
Abstract
One drawback of existing artificial intelligence (AI)-based histopathological prediction models is the lack of interpretability. The objective of this study is to extract p16-positive oropharyngeal squamous cell carcinoma (OPSCC) features in a form that can be interpreted by pathologists using AI model. We constructed a model for predicting p16 expression using a dataset of whole-slide images from 114 OPSCC biopsy cases. We used the clustering-constrained attention-based multiple-instance learning (CLAM) model, a weakly supervised learning approach. To improve performance, we incorporated tumor annotation into the model (Annot-CLAM) and achieved the mean area under the receiver operating characteristic curve of 0.905. Utilizing the image patches on which the model focused, we examined the features of model interest via histopathologic morphological analysis and cycle-consistent adversarial network (CycleGAN) image translation. The histopathologic morphological analysis evaluated the histopathological characteristics of image patches, revealing significant differences in the numbers of nuclei, the perimeters of the nuclei, and the intercellular bridges between p16-negative and p16-positive image patches. By using the CycleGAN-converted images, we confirmed that the sizes and densities of nuclei are significantly converted. This novel approach improves interpretability in histopathological morphology-based AI models and contributes to the advancement of clinically valuable histopathological morphological features.
Similar content being viewed by others

Introduction
Oropharyngeal squamous cell carcinoma (OPSCC) is a cancer that affects the tonsils, root of the tongue, soft palate, and uvula1. Human papillomavirus (HPV) infection has recently emerged as an important risk factor for OPSCC1. The most recent American Joint Committee on Cancer (AJCC) staging system separates HPV-positive and HPV-negative OPSCC because of their different tumor characteristics and outcomes1,2.
p16 overexpression, as assessed by p16 immunohistochemistry (IHC), has emerged as a surrogate marker for HPV-mediated carcinogenesis and, therefore, as a way to identify HPV-positive oropharyngeal cancer2. While p16 IHC is considered a sufficient standalone test for HPV status, there remains discordance between the results of p16 IHC and DNA in situ hybridization or polymerase chain reaction3, with discordance rates ranging from 4 to 20%3. For this reason, in addition to p16 IHC, the American Society of Clinical Oncology (ASCO) guidelines state that HPV-specific testing may be performed at the discretion of the pathologist and/or treating clinician3. Therefore, recognizing the typical histopathological features of p16-positive cases is important for pathologists.
Clinically, HPV-positive OPSCC is associated with a more favorable prognosis than HPV-negative OPSCC1,4. Some trials are examining the potential for de-escalating the intensity of OPSCC treatments to improve quality of life while maintaining an acceptable survival rate1. However, clinical trials using HPV status as a single stratification biomarker cannot show the utility of such de-escalation treatments4. This is because HPV-positive patients cannot be considered a single group, as the characteristics of patients and the disease can vary5. For this reason, further stratification of OPSCC has been proposed6,7.
In recent years, artificial intelligence (AI) has been applied to various types of medical images, including histopathological images, to achieve largely improved diagnostic accuracy over human assessment8. In the field of histopathology, deep learning algorithms have been applied for tasks such as tumor detection, grading, subtyping, and biomarker prediction9,10,11,12,13,14,15. For HPV-positive OPSCC, some AI-based approaches have been developed for HPV infection prediction and prognosis4,16,17. Klein et al. reported a model for predicting the association of HPV infection with OPSCC using hematoxylin eosin (HE)-stained slides and indicated the utility of their model for stratifying patient prognosis4. However, attempts have been made to understand the histopathologic features that are important for AI prediction, but they are not clear.
The interpretability of AI prediction models is significant in the context of AI development and model evaluation18. The ability to interpret the basis for the model's decisions facilitates an assessment of whether the predictions rely on clinically relevant features. Furthermore, such interpretability enables the model to uncover novel features18. Various methods, such as gradient class activation mapping (Grad-CAM), have been proposed to visualize regions crucial for prediction8,18. These existing methods do not directly approach the important feature for prediction. Cycle-consistent generative adversarial network (CycleGAN) is an approach for unpaired image-to-image translation19. Specifically, when converting images, CycleGAN captures the features of one image group and translates the features of another image group20. In histopathological images, the utility of CycleGAN has been reported for stain normalization and stain transformation to special stains20,21,22. However, the effectiveness of CycleGAN in enhancing the interpretability of the AI model by employing it for the visualization of features on which the AI model focused has not been evaluated.
One problem with existing deep learning algorithms for making predictions from histopathological images is the heterogeneity of the tissue contained in a single slide13. That is, a single tissue slide usually contains an abundance of nontumor tissue in addition to tumor tissue, which dilutes the overall information content13. Two main approaches are available for handling the heterogeneity of tissue samples: fully supervised and weakly supervised approaches (Supplementary Table 1)23. Fully supervised approaches require the tumor region to be manually annotated on all slides used for training and testing, which imposes a heavy burden on the annotator23. In contrast, weakly supervised approaches reduce the annotation burden by labeling each slide with a single label instead of manually annotating the tumor region11. This type of approach also makes it possible to understand the predictive basis of the model by using images that the model focused on when making its predictions18. Using this predictive basis, we can confirm whether the trained model had captured reasonable features.
