
Abstract
The aim of this study was to investigate the variation in gene expression in the complete transcripts of Congenitalpulmonary airwaymalformation (CPAM) of the lung using Next Generation Sequencing (NGS) technology. There were 20 cases involving children with CPAM were used for selection of study sample. NGS was used to establish RNA-Seq libraries for the two groups of samples separately, and both groups were conducted to differential expression analysis and Gene Ontology (GO) functional enrichment analysis. The pathways of the differential genes were analyzed to find the enriched target pathways. A total of 592 genes were expressed with significant differences (CPAM vs. normal tissue, P < 0.05). GO functional analysis of DEGs indicated that abnormal ciliary function played a role in the development of CPAM. Subsequently, analysis of these genes pathways showed the TGF-β signaling pathway was significantly enriched. Finally, the results of immunohistochemical analysis of some DEGs showed that a significant reduction in the expression of SMAD6, a gene related to the TGF-β signaling pathway, led to abnormal activation of the pathway. TGF-β signaling pathway involved in the evolution of the disease obtained by DEGs enrichment pathway analysis. SMAD6, a gene involved in this pathway, might be a potential biomarker for the diagnosis and treatment of CPAM.
Similar content being viewed by others


Introduction
Congenital pulmonary malformation (CPAM) is a rare lung malformation, and accounts for approximately 30–40% of congenital lung disease and 1/25,000 to 1/35,000 in live births1,2. It is characterized by an unusual airway patterning and different degrees of pseudo-cystic dilatation of airways with unclear nosogenesis3. It leads to defective development of fetal lungs with mediastinal shift, neonatal respiratory distress, pulmonary edema, lung infections, and even the development of malignant tumors4. However, as prenatal diagnostic techniques continue to progress, the rate of their detection has been increasing5. Most patients with CPAM are detected at prenatal diagnosis or in the neonatal period6. Currently, routine prenatal US screening complemented with magnetic resonance imaging (MRI) has become increasingly valuable in detecting CPAMs, with an accuracy of 65–91%7.
With the rapid development of genomics and bioinformatics, a large amount of genomic data is being analyzed. Due to the complex pathophysiology and the heterogeneity of cells in lung tissues, it is necessary to have a comprehensive insight into the mRNA expression of CPAM. Many researchers have devoted themselves to the study of CPAM, several factors such as fibroblast growth factor-10 (FGF-10)8, FGF-29, transforming growth factor-β (TGF-β)10 and other factors associated with lung development have been identified. However, the genetic landscape of CPAM is poorly known compared to other congenital lesions, and its introduction into biologically-driven risk stratification approaches and new drug development has been relatively slow.
This study aimed to analyze the genomic characteristics and the target signaling pathway involved in the development of CPAM. Potential biomarkers were also explored in the development of CPAM.
Material and methods
The study was approved by The Third Affiliated Hospital of Guangzhou Medical University (NO. 202030). All described experiments were performed in accordance to the relevant guidelines and regulations set forth by the IRB. Written informed consents were obtained from both groups.
Sample collection
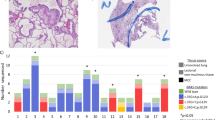
We collected tissue samples from a total of 20 patients, including 10 cases of histopathologically confirmed CPAM and 10 cases of normal lung tissue (NL) adjacent to other lung lesions from patients who underwent surgery due to pulmonary diseases since 2019. In case 4, a 3-month-old male infant were diagnosed with a suspected right-sided CPAM based on an abnormal 20-week prenatal ultrasound. A chest computed tomography (CT) scan was performed prior to surgery (Fig. 1A), revealing a massive right-sided cystic lesion measuring 63 × 52 × 49 mm, along with poor ventilation of the remaining lungs. The patient underwent a thoracoscopic procedure, wherein the dorsal and basal segments of the right lower lobe were excised (Fig. 1B). Postoperative histopathological examination confirmed the diagnosis of CPAM.

In case 4, a 3-month-old male infant were diagnosed with a suspected right-sided CPAM based on an abnormal 20-week prenatal ultrasound. A chest computed tomography (CT) scan was performed prior to surgery (A), revealing a massive right-sided cystic lesion measuring 63 × 52 × 49 mm, along with poor ventilation of the remaining lungs. The patient underwent a thoracoscopic procedure, wherein the dorsal and basal segments of the right lower lobe were excised (B).
