process consisted of 3 recrystallizations from acetone and drying until a constant weight was measured. The selected properties of the TNT used in the tests are presented in Table S2, SI 2. A stock solution of TNT, in the concentration of 25 g L−1, was prepared using 625 mg of TNT dissolved in a 25-mL volumetric flask using acetonitrile (gradient grade for HPLC, purchased from ChemSolve). The remaining chemicals are described in SI 2.
AO effectiveness control
Chemical analysis, including measurements of pH, conductivity, oxidation reduction potential (ORP), voltage and specific electrical charge, was performed during the electrooxidation process to monitor the anodic oxidation (Table S4, SI 3). For this purpose, 2 mL of solution was sampled every hour (except experiments with real matrix, then 4 mL was sampled). pH and ORP were measured in-situ. The energy consumption (ECon) was calculated using Eq. (10)51 and expressed in kWh m−3.
where Q corresponds to specific electrical charge [kAh m−3], U reflects the average cell voltage [V], j applied current density [A cm−2], A is geometric electrode area [cm2], t refers to the electrolysis time, and V is the volume of the electrolyte [m3].
High-performance liquid chromatography with a photodiode array detector, HPLC–PDA, (Prominence-i LC-2030 3D plus, Shimadzu, Japan) and electrochemical sensing were used to analyse the effectiveness of TNT removal during the AO process. HPLC–PDA details are shown in SI 3. Electrochemical sensing (ES) was conducted using sp2-rich boron-doped diamond/few-layer graphene nanowall electrodes (BDGNW). BDGNW electrodes were deposited following the previously reported synthesis46. Fabrication and characterization of BDGNW are shown in SI 4. ES was performed using differential pulse voltammetry (DPV). The DPV parameters (pH = 25 mV, pW = 200 ms, st = 500 ms, and a scan rate of 10 mV s−1) were based on our previous work46.
Gas chromatography equipped with a tandem mass spectrometer (GC–MS/MS) was utilized to detect by-product compounds formed during electro-oxidation. Details can be found in SI 3.
Data availability
Correspondence and requests for materials should be addressed to A.D.
References
Ayoub, K., van Hullebusch, E. D., Cassir, M. & Bermond, A. Application of advanced oxidation processes for TNT removal: A review. J. Hazard. Mater. 178, 10–28 (2010).
Li, K., Sherman, C. D., Beaumont, J. & Sandy, M. S. The Office of Environmental Health Hazard Assessment: Evidence on the Carcinogenicity of 2,4,6-trinitrotoluene. (2010).
Yinon, J. Toxicity and Metabolism of Explosives (CRC Press, 1990).
United States Environmental Protection Agency. Fact Sheet - 2,4,6-Trinitrotoluene. 2 (2021).
United States Environmental Protection - Agency Federal Facilities Restoration and Reuse Office: Emerging Contaminants and Federal Facility Contaminants of Concern. https://www.epa.gov/fedfac/emerging-contaminants-and-federal-facility-contaminants-concern (Available 24. 01. 2023) (2022).
Gledhill, M., Beck, A. J., Stamer, B., Schlosser, C. & Achterberg, E. P. Quantification of munition compounds in the marine environment by solid phase extraction—Ultra high performance liquid chromatography with detection by electrospray ionisation—Mass spectrometry. Talanta 200, 366–372 (2019).
Koske, D. et al. First evidence of explosives and their degradation products in dab (Limanda limanda L.) from a munition dumpsite in the Baltic Sea. Mar. Pollut. Bull. 155, 111131 (2020).
Bhanot, P. et al. Application of integrated treatment strategies for explosive industry wastewater—A critical review. J. Water Process Eng. 35, 101232 (2020).
Barreto-Rodrigues, M., Silva, F. T. & Paiva, T. C. B. Characterization of wastewater from the Brazilian TNT industry. J. Hazard. Mater. 164, 385–388 (2009).
Maloney, S. W., Adrian, N. R., Hickey, R. F. & Heine, R. L. Anaerobic treatment of pinkwater in a fluidized bed reactor containing GAC. J. Hazard. Mater. 92, 77–88 (2002).
Guz, R., de Moura, C., da Cunha, M. A. A. & Rodrigues, M. B. Factorial design application in photocatalytic wastewater degradation from TNT industry—red water. Environ. Sci. Pollut. Res. 24, 6055–6060 (2017).
The European Parliament and the Council of the European Union. Directive (EU) 2020/2184 of the European Parliament and of the Council. Off. J. Eur. Union 2019, 1–62 (2020).
Cortes, L. G. et al. JRC Technical Report: Selection of substances for the 3rd Watch List under WFD. (2020). doi:https://doi.org/10.2760/194067.
Council, O. F. T. H. E. Directive 2013/11/EU of the European Parliament and of the Council. Fundam. Texts Eur. Priv. Law 2013, 1–17 (2020).