There is a drawback to the use of weakly supervised approaches, however. For small cancer datasets, weakly supervised approaches are uncapable of fully capturing the morphological variability of tumor tissue24. Indeed, the advantage of weakly supervised approaches over fully supervised approaches is that they enable training to be performed on large-scale datasets18,24. For example, in the weakly supervised learning of pathology images, Lu et al. reported clustering-constrained attention-based multiple-instance learning (CLAM)9,10. The CLAM model uses an attention mechanism to extract representative images for prediction, aiding in the interpretation of the features on which the AI model focused15. In one study using the CLAM model, a large-scale dataset with 22,833 slides was used for training, and the utility of the model for assessing the tumor origin for cancer of unknown origin was demonstrated9.
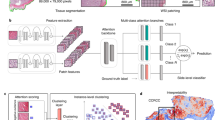
In this study, we aimed to extract interpretable histopathologic morphological features of p16-positive OPSCC by using an AI prediction model (Fig. 1, Supplementary Fig. 1). We used the weakly supervised CLAM model for the prediction. To improve the performance of CLAM on small datasets, we modified the model to use the annotated tumor area (Annot-CLAM). Furthermore, to confirm the predictive features in a form that is easy for pathologists to understand, we attempted to visualize the features that our prediction model focused on by utilizing CycleGAN image translation. Then, to examine the reason for the discrepancy in the p16 IHC results and our model prediction results, we evaluated histopathologic morphology and gene expression using the Cancer Genome Atlas (TCGA)-Head and Neck Squamous Cell Carcinoma (HNSC) dataset. Here, we describe a novel approach for interpreting the predictive basis of weakly supervised prediction models for histopathological images. This approach introduces a new direction to utilize generative adversarial network (GAN) for model interaction.

Overview of the study The Annot-CLAM model, a version of the CLAM model modified to use annotated ROIs, was applied. Two analysis approaches were used to interpret the features that the prediction model focused on.
Results
CLAM model performance and generated heatmaps
To predict p16 expression, we attempted to construct a model based on CLAM using the tissues of patients with OPSCC using the National Cancer Center Hospital East dataset. The dataset consisted of 116 whole-slide images (WSIs) from 114 primary OPSCC biopsy cases (50 p16-positive cases and 64 p16-negative cases). We first extracted 256 × 256 pixel patches at × 10, × 20, and × 40 magnification levels and trained CLAM with images from each magnification level separately (Supplementary Fig. 2). We evaluated the slide-level prediction performance using tenfold cross-validation; for each fold, the performance was evaluated using a test set after the training process was completed.
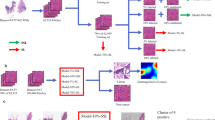
The mean area under the receiver operating characteristic (ROC) curve (AUC) in predicting p16 expression in OPSCC tissue ranged from 0.802 to 0.834 for CLAM models using patches with different magnifications from the whole tissue area (Table 1). The CLAM model produced interpretable heatmaps based on the contribution of each patch to the prediction process, examples of which are shown in Fig. 2a.

Heatmaps and highly predictive patches (a, b), Heatmaps obtained using the CLAM (a) and Annot-CLAM models (b). The heatmaps were generated by computing the attention score for the model prediction. The levels of attention given to the tumor areas by the CLAM and Annot-CLAM models differed. (c), Patches from p16-negative cases at 20 × magnification. The patches have low nuclear density and intercellular bridges. (d), Patches from p16-positive cases at 20 × magnification. The patches demonstrate high nuclear density and small nucleus size.
Annot-CLAM model performance and generated heatmaps
We assumed that the predictable patches would be mainly in the tumor cell areas. Thus, improve the performance of the model, we developed the Annot-CLAM model to use regions of interest (ROIs) in those areas. A pathologist manually annotated the tumor areas for all WSIs. The mean AUC of the Annot-CLAM models constructed with images of different magnifications using the annotated tumor areas ranged from 0.900 to 0.905, an improvement over the CLAM model results obtained using the whole tumor tissues (Table 1). The model using 20 × magnification was the best AUC. Thus, the model with magnification of 20 × was used for the rest of the study. The heatmaps indicate the differences between the levels of attention given to the tumor areas by the CLAM and Annot-CLAM models. Specifically, in the CLAM model, attention was focused on the tumor as a whole, whereas in the Annot-CLAM model, attention further stratified importance for prediction within the tumor area (Fig. 2b). Of note, the model performance improved by using the annotated tumor ROIs.