NL samples were acquired from patients who underwent surgery for non-malignant lung conditions, with histopathological confirmation of normal lung tissue. Tissue samples were immediately preserved using two different methods: Rapid freezing in liquid nitrogen and storage at − 80 °C. Fixation in formalin followed by paraffin embedding for histological analysis.
RNA seqencing
The 6 groups of samples collected in the early stage were offered RNA sequencing. The total RNA extraction was performed using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Subsequently, the concentration and purity of the extracted RNA were assessed using a Nanodrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). RNA quality standards were set as follows: a quantity greater than 5 µg and a concentration of at least 200 ng/µL. The RNA integrity number (RIN) value, exceeding 7, was determined using an Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA).
To eliminate ribosomal RNA (rRNA) from the total RNA samples, the Ribo-Zero Gold rRNA removal kit (Illumina) was employed. Subsequently, the RNA was fragmented into 200 bp fragments using the total RNA SEQ (H/M/R) library prep kit (Illumina, San Diego, CA, USA). The fragmented RNA was then used for complementary DNA (cDNA) synthesis. After purification, end repair, adapter ligation, A-tail addition, product purification, and fragment size sorting, library amplification was carried out. Following amplification, the RNA SEQ library underwent purification and recovery using magnetic beads. The constructed library was subjected to inspection, and the qualified libraries were pooled based on data size and effective cDNA concentration.
The sequencing of the library was performed on the IlluminaNovaSeq 6000 platform in paired-end 150-bp mode. Raw data underwent filtration to eliminate low-quality reads and reads containing adapter or Poly-N sequences. Clean reads were then aligned to the Ribosomal Database Project using Bowtie, and reads corresponding to ribosomal DNA (rRNA) were subsequently removed. The reads mapped to the reference sequence were utilized for mapping, sequence prediction, calculating expression values, and conducting expression difference analyses.
Differential gene expression analysis
With the utilization of the R package limma, we detected a total of 592 differentially expressed genes (DEGs) in CPAM. Among these, 572 genes exhibited upregulation, while 20 were observed to be downregulated (Fig. 2A, B).

Differentially expressed genes (DEG) analysis. (A) The Volcano Plot illustrates the relationship between gene expression fold change and significance, highlighting significantly upregulated (red) and downregulated (blue) genes; (B) the Heatmap displays gene expression levels across various conditions, with red colors indicating higher expression and blue colors indicating lower expression.
GO and KEGG pathway analysis of DEGs
GO (geneontology.org) and KEGG (genome.jp/kegg) analyses of differentially expressed lncRNA-associated genes were performed using the online Database for Annotation, Visualization and Integrated Discovery tool. The top 10 enriched GO terms among the two groups were identified. GO analysis results consisted of ‘biological process’ (BP), ‘cell composition’ (CC) and ‘molecular function’ (MF). The adjusted P value was also obtained using the Benjamini & Hochberg method and an adjusted P < 0.05 was considered to indicate a statistically significant difference. The pathways associated with lncRNA-targeted mRNAs were identified by KEGG pathway analysis11.
Quantitative real-time PCR
RNA extraction from 5 CPAM samples and 5 normal lung tissues using TRIzol (Tel-Test, Friendswood, Tex). cDNA synthesis from 1 μg RNA using Superscript III (Invitrogen, Carlsbad, Calif). Real-time quantitative PCR was performed on cDNAs using the Quanti-Tect SYBR Green PCR Kit (Qiagen, Valencia, Calif) and an ABI Prism 7000 (Applied Biosystems, Foster City, Calif). The following primers were used:
SMAD6-F:AGTGACTGCGAGACGGTGACC R:GCAGCGAGTACGTGACGGTTT;
RANBP9-F: ACGAGCAGGAGAAGGAGTTGC R: TGGTATTGGATGCGTGGCTCG;
DNAH2-F: GCTGTCAGTGAGCCAGAGCTG R: CTGTGTCCACACTGCATCCGC;
DNAH11-F: CGGAATTCCCACAGACACGCA R: CAGCCACCTGCACCTTTTCCA;
CFAP43-F: TTGTGGGCGTCATGGCAACTA R: GCCAGGTAGGTGCCACAGTAA;
CFAP46-F: CCCATCACCCAGTTTCTGGGC R: CTTGAGGAACGGCCTCACCAT;
RP1-F: AAGAGCGGAGACCCCCAATTC R: TAGGTAGGACTCGCCGTCCTC;
CFAP77-F: ATCCGTTCCGGCATGGAGAAC R: GATGGCTTCAGGGACTCCTCC;
RSPH4A-F: GCCAGAAACTGGACGCCAGTC R: CTCGTGGTCCTGTCCGACTGA;
DRC3-F: CCGAGGGTGATGGACGATGAC R: CTGGGAATCCTCCTGGCCATC.