United States Environmental Protection Agency. 2006 Edition of the Drinking Water Standards and Health Advisories. Environ. Prot. 18 (2006).
Moreira, F. C., Boaventura, R. A. R., Brillas, E. & Vilar, V. J. P. Electrochemical advanced oxidation processes: A review on their application to synthetic and real wastewaters. Appl. Catal. B Environ. 202, 217–261 (2017).
McBeath, S. T., Wilkinson, D. P. & Graham, N. J. D. Application of boron-doped diamond electrodes for the anodic oxidation of pesticide micropollutants in a water treatment process: A critical review. Environ. Sci. Water Res. Technol. 5, 2090–2107 (2019).
Cobb, S. J., Ayres, Z. J. & Macpherson, J. V. Boron doped diamond: A designer electrode material for the twenty-first century. Annu. Rev. Anal. Chem. 11, 463–484 (2018).
Macpherson, J. V. A practical guide to using boron doped diamond in electrochemical research. Phys. Chem. Chem. Phys. 17, 2935–2949 (2015).
Dettlaff, A., Sobaszek, M., Klimczuk, T. & Bogdanowicz, R. Enhanced electrochemical kinetics of highly-oriented (111)-textured boron-doped diamond electrodes induced by deuterium plasma chemistry. Carbon N. Y. 174, 594–604 (2021).
Borràs, N., Arias, C., Oliver, R. & Brillas, E. Mineralization of desmetryne by electrochemical advanced oxidation processes using a boron-doped diamond anode and an oxygen-diffusion cathode. Chemosphere 85, 1167–1175 (2011).
Henke, A. H., Saunders, T. P., Pedersen, J. A. & Hamers, R. J. Enhancing electrochemical efficiency of hydroxyl radical formation on diamond electrodes by functionalization with hydrophobic monolayers. Langmuir 35, 2153–2163 (2019).
Garcia-Segura, S., Keller, J., Brillas, E. & Radjenovic, J. Removal of organic contaminants from secondary effluent by anodic oxidation with a boron-doped diamond anode as tertiary treatment. J. Hazard. Mater. 283, 551–557 (2015).
Polcaro, A. M., Vacca, A., Mascia, M. & Ferrara, F. Product and by-product formation in electrolysis of dilute chloride solutions. J. Appl. Electrochem. 38, 979–984 (2008).
Bergmann, M. E. H., Rollin, J. & Iourtchouk, T. The occurrence of perchlorate during drinking water electrolysis using BDD anodes. Electrochim. Acta 54, 2102–2107 (2009).
Cavalcanti, E. B., Garcia-Segura, S., Centellas, F. & Brillas, E. Electrochemical incineration of omeprazole in neutral aqueous medium using a platinum or boron-doped diamond anode: Degradation kinetics and oxidation products. Water Res. 47, 1803–1815 (2013).
Budil, J. et al. Impact of electrolyte solution on electrochemical oxidation treatment of Escherichia coli K-12 by boron-doped diamond electrodes. Lett. Appl. Microbiol. 74, 924–931 (2022).
Saha, P. et al. Effect of electrolyte composition on electrochemical oxidation: Active sulfate formation, benzotriazole degradation, and chlorinated by-products distribution. Environ. Res. 211, 113057 (2022).
Chen, W. S., Juan, C. N. & Wei, K. M. Mineralization of dinitrotoluenes and trinitrotoluene of spent acid in toluene nitration process by Fenton oxidation. Chemosphere 60, 1072–1079 (2005).
He, X. et al. A DFT study toward the reaction mechanisms of TNT with hydroxyl radicals for advanced oxidation processes. J. Phys. Chem. A 120, 3747–3753 (2016).
Ayoub, K. et al. Electro-Fenton removal of TNT: Evidences of the electro-chemical reduction contribution. Appl. Catal. B Environ. 104, 169–176 (2011).
Lamba, J. et al. Study on aerobic degradation of 2,4,6-trinitrotoluene (TNT) using Pseudarthrobacter chlorophenolicus collected from the contaminated site. Environ. Monit. Assess. https://doi.org/10.1007/s10661-021-08869-7 (2021).
Pervukhin, V. V. & Sheven, D. G. Photolysis by UVA-visible light and thermal degradation of TNT in aqueous solutions according to aerodynamic thermal breakup droplet ionization mass spectrometry. J. Photochem. Photobiol. A Chem. 432, 114079 (2022).
Esteve-Nunez, A., Caballero, A. & Ramos, J. Biological degradation of 2,4,6-trinitrotoluene. Microbiol. Mol. Biol. Rev. 65, 335–352 (2001).
George, S. E., Huggins-Clark, G. & Brooks, L. R. Use of a Salmonella microsuspension bioassay to detect the mutagenicity of munitions compounds at low concentrations. Mutat. Res. Genet. Toxicol. Environ. Mutagen 490, 45–56 (2001).
Hernandez, R., Zappi, M. & Kuo, C. H. Chloride effect on TNT degradation by zerovalent iron or zinc during water treatment. Environ. Sci. Technol. 38, 5157–5163 (2004).