Comparison between the Annot-CLAM model and a review performed by pathologists
Given the performance of the Annot-CLAM model described above, we next sought to compare this performance with that of pathologists in predicting p16 IHC result using HE slides. We first created a dataset containing 30 cases (15 p16-negative cases and 15 p16-positive cases) distinct from the cases used for model training and testing. The Annot-CLAM model developed with the × 20 magnification images yielded an AUC of 0.871, accuracy (ACC) of 0.800, sensitivity of 0.800, and specificity of 0.800 in this new dataset; on the other hand, the pathologist assessment of the cases yielded an average ACC of 0.692, sensitivity of 0.633 and specificity of 0.750 (Supplementary Table 2). The ACC, sensitivity, and specificity of the Annot-CLAM model were higher than those of the pathologists.
External validation of the Annot-CLAM model
To validate our model, we used WSIs from the TCGA- HNSC project as an external and independent dataset25. A total of 22 oropharyngeal cancer cases used in this project underwent p16 IHC; among them, 17 yielded WSIs that were suitable for use in the model. The TCGA-datasets consisted of 12 p16-positive cases and 5 p16-negative cases. Similar to the steps performed in the comparison with the pathologists, we annotated the tumor areas of the slides and evaluated them using the Annot-CLAM model developed with the × 20 magnification images, yielding an AUC of 0.874, ACC of 0.824, and F1 score 0.889 (Supplementary Table 3). This result indicates that our Annot-CLAM prediction model can make predictions with external datasets.
Histopathologic morphological feature analysis of highly predictive patches
The CLAM and Annot-CLAM models calculate an attention score for each patch depending on its contribution to the prediction. We extracted the highly predictive patches—those that achieved high attention scores—using the Annot-CLAM models developed with the × 20 magnification images that achieved the best performance among the models (Fig. 2c,d). From each of the test cases predicted correctly using the top three models, 5 highly predictive patches were extracted, yielding a total of 140 patches (95 p16-negative patches and 45 p16-positive patches), whose characteristics were subsequently determined. The numbers of nuclei, the perimeters of the nuclei, and the intercellular bridges of the p16-negative and p16-positive patches were significantly different (all P < 0.001) (Table 2).
Feature evaluation using CycleGAN
Although the previous set of findings demonstrated the patch characteristics that were assessed by pathologists differed between p16-positive and p16-negative tissues, the features used by our model itself for prediction remain unknown. To interpret the features that our prediction model focused on, we evaluated the feature differences between p16-positive and p16-negative patches using CycleGAN image translation. The generator for converting between p16-positive and p16-negative patches was trained using the 1266 highly predictive patches (785 p16-negative patches and 481 p16-positive patches) extracted from the training cases predicted correctly using the top three models, and 5 highly predictive patches were extracted from each case. The feature-converted images obtained after 50 epochs of training are shown in Fig. 3. To evaluate the features changed in the patches converted using CycleGAN, we reviewed the converted features in the 140 patches used earlier for histopathologic morphological feature analysis. When p16-negative patches were converted to fake p16-positive-like patches, the mean numbers of nuclei (P < 0.001), the calipers of the nuclei (P < 0.001), the circularity of the nuclei (P < 0.001), the numbers of distinct nucleoli (P < 0.001), the intercellular bridges (P = 0.029), and the keratin pearls (P = 0.023) significantly differed. There were no significant differences in the mean numbers of nuclei, the calipers of the nuclei (P = 0.253), the circularity of the nuclei (P = 0.066), and the keratin pearls (P = 0.242) between the original p16-positive patches and the fake p16-positive-like patches (Table 3). When the original p16-positive patches were converted to fake p16-negative-like patches, the mean number of nuclei and the calipers of the nuclei were significantly changed (all P < 0.001), but these features were not significantly different between the original p16-negative patches and the fake p16-negative-like patches (Table 4). The mean number of nuclei and the calipers of the nuclei changed significantly in both directions of image conversion.

Image conversion results using CycleGAN. (a), Image conversion from p16-negative patches to p16-positive patches. The first row shows the original patches, and the second row shows the corresponding CycleGAN-converted patches. Small and dense nuclei were produced in the fake patches. (b), Image conversion from p16-positive patches to p16-negative patches. The number of nuclei decreased and the size increased in the fake patches.
Analysis of the incorrectly predicted cases using pathological review and clustering
The Annot-CLAM model was unable to correctly predict the labels for five cases (four p16-positive cases and one p16-negative case) in the National Cancer Center Hospital East test set. Compared to those of the correctly predicted cases, the numbers of nuclei, the perimeters of the nuclei, and the intercellular bridges of the incorrectly predicted p16-positive cases significantly differed (Supplementary Fig. 3, Supplementary Table 4). A previous report indicated the utility of the HPV prediction result using deep learning as a single biomarker with a favorable prognosis4. We assumed that our prediction result would correlate with prognostic factors such as gene expression. To determine the correlations between Annot-CLAM model prediction and gene expression, we performed clustering using the RNA sequence results obtained for the TCGA dataset. Three cases were found in which our model predicted false labels, all of which were p16 negative; of these, one was HPV positive. The result of hierarchical clustering is demonstrated in Fig. 4a. The p16-negative cases that the Annot-CLAM model correctly predicted were clustered into one cluster, while the p16-negative cases incorrectly predicted by the Annot-CLAM model as p16 positive were clustered separately. Principal component analysis (PCA) indicated that the cases were separated into two clusters by the Annot-CLAM model prediction method (Fig. 4b). PCA also showed that the p16-negative cases and those incorrectly predicted by the Annot-CLAM model were among the cluster of p16-positive cases. That is, the cases with a distinct histopathological morphology and which showed discrepancies between the p16 IHC results and Annot-CLAM predictions, showed different patterns of gene expression.