Relative abundance of RNA was calculated by the ΔΔCt method. Primers were designed using Primer Express v2.0 (Applied Biosystems, Foster City, Calif). All primers were between 90 and 110% efficient, as assessed by standard curve, and all displayed only 1 dissociation peak.
Immunohistochemistry
The remaining portion of the sample was analyzed utilizing immunohistochemistry. Immunohistochemical staining procedures were carried out in accordance with the manufacturer's provided instructions. Primary antibodies utilized for staining were as follows: rabbit anti-SMAD6 (dilution 1:100, Abcam, Cambridge, UK), rabbit anti- InhibinB (dilution 1:100, Abcam, Cambridge, UK), and rabbit anti-GDF7 (dilution 1:2000, Abcam, Cambridge, UK).
The mean optical density of positively stained cells was quantified using ImageJ software (version 1.53e). This quantification was based on the optical density of stained positive cells, which was then normalized to the total cell area within each field of view at 200× magnification.
Statistical analysis
Statistical analyses were conducted utilizing GraphPad Prism (version 7) and R packages. For comparisons between independent groups, the Student's t test was applied, while paired groups were assessed using the paired t test. Continuous variable results were expressed as mean ± standard deviation. A significance threshold of P ≤ 0.05 was used to determine statistical significance.
All the data collected in this study were analyzed using GraphPad Prism (version 7) and R software 4.0. Normally distributed measurement data were expressed as mean ± standard deviation (SD), and the comparisons were examined by Student-t test. P < 0.05 was considered statistically significant.
Ethics approval and consent to participate
The study was approved by The Third Affiliated Hospital of Guangzhou Medical University. (NO. 202030). All described experiments were performed in accordance to the relevant guidelines and regulations set forth by the IRB. Written informed consents were obtained from all patients.
Results
Identification of differentially expressed genes in CPAM
All cases of CPAM with detailed clinical information were recorded in Table 1. In this study, we conducted RNA-seq analysis on CPAM tissue samples and normal lung tissues, identifying a total of 592 differentially expressed genes (|FoldChange|> 2 and q-value < 0.05. DEGs included both upregulated (572) and downregulated (20) genes in CPAM compared to normal lung tissues (Supplementary table 1).
The gene expression patterns were represented by heat maps (Fig. 2A), and the distinct gene expression profiles in CPAM were displayed by volcano plots (Fig. 2B).
GO and KEGG function enrichment analyses for the differentially expressed genes
GO enrichment and KEGG pathway analyses were conducted on the 592 DEGs, employing an adjusted P value threshold of < 0.05 for screening (Supplementary table 2). In the CC enrichment analysis, these genes exhibited significant enrichment in signaling pathways related to 'cillium' and 'cilliary part' pathways. Regarding BP, the genes were prominently associated with responses to 'cillium movement,' 'axoneme assembly,' and 'cillium organization.' In terms of MF, the genes were primarily involved in binding activities, especially 'heme binding' (Fig. 3A, B. Additionally, KEGG pathway analysis highlighted the 'TGF-β' and 'cAMP' signaling pathways as the most pertinent pathways linked to CPAM (Fig. 3C, D).

GO and KEGG analysis. (A) GO enrichment bubble plot; (B) Top 20 GO enrichment terms; (C) KEGG pathway enrichment; (D) pathway functional classification.
Validation of ciliated epithelium and TGF-β signal pathway related gene in CPAM
Based on the transcriptome sequencing results, which indicated elevated expression of and dysregulated expression of TGF-β signal pathway in CPAM (Supplementary table 3), further validation experiments were conducted to provide additional support for these findings in tissue samples (Figs. 4A, 5A). We assessed the expression of a panel of genes associated with ciliated epithelium in CPAM tissues. Including DNAH2, DNAH11, CFAP43, CFAP46, RP1, CFAP77, RSPH4A, DRC3, exhibited upregulation in CPAM compared to normal lung tissues (Fig. 4B). These findings suggest these intriguing findings strongly suggest a potential association between the dysregulation of ciliated epithelium-related genes and the pathogenesis of CCAM.