Gao, S., Zhou, X. & Wu, F. Experimental Study on the Treatment of Ammunition Destroyed Wastewater by Electrolysis Combined with H2O2. Proc. 2015 7th Int. Conf. Meas. Technol. Mechatronics Autom. ICMTMA 2015 481–484 doi:https://doi.org/10.1109/ICMTMA.2015.122, (2015).
Pierpaoli, M. et al. Electrochemical oxidation of PFOA and PFOS in landfill leachates at low and highly boron-doped diamond electrodes. J. Hazard. Mater. 403, 123606 (2021).
Wilk, B. K. et al. Electrochemical oxidation of landfill leachate using boron-doped diamond anodes: Pollution degradation rate, energy efficiency and toxicity assessment. Environ. Sci. Pollut. Res. 29, 65625–65641 (2022).
Chen, W. S. & Huang, S. C. Sonophotocatalytic degradation of dinitrotoluenes and trinitrotoluene in industrial wastewater. Chem. Eng. J. 172, 944–951 (2011).
Shukla, N. et al. 2, 4-Dinitrotoluene (DNT) and 2, 4, 6-Trinitrotoluene (TNT) removal kinetics and degradation mechanism using zero valent iron-silica nanocomposite. J. Environ. Chem. Eng. 6, 5196–5203 (2018).
Barreto-Rodrigues, M., Silva, F. T. & Paiva, T. C. B. Optimization of Brazilian TNT industry wastewater treatment using combined zero-valent iron and fenton processes. J. Hazard. Mater. 168, 1065–1069 (2009).
Ludwichk, R. et al. Characterization and photocatalytic treatability of red water from Brazilian TNT industry. J. Hazard. Mater. 293, 81–86 (2015).
European Green Deal. (Available 29. 11. 23) https://www.gov.pl/web/rolnictwo/europejski-zielony-lad-european-green-deal (2019).
UN Sustainable Development Goals. (Available 29. 11. 23) https://sdgs.un.org/goals (2015).
Dettlaff, A. et al. Electrochemical determination of nitroaromatic explosives at boron-doped diamond/graphene nanowall electrodes: 2,4,6-trinitrotoluene and 2,4,6-trinitroanisole in liquid effluents. J. Hazard. Mater. 387, 121672 (2020).
Li, P., Li, X. & Chen, W. Recent advances in electrochemical sensors for the detection of 2, 4, 6-trinitrotoluene. Curr. Opin. Electrochem. 17, 16–22 (2019).
Dettlaff, A. et al. Electrochemical detection of 4,4’,5,5’-tetranitro-1H,1’H-2,2’-biimidazole on boron-doped diamond/graphene nanowall electrodes. IEEE Sens. J. 20, 9637–9643 (2020).
Saǧlam, Ş, Üzer, A., Erçaǧ, E. & Apak, R. Electrochemical determination of TNT, DNT, RDX, and HMX with gold nanoparticles/poly(carbazole-aniline) film-modified glassy carbon sensor electrodes imprinted for molecular recognition of nitroaromatics and nitramines. Anal. Chem. 90, 7364–7370 (2018).
Pierpaoli, M. et al. Carbon nanoarchitectures as high-performance electrodes for the electrochemical oxidation of landfill leachate. J. Hazard. Mater. 401, 123407 (2021).
Anglada, Á., Urtiaga, A. & Ortiz, I. Contributions of electrochemical oxidation to waste-water treatment: Fundamentals and review of applications. J. Chem. Technol. Biotechnol. 84, 1747–1755 (2009).
Acknowledgements
The research leading to these results has received funding from the Norway Grants 2014-2021 via the National Centre for Research and Development [NOR/SGS/NITROsens/0011/2020-00]. The DS funds of the Faculty of Electronics, Telecommunications, and Informatics of the Gdansk University of Technology are also acknowledged. A.D. thanks Daniel Houghton, Joshua Tully and Sławomir Makowiec for fruitful discussions.
Author information
Authors and Affiliations
Contributions
M.S.: Methodology, Investigation, Formal analysis, Writing—original draft; P.P.: Resources, Writing—original draft; P.B.: Investigation; I.K.: Investigation, Formal analysis, Writing—original draft; M.P.: Investigation, Visualization; J.N.: Investigation; M.S.: Resources; S.F.-K.: Methodology; A.K.-D.: Resources; A.D.: Conceptualization, Investigation; Formal analysis; Supervision; Methodology, Visualization, Project administration, Funding acquisition, Writing—original draft, Writing—review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szopińska, M., Prasuła, P., Baran, P. et al. Efficient removal of 2,4,6-trinitrotoluene (TNT) from industrial/military wastewater using anodic oxidation on boron-doped diamond electrodes. Sci Rep 14, 4802 (2024). https://doi.org/10.1038/s41598-024-55573-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55573-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