RNA sequence data from the TCGA dataset. (a), Hierarchical clustering and heatmaps. The top 100 variance genes were used for heatmap generation. The Euclidean distance and average variance methods were used to generate a hierarchical clustering dendrogram. (b), PCA plots presenting clusters of samples based on p16 IHC and Annot-CLAM prediction. p16-negative cases and those incorrectly predicted by the Annot-CLAM model were among the cluster of p16-positive cases.
Discussion
In this study, we aimed to extract features for predicting p16 expression in OPSCC tissue by using an AI model. To improve model performance, we constructed Annot-CLAM by modifying CLAM to use annotated ROIs. We interpreted the features that our prediction model focused on with two analytic approaches: a histopathologic morphological analysis of the patches with high attention scores and CycleGAN image translation analysis. Our analysis suggested that our prediction model possibly used the morphological features from histopathological images, especially the nucleus size and the number of nuclei, for p16 IHC prediction. CycleGAN could then be used to visualize these features. This visualization process is a novel and simple method for pathologists to visualize the features that the weakly supervised model focused on.
Nonkeratinizing squamous cell carcinoma has been reported as a histopathological feature of HPV-positive OPSCC26,27. However, it is difficult to predict HPV infections by using keratinization because only approximately 30% of HPV-positive OPSCC cases demonstrate keratinization27,28. In our study, the highly predictive patches indicated that the presence of an intercellular bridge, a feature of keratinization, was a significant factor in predicting p16 IHC. On the other hand, keratin pearls, another typical feature of keratinization, were not a significant factor. Our results revealed that the nuclear size and the density of the tumor cells were also important pathological features for p16 IHC prediction. Consistent with our findings, previous reports have stated that the nucleus size of HPV-related SCC is significantly smaller than that of non-HPV-related SCC, which is in accordance with the DNA ploidy of these tumors29,30. Overall, our results suggest that in addition to keratinization, nuclear size and density are key features for predicting p16 expression in IHC.
In this study, in addition to an attention network, we used CycleGAN to visualize the features that our prediction model focused on. Previous studies have reported methods for interpreting the reasons underlying model predictions8,15.Grad-CAM was reported as the method for revealing the factors that contribute to model prediction8,31. Grad-CAM highlights the important region for prediction, allowing CNN-based model to become more transparent by producing visual explanations27. The output of the important parts for prediction is different between CycleGAN and Grad-CAM, with Grad-CAM extracting important regions and CycleGAN extracting the important features themselves, not the regions. By using CycleGAN with feature translation, we confirmed the features itself extracted in the histopathologic morphological analysis. In addition, by showing the differences in features through visual information, pathologists, who routinely use visual information for diagnosis, will find it easier to recognize the differences. The use of the images produced using CycleGAN facilitates recognition of features and may improve pathologists' diagnosis; in other words, pathologists may be able to learn from the findings discovered by AI and improve their own diagnostic abilities.
From the results of our study, the histopathologic and morphologic features of incorrectly predicted p16-positive cases differed from those our model used for prediction, suggesting that there were histopathologic morphologically distinct cases. In addition, using TCGA datasets, we showed that cases with discrepancies between our predictions and the p16 IHC results had different gene expression levels from those of the correctly predicted cases. This result suggests that our histopathologic morphology-based prediction model likely focuses on features that are correlated with gene expression changes, which cannot be identified by IHC. Other studies have also investigated the relationship between histopathological morphology and molecular profile15,32; for example, Chen et al. reported a change in the highly predictive area when the molecular profile was combined with a WSI-based prognostic model15. Their findings indicated that histopathological morphology is correlated with the molecular profile, similar to the findings of our study. We believe that the advantage of AI-based histopathological evaluation lies in its ability to compare histopathological morphology with molecular features by objectively evaluating morphological features.
In this study, we employed CLAM model, a method of weakly supervised learning. Several methods of weakly supervised learning have been reported in the context of histopathological images18,33,34. Most weakly supervised learning models utilize methods such as multiple-instance learning and vision transformer18. Ghaffari Laleh et al. compared six weakly supervised learning pipelines, including the CLAM method, and reported that the accuracy of predictions varied depending on the task, with some models demonstrating superior accuracy to CLAM34. It is possible that other prediction models may yield more accurate results, and changes to the model may alter the important features for prediction. In many weakly supervised models, the relative importance of each image patch can be calculated18. Therefore, CycleGAN visualization used in this study could be applied to other weakly supervised learning models, potentially contributing to the improvement of interpretability of weakly supervised learning models for histopathological images.