Epithelial gene expression. (A) The heatmap displays expression of epithelial-related genes in Non-Lesion (NL) and Lesion (LA) conditions. Rows represent genes, columns represent samples. (B) Validation of mRNA expression in tissue samples for epithelial-related genes.

TGF-β signaling pathway differential expression. (A) The heatmap displays differential expression of genes in the TGF-β signaling pathway; (B) validation of mRNA expression in tissue samples for SMAD6; (C) results of the representative immunohistochemistry staining showing the protein level expressions of SMAD6 in NL and LA tissues.
Our investigation also included an examination of genes involved in the TGF-β signaling pathway within CPAM tissues. We observed in the expression of TGF-β signal pathway-related genes, such as SMAD6, when comparing CPAM to adjacent normal lung tissues (Fig. 5B). The downregulation of SMAD6, a critical negative regulator that inhibits the transmission of TGF-β signals, implies the potential for excessive activation of the TGF-β signaling pathway, thesubsequently influencing cell function and tissue development.
Immunohistochemical analysis of TGF-β signal pathway related genes expression in CPAM
Immunohistochemical analysis was conducted to assess the expression of SMAD6, GDF7, and InhibinB in CPAM tissues, with a particular focus on their potential involvement in the TGF-β signaling pathway. The results revealed a significant decrease in SMAD6 expression, which was a critical negative regulator of the TGF-β signaling pathway. This downregulation of SMAD6 implied a potential for excessive activation of the TGF-β pathway in CPAM (Fig. 5C).
While GDF7 exhibited a slight increase in expression, but without statistical significance, suggesting a possible association with the TGF-β pathway that warrants further investigation (Fig. 6A). In the case of InhibinB, there was a decreasing trend in expression within CPAM tissues, although statistical significance was not achieved (Fig. 6B). This trend may suggest a role for InhibinB in modulating the TGF-β signaling pathway in CPAM, but additional studies are required to confirm this.

TGF-β signaling pathway related genes expression. (A) Results of the representative immunohistochemistry staining showing the protein level expressions of GDF7 in NL and LA tissues; (B) results of the representative immunohistochemistry staining showing the protein level expressions of InhibinB in Non-Lesion (NL) and Lesion (LA) tissues.
Discussion
Our study has unveiled a comprehensive overview of the molecular alterations present in Congenital pulmonary airway malformation (CPAM). Through RNA-seq analysis, we identified a substantial number of differentially expressed genes (DEGs) between CPAM and normal lung tissues. These DEGs, comprising both upregulated and downregulated genes, provide a foundation for understanding the underlying genetic landscape of CPAM.
Congenital pulmonary airway malformation has five major subtypes that have been described and assigned by the Stocker classification. Each type originates from a different part of the bronchial tree, subsequently leading to distinct histopathological differentiation, clinical features, malignant potential, and prognosis1,12. The overall prognosis for congenital pulmonary airway malformation, when diagnosed prenatally, is excellent. Infection is the most common known complication of congenital pulmonary airway malformation and usually occurs in the first few years of the lives of asymptomatic infants, especially if surgical resection does not take place. The most commonly known malignant complication associated with CPAM is pleuropulmonaryblastoma13. Currently, routine prenatal US screening complemented with magnetic resonance imaging (MRI) has become increasingly valuable in detecting CPAMs, with an accuracy of 65–91%7. Imaging evaluation and clinical follow-up after birth is required in all cases to confirm the diagnosis and to initiate adequate treatment14. Prenatal treatment options include the maternal administration of steroids, minimally invasive procedures or open foetal surgery.
Functional enrichment analyses, including Gene Ontology (GO) and KEGG pathway analyses, have shed light on the potential biological processes and pathways that are dysregulated in CPAM. Notably, the enrichment of ciliated epithelium-related pathways and the TGF-β signaling pathway underscores their potential roles in CPAM pathogenesis.