This study had several limitations. First, the labels used for our model were the p16 IHC results, which, although suitable for clinical use, were in some instances incorrect, leading to false-negative and false-positive cases that affected the prediction results. For example, in the predictions obtained for the TCGA dataset, Annot-CLAM predicted positivity for p16 IHC-negative but HPV-positive cases. This means that our model was trained not on features related to p16 expression but on those related to HPV infection, which most p16-positive cases have. Labeling HPV results may therefore increase the accuracy of the model. Second, the slide images that we used were not color normalized, and therefore, we excluded features related to the color information. By using color-normalized images in the future, we may be able to improve the prediction models and identify useful color features in tissue slides.
In conclusion, we demonstrated important features for predicting p16 expression using interpretation of the AI mode. In the analysis approach using CycleGAN, features can be presented in an easily recognized form by pathologists through visualization. This is a novel approach for interpreting the predictive basis of weakly supervised prediction models for histopathological images by utilizing GAN for model interaction. This approach improves the interpretability of AI models that focus on histopathological morphology and contributes to the advancement of clinically valuable histopathological morphological features.
Methods
Patients and WSI datasets
We included 116 slides from 114 primary OPSCC samples for model training. These samples were obtained from patients who underwent biopsies between January 2018 and March 2021 at the National Cancer Center Hospital East. The inclusion criteria were as follows: histologically diagnosed squamous cell carcinoma from the biopsy sample and p16 IHC. The HE slides from the biopsy samples were scanned at 40 × magnification into digital slides using a NanoZoomer2.0HT digital slide scanner (Hamamatsu Photonics, Hamamatsu, Japan) and used as WSIs of this study.
p16 IHC was performed using mouse monoclonal anti-p16 antibody clone E6H4 (Roche Diagnostics, Mannheim, Germany). Subsequent steps were performed with the OptiView DAB IHC detection kit (Roche Diagnostics, Mannheim, Germany). We assessed the p16 status of each patient based on the nucleic p16 expression levels in 70% of the tumor cells in p16 IHC2. The p16 expression was evaluated by one pathologist and then double-checked by another pathologist. The p16 status was used for the label of this study.
The study was performed according to the Declaration of Helsinki and was approved by the institutional review board (IRB) of the National Cancer Center Hospital East (approval number 2022–142). Informed consent was waived by IRB of the National Cancer Center Hospital East. An overall flowchart of the study is presented in Fig. 1 and Supplementary Fig. 1.
Deep learning model
To verify model performance using a weakly supervised learning approach, we implemented the CLAM model, which is easy to use and performs digital pathology on WSIs (code available at https://github.com/mahmoodlab/CLAM)10. The CLAM model can make predictions using a dataset of slide images and their labels. It also uses an attention mechanism to produce the images on which the model focuses when making its predictions. For this reason, this model was used in this study.
When preprocessing the slide images, CLAM segments the tissue area, which is then cropped into small patches. To create the patches, in this study, each slide is cropped into nonoverlapping 256 × 256 pixel regions from segmented images at 10x, 20x, and 40 × magnification. The training was performed separately for each magnification patch. After patch creation, CLAM extracts features from each patch by encoding into a 1024 length one-dimensional feature vector. When encoding the patches, CLAM uses a convolutional neural network (CNN) of the imagenet pretrained ResNet50 architecture. During training, the model examines and ranks all patches, assigning an attention score to each patch, which informs its contribution or importance to the collective slide-level representation for prediction10. The attention score is reflected in the rule of attention-based pooling for slide-level prediction, which computes the slide-level representation as the average of all patches in the slide weighted by their respective attention score10. During training, CLAM learns from an additional learning task of clustering the top high- and bottom low-attention scored patches into distinct clusters10. The total loss for a slide is calculated by summing both the slide-level prediction loss and the patch-level clustering loss10. For model development and evaluation purposes, a tenfold Mote Carlo cross-validation strategy was implemented in which the training/validation/testing subsets were randomly derived from the entire cohort. Specifically, for each fold, the dataset was randomly split into training (60% of cases)/validation (10%)/testing (10%) sets. Performance was assessed using the AUC and ACC. The model was trained using the adaptive moment estimation (Adam) optimizer with a learning rate of 2 × 10–4. We used the default algorithm for the other parameters and did not perform data augmentation10. The training process ended at the 200th epoch if the validation loss did not decrease from its previous minimum for 20 consecutive epochs.
Attention heatmaps
The CLAM model calculates an attention score for each patch, allowing slide-level predictions. The attention scores are calculated by the attention branches that contribute to the prediction process and then converted between 0 and 1, with 1 being most predictive and 0 being least predictive10. Based on the attention scores, CLAM then produces heatmaps that allow the interpretation of the contribution of the tissue area to the model prediction process10. The attention scores are converted to RGB colors; patches that receive high attention scores are displayed in red (highly predictive), and patches with low attention scores are displayed in blue (less predictive). We tiled the slides into 256 × 256 pixel patches at the magnification level used for model development10.
Annot-CLAM model
When training on relatively small datasets, which is an issue for weakly supervised approaches, we considered that incorporating annotations could improve the performance of the model. Therefore, we developed a CLAM model with annotations (Annot-CLAM) to extract interpretable features more efficiently. In this study, we constructed the deep learning model using the ROIs of annotations based on the CLAM model. In the segmentation and patch image generation part, we modified the CLAM model. The segmentation process was modified to use the ROI of the annotated tumor area on each slide. The tumor area was annotated by one pathologist using QuPath version 0.3.2, a publicly available annotation tool for digital slides35.We used the same algorithm for the parameters as the CLAM model when developing Annot-CLAM.
Comparison of the performances of the annot-CLAM model and pathologists
We used 30 primary OPSCC biopsy cases (30 slides) from the National Cancer Center Hospital East as the dataset to compare the performance of Annot-CLAM with that of pathologists in identifying p16 expression from HE slides. The dataset consisted of 15 p16-positive cases and 15 p16-negative cases, all distinct from the cases used for model development. The HE slides were scanned into digital slides using a NanoZoomer2.0HT digital slide scanner and cropped into patches at 20 × magnification. The digital slides were assessed by the Annot-CLAM developed with 20 × magnification images with the top AUC. Eight pathology residents reviewed the slides contained in the dataset under a microscope. The analysis was performed blindly, and the assessment was based on the histopathologic morphological features of HPV-positive oropharyngeal cancer, as reported in the WHO classification26: (i) distinctive nonkeratinizing morphology, (ii) little surface dysplasia, (iii) growth beneath the surface epithelium lining as nests and lobules with central necrosis, (iv) tumor nests embedded in the lymphoid stroma, and (v) a high nuclear/cytoplasmic (N/C) ratio and high mitotic and/or high apoptotic ratios.
External validation of the model
To externally validate the performance of the model with the highest AUC—the one developed with 20 × magnification images—we used the data from the public TCGA-HNSC dataset. The inclusion criteria were as follows: oropharyngeal carcinoma; available p16 IHC; and available digital histological slides. The p16 IHC data were obtained from the work published by the TCGA network25. WSIs of HE-stained tissue were obtained from the TCGA-HNSC dataset. Each WSI was reviewed and annotated to create the ROI of its tumor area using QuPath version 0.3.235. The annotated tumor area was split into patches of 256 × 256 pixels at 20 × magnification. If the 20x-magnified image was not found in the dataset, 512 × 512 pixel patches were instead split from the 40x-magnified image and downscaled to 256 × 256 pixel patches.
Histopathologic morphological feature analysis of highly predictive patches
To compare the histopathological features between the highly predictive patches of p16-positive and p16-negative samples, the five highly predictive patches with the highest attention scores were extracted from each of the slides. We used the slides that were correctly predicted by the three models with the highest AUCs in the test dataset used for tenfold cross-validation. These patches were reviewed by pathologists, and the features that were objectively assessable and useful for predicting the p16 IHC results were selected. The following features were systematically recorded for each patch: the number of nuclei, the circularity of the nuclei, the maximum and minimum nucleus calipers, perinuclear halos, the number of distinct nucleoli, intercellular bridges, keratin pearls, and necrosis (Supplementary Fig. 4). The number of nuclei, the circularity of the nuclei, and the maximum and minimum nucleus calipers were measured using QuPath version 0.3.235. We also compared the patch features between correctly and incorrectly predicted slides.
Feature evaluation using CycleGAN
We applied CycleGAN to translate p16-negative and p16-positive patches19. CycleGAN is an approach for unpaired image-to-image translation19. Specifically, when converting images, CycleGAN captures the features of one image group and translates them into the features of another image group20. CycleGAN is a GAN method that relies upon an unsupervised approach19. We applied CycleGAN to capture the features from the highly predictive patches of p16-positive and p16-negative samples and visualize these features by translating them. To build the training dataset for CycleGAN, we first made predictions and calculated attention scores for all slides in the dataset using the three models with the top AUC values. We then extracted the five patches with the highest attention scores from the correctly predicted slides. Duplicate extracted patches were removed to create a dataset consisting of a total of 785 p16-negative patches and 481 p16-positive patches. Finally, we applied this dataset to the CycleGAN model19. The model is trained for 50 epochs, and the number of epochs is chosen based on model loss and learning efficiency. We evaluated the differences between the original images and the images produced by CycleGAN using the highly predictive patches extracted from each of the correctly predicted slides in the test dataset, which were used for the pathological review discussed above. We also compared the changes in the histopathological features between the original and artificially produced images.
RNA sequence data processing and clustering analysis
Read count data were obtained from the TCGA-HNSC dataset. We used single-stranded first RNA count data for the analysis. Normalization and analysis were performed using iDEP 0.9636. The read counts for all samples were normalized using EdgeR. Weakly expressed genes were excluded if they did not have more than 0.5 reads per million in at least three samples. We performed heatmap production, hierarchical clustering, and PCA using RNA count data. The top 100 variance genes were used for heatmap generation. The Euclidean distance and average variance methods were used to generate a hierarchical clustering dendrogram.