Furthermore, our validation studies have confirmed the upregulation of ciliated epithelium-associated genes, strengthening the hypothesis that aberrant ciliated epithelium function may contribute to the development of CPAM. Additionally, the downregulation of SMAD6, a critical negative regulator of the TGF-β pathway, suggests the possibility of TGF-β pathway dysregulation in CPAM. The upregulation of genes associated with ciliated epithelium suggests that abnormalities in cilia function may play a role in CPAM development. Dysfunctional cilia can disrupt normal respiratory processes and contribute to lung developmental disorders. Among them, such genes as Dnah2, DNAH11, CFAP43, CFAP77, RSPH4A, and DRC3 play important roles in the driving and regulation of cilia movement15,16,17,18,19,20. The continuous coordinated beating of cilia transports mucus and foreign matter out of the airway to help maintain a sterile environment in the airways, and it takes an essential part in protecting the respiratory tract. Deletion or mutation of one of these genes can lead to alterations in ciliary ultrastructure and ciliary motility, which will lead to poor fiber motility and clearance dysfunction, resulting in severe accumulation of respiratory mucus, eventually leading to inflammation and affecting lung function and development. CFAP46 has also been reported to be associated with airway and lung diseases by modulating inflammatory responses21. Moreover, a study showed that the downregulation of ACSL5 and Wnt2B could play an important role in the development of bronchial-alveolar structures in CPAM22.
The downregulation of SMAD6, a key negative regulator of the TGF-β signaling pathway, implies excessive activation of this pathway in CPAM17. Dysregulated TGF-β signaling can influence cell proliferation, differentiation, and extracellular matrix production, potentially contributing to CPAM pathogenesis18. A study showed that TGF-β signaling in prenatal lung mesenchyme is essential for lung development and maturation, and defective TGF-β signaling in lung mesenchyme may be related to abnormal airway branching morphogenesis and congenital airway cystic lesions23. Our study lays the foundation for future investigations into CPAM. Detailed functional studies are needed to explore the precise roles of upregulated ciliated epithelium-associated genes in CPAM pathogenesis. Further investigations into the TGF-β signaling pathway in CPAM are essential. This could include assessing downstream signaling events, identifying potential therapeutic targets within the pathway, and elucidating the mechanisms responsible for SMAD6 downregulation. Exploring genetic and epigenetic factors contributing to CPAM susceptibility could provide valuable insights. Our study holds significant implications for the understanding and potential treatment of CPAM. By identifying dysregulated genes and pathways, we offer a stepping stone towards targeted therapies for this congenital lung disorder. Improved clinical management and interventions may arise from unraveling the precise molecular mechanisms driving CPAM.
Conclusion
In conclusion, this study serves as a crucial initial exploration of the molecular landscape of CPAM, and the downregulation of SMAD6, a critical negative regulator of the TGF-β pathway, suggested the possibility of TGF-β pathway dysregulation in CPAM. In addition, the upregulation of ciliated epithelium-associated genes, strengthening the hypothesis that aberrant ciliated epithelium function may contribute to the development of CPAM.
Limitation
This study is limited by the relatively small sample size; larger cohorts were considered in the future to enhance the generalizability of the findings. In addition, more validation experiments of Smad-6 in TGF signal pathway in children with CPAM was need.
Data availability
The data could be shared after publication upon request for use via communication with the corresponding authors.
Abbreviations
- CPAM:
-
Congenital pulmonary malformation
- cDNA:
-
Complementary DNA
- DEGs:
-
Differentially expressed genes
- FGF-10:
-
Fibroblast growth factor-10
- GO:
-
Gene ontology
- NGS:
-
Next Generation Sequencing
- NL:
-
Normal lung tissue
- qRT-PCR:
-
Quantitative reverse transcriptase polymerase chain reaction
- RIN:
-
RNA integrity number
- rRNA:
-
Ribosomal RNA
- TGF-β:
-
Transforming growth factor-β
References
Leblanc, C. et al. Congenital pulmonary airway malformations: State-of-the-art review for pediatrician’s use. Eur. J. Pediatr. 176(12), 1559–1571 (2017).
Szaflik, K. et al. Fetal therapy–evaluation of intrauterine therapy in congenital cystic adenomatoid malformation of the lung (CCAM). Ginekol. Pol. 86(1), 8–15 (2015).
Boucherat, O., Jeannotte, L., Hadchouel, A., Delacourt, C. & Benachi, A. Pathomechanisms of congenital cystic lung diseases: Focus on congenital cystic adenomatoid malformation and pleuropulmonary blastoma. Paediatr. Respir. Rev. 19, 62–68 (2016).
Hamanaka, R., Yagasaki, H., Kohno, M., Masuda, R. & Iwazaki, M. Congenital cystic adenomatoid malformation in adults: Report of a case presenting with a recurrent pneumothorax and a literature review of 60 cases. Respir. Med. Case Rep. 26, 328–332 (2019).