Environmental and statistical analysis
The analysis of this study was executed on an Ubuntu 20.04 Linux system with an A100 GPU (NVIDIA, Santa Clara, CA). All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria)37. A P value < 0.05 was considered to indicate statistical significance. We performed between-group comparisons using Fisher’s exact test and the t test for categorical and continuous variables, respectively. Image patches before and after CycleGAN conversion were statistically compared using the McNemar and paired t tests for categorical characteristics and continuous variables, respectively.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and that the source data for the figures are provided with this paper. The raw patient data are under restricted access for privacy reasons. Data from the TCGA, including digital histology and RNA sequence data, are available from https://portal.gdc.cancer.gov. The HPV infection and p16 IHC results are available from the published work of The Cancer Genome Atlas Network19 (https://doi.org/https://doi.org/10.1038/nature14129). All other results in support of this manuscript are available from the corresponding author upon reasonable request.
Code availability
Our code used for the CLAM prediction and CycleGAN image translation processes is available from https://github.com/epocaipath/Annot-CLAM-CycleGAN.
References
Lechner, M., Liu, J., Masterson, L. & Fenton, T. R. HPV-associated oropharyngeal cancer: epidemiology, molecular biology and clinical management. Nat. Rev. Clin. Oncol. 19, 306–327. https://doi.org/10.1038/s41571-022-00603-7 (2022).
AJCC Cancer Staging Manual, 8th Edition (eds Amin, M. B. et al.) (Springer, New York, 2017).
Fakhry, C. et al. Human papillomavirus testing in head and neck carcinomas: ASCO clinical practice guideline endorsement of the college of American pathologists guideline. J. Clin. Oncol. 36, 3152–3161. https://doi.org/10.1200/JCO.18.00684 (2018).
Klein, S. et al. Deep learning predicts HPV association in oropharyngeal squamous cell carcinomas and identifies patients with a favorable prognosis using regular H&E stains. Clin. Cancer Res. 27, 1131–1138. https://doi.org/10.1158/1078-0432.CCR-20-3596 (2021).
Petrelli, F. et al. Treatment de-escalation for HPV+ oropharyngeal cancer: A systematic review and meta-analysis. Head Neck 44, 1255–1266. https://doi.org/10.1002/hed.27019 (2022).
Gillison, M. L. et al. Tobacco smoking and increased risk of death and progression for patients with p16-positive and p16-negative oropharyngeal cancer. J. Clin. Oncol. 30, 2102–2111. https://doi.org/10.1200/JCO.2011.38.4099 (2012).
Wotman, M. T., Miles, B. A., Bakst, R. L. & Posner, M. R. A proposal for risk-based and strategy-adapted de-escalation in human papillomavirus-positive oropharyngeal squamous cell carcinoma. Cancer 127, 4330–4338. https://doi.org/10.1002/cncr.33851 (2021).
Komura, D. & Ishikawa, S. Machine learning approaches for pathologic diagnosis. Virchows. Arch. 475, 131–138. https://doi.org/10.1007/s00428-019-02594-w (2019).
Lu, M. Y. et al. AI-based pathology predicts origins for cancers of unknown primary. Nature 594, 106–110. https://doi.org/10.1038/s41586-021-03512-4 (2021).
Lu, M. Y. et al. Data-efficient and weakly supervised computational pathology on whole-slide images. Nat. Biomed. Eng. 5, 555–570. https://doi.org/10.1038/s41551-020-00682-w (2021).
van der Laak, J., Litjens, G. & Ciompi, F. Deep learning in histopathology: The path to the clinic. Nat. Med. 27, 775–784. https://doi.org/10.1038/s41591-021-01343-4 (2021).
Uegami, W. et al. MIXTURE of human expertise and deep learning-developing an explainable model for predicting pathological diagnosis and survival in patients with interstitial lung disease. Mod. Pathol. 35, 1083–1091. https://doi.org/10.1038/s41379-022-01025-7 (2022).
Echle, A. et al. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 124, 686–696. https://doi.org/10.1038/s41416-020-01122-x (2021).
Zeng, Q. et al. Artificial intelligence predicts immune and inflammatory gene signatures directly from hepatocellular carcinoma histology. J. Hepatol. 77, 116–127. https://doi.org/10.1016/j.jhep.2022.01.018 (2022).
Chen, R. J. et al. Pan-cancer integrative histology-genomic analysis via multimodal deep learning. Cancer Cell. 40(8), 865–878 (2022).
Wilde, D. C. et al. Oropharyngeal cancer outcomes correlate with p16 status, multinucleation and immune infiltration. Mod. Pathol. 35, 1045–1054. https://doi.org/10.1038/s41379-022-01024-8 (2022).
Koyuncu, C. F. et al. Computerized tumor multinucleation index (MuNI) is prognostic in p16+ oropharyngeal carcinoma. J. Clin. Invest. https://doi.org/10.1172/JCI145488 (2021).
Lipkova, J. et al. Artificial intelligence for multimodal data integration in oncology. Cancer Cell 40, 1095–1110. https://doi.org/10.1016/j.ccell.2022.09.012 (2022).
Zhu, J.-Y., Park, T., Isola, P. & Efros, A. A. Unpaired image-to-image translation using cycle-consistent adversarial networks. Preprint at https://doi.org/10.48550/arXiv.1703.10593 (2017).
Tschuchnig, M. E., Oostingh, G. J. & Gadermayr, M. Generative adversarial networks in digital pathology: A survey on trends and future potential. Patterns 1, 100089 (2020).
Shi, J. Y. et al. Exploring prognostic indicators in the pathological images of hepatocellular carcinoma based on deep learning. Gut 70, 951–961. https://doi.org/10.1136/gutjnl-2020-320930 (2021).
de Haan, K. et al. Deep learning-based transformation of H&E stained tissues into special stains. Nat. Commun. 12, 4884. https://doi.org/10.1038/s41467-021-25221-2 (2021).
Brendel, M. et al. Weakly-supervised tumor purity prediction from frozen H&E stained slides. EBioMedicine 80, 104067. https://doi.org/10.1016/j.ebiom.2022.104067 (2022).
Campanella, G. et al. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 25, 1301–1309. https://doi.org/10.1038/s41591-019-0508-1 (2019).
The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. https://doi.org/10.1038/nature14129 (2015).
WHO classification of Head and Neck Tumours, Fourth Edition (eds El-Naggar, F. T., Chan J.K.C., Grandis, J.R., Takata. T. & Slootweg, P.J.). (IARC Publications, Lyon, 2017).
Chernock, R. D., El-Mofty, S. K., Thorstad, W. L., Parvin, C. A. & Lewis, J. S. Jr. HPV-related nonkeratinizing squamous cell carcinoma of the oropharynx: utility of microscopic features in predicting patient outcome. Head. Neck. Pathol. 3, 186–194. https://doi.org/10.1007/s12105-009-0126-1 (2009).
Lewis, J. S. Jr. et al. Recognition of nonkeratinizing morphology in oropharyngeal squamous cell carcinoma—a prospective cohort and interobserver variability study. Histopathology 60, 427–436. https://doi.org/10.1111/j.1365-2559.2011.04092.x (2012).
Kotb, W. F. et al. Core classification of head and neck squamous cell carcinomas: correlations between morphology, DNA ploidy and HPV infection. Pathol. Res. Pract. 206, 768–771. https://doi.org/10.1016/j.prp.2010.07.011 (2010).
Kotb, W. F. & Petersen, I. Morphology, DNA ploidy and HPV in lung cancer and head and neck cancer. Pathol. Res. Pract. 208, 1–8. https://doi.org/10.1016/j.prp.2011.10.009 (2012).
R.R. Selvaraju, et al. Grad-CAM: Visual explanations from deep networks via gradient-based localization. Preprint at https://doi.org/10.48550/arXiv.1610.02391 (2016).
Komura, D. et al. Universal encoding of pan-cancer histology by deep texture representations. Cell Rep. 38, 110424. https://doi.org/10.1016/j.celrep.2022.110424 (2022).
Wang, X. et al. Transformer-based unsupervised contrastive learning for histopathological image classification. Med. Image Anal. 81, 102559 (2022).
Ghaffari Laleh, N. et al. Benchmarking weakly-supervised deep learning pipelines for whole slide classification in computational pathology. Med. Image Anal. 79, 102474 (2022).
Bankhead, P. et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878. https://doi.org/10.1038/s41598-017-17204-5 (2017).
Ge, S. X., Son, E. W. & Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics 19, 534. https://doi.org/10.1186/s12859-018-2486-6 (2018).
Kanda, Y. Investigation of the freely available easy-to-use software “EZR” for medical statistics. Bone Marrow Transp. 48, 452–458. https://doi.org/10.1038/bmt.2012.244 (2013).
Acknowledgements
This work was supported by JSPS KAKENHI (grant numbers JP20K22859 and JP21K06899) and the National Cancer Center Research and Development Fund, Grant/Award Number: 2021-A-7. The authors thank AJE (https://www.aje.com/) for scientific editing.
Author information
Authors and Affiliations
Contributions
M.A. and S.S. were responsible for the concept proposal and study design. M.A. and A.H. performed essential programming work. T.T., N.S., M.K., K.M., R.H. and K.T. contributed to the data interpretation process and statistical approaches. S.I. and G.I. supervised the research. The first draft of the manuscript was written by M.A., and all authors commented on the previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Adachi, M., Taki, T., Sakamoto, N. et al. Extracting interpretable features for pathologists using weakly supervised learning to predict p16 expression in oropharyngeal cancer. Sci Rep 14, 4506 (2024). https://doi.org/10.1038/s41598-024-55288-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-55288-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