Hardee, S. et al. Congenital cystic lung lesions: Evolution from in-utero detection to pathology diagnosis-a multidisciplinary approach. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 20(5), 403–410 (2017).
Mann, S., Wilson, R. D., Bebbington, M. W., Adzick, N. S. & Johnson, M. P. Antenatal diagnosis and management of congenital cystic adenomatoid malformation. Semin. Fetal Neonatal. Med. 12(6), 477–481 (2007).
Tsai, A. Y. et al. Outcomes after postnatal resection of prenatally diagnosed asymptomatic cystic lung lesions. J. Pediatr. Surg. 43(3), 513–517 (2008).
Bellusci, S., Grindley, J., Emoto, H., Itoh, N. & Hogan, B. L. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development (Cambridge, England) 124(23), 4867–4878 (1997).
De Moerlooze, L. et al. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development (Cambridge, England) 127(3), 483–492 (2000).
Peng, D., Fu, M., Wang, M., Wei, Y. & Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 21(1), 104 (2022).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Fowler, D. J. & Gould, S. J. The pathology of congenital lung lesions. Semin. Pediatr. Surg. 24(4), 176–182 (2015).
Wong, A., Vieten, D., Singh, S., Harvey, J. G. & Holland, A. J. Long-term outcome of asymptomatic patients with congenital cystic adenomatoid malformation. Pediatr. Surg. Int. 25(6), 479–485 (2009).
Raychaudhuri, P., Pasupati, A., James, A., Whitehead, B. & Kumar, R. Prospective study of antenatally diagnosed congenital cystic adenomatoid malformations. Pediatr. Surg. Int. 27(11), 1159–1164 (2011).
Rachev, E. et al. CFAP43 modulates ciliary beating in mouse and Xenopus. Dev. Biol. 459(2), 109–125 (2020).
Dougherty, G. W. et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am. J. Respir. Cell Mol. Biol. 55(2), 213–224 (2016).
Zhang, Y. et al. Vertebrate Dynein-f depends on Wdr78 for axonemal localization and is essential for ciliary beat. J. Mol. Cell Biol. 11(5), 383–394 (2019).
Kubo, S. & Black, C. S. Native doublet microtubules from Tetrahymena thermophila reveal the importance of outer junction proteins. Nat. Commun. 14(1), 2168 (2023).
De Jesús-Rojas, W. & Meléndez-Montañez, J. The RSPH4A gene in primary ciliary dyskinesia. Int. J. Mol. Sci. 24(3), 1936 (2023).
Song, K. et al. In situ localization of N and C termini of subunits of the flagellar nexin-dynein regulatory complex (N-DRC) using SNAP tag and cryo-electron tomography. J. Biol. Chem. 290(9), 5341–5353 (2015).
Li, L. & Holloway, J. W. Newborn DNA methylation and asthma acquisition across adolescence and early adulthood. Clin. Exp. Allergy 52(5), 658–669 (2022).
Qu, Y., Liu, D., Jia, H. & Zhou, X. Expression analysis of ACSL5 and Wnt2B in human congenital pulmonary airway malformations. J. Surg. Res. 232, 128–136 (2018).
Miao, Q. et al. Abrogation of mesenchyme-specific TGF-β signaling results in lung malformation with prenatal pulmonary cysts in mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 320(6), L1158–L1168 (2021).
Funding
This project was funded by Science and Technology Projects in Guangzhou (202201020395), and Guangdong Provincial Science and Technology Plan Project (2014A020213021).
Author information
Authors and Affiliations
Contributions
G.Z., R.W. and D.L. participated in study design; G.Z., L.L., L.S., H.Z., C.C. and D.L. performed laboratory experiments and data analysis; G.Z., L.L., L.S., H.Z., C.C., R.W. and D.L. recruited patients; G.Z., L.L., L.S. and D.L. drafted the manuscript; R.W. and D.L. provided critical review of the manuscript. All authors approved the final draft for publication.
Corresponding author
Ethics declarations
Competing interests
All authors have no potential conflicts of interest with any companies/organizations whose products or services may be discussed in this article.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, G., Lou, L., Shen, L. et al. The underlying molecular mechanism of ciliated epithelium dysfunction and TGF-β signaling in children with congenital pulmonary airway malformations. Sci Rep 14, 4430 (2024). https://doi.org/10.1038/s41598-024-54924-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54924-x
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
