
Abstract
Canine bufavirus (CBuV) was reported in domestic dogs worldwide. We conducted a survey of canine bufavirus in domestic dogs in Thailand from September 2016 to October 2022. Rectal swab samples (n = 531) were collected from asymptomatic dogs and dogs with gastroenteritis signs. The samples were tested for CBuV using PCR with specific primers to the VP1/VP2 gene, and 9.42% (50/531) was CBuV positive. Our findings showed that CBuVs could be detected in both symptomatic and healthy dogs. The Thai CBuVs were found in dogs from different age groups, with a significant presence in those under 1 year (12.60%) and dogs aged 1–5 years (7.34%) (p < 0.05), suggesting a high prevalence of Thai CBuVs in dogs under 5 years of age. We performed complete genome sequencing (n = 15) and partial VP1/VP2 sequencing (n = 5) of Thai CBuVs. Genetic and phylogenetic analyses showed that whole genomes of Thai CBuVs were closely related to Chinese and Italian CBuVs, suggesting the possible origin of Thai CBuVs. The analysis of VP1 and VP2 genes in Thai CBuVs showed that 18 of them were placed in subgroup A, while only 2 belonged to subgroup B. This study is the first to report the detection and genetic characterization of CBuVs in domestic dogs in Thailand. Additionally, surveillance and genetic characterization of CBuVs in domestic animals should be further investigated on a larger scale to elucidate the dynamic, evolution, and distribution of CBuVs.
Similar content being viewed by others


Introduction
Bufavirus (BuV) is a small, non-enveloped, non-segmented, single-stranded linear DNA virus with a genome size of 4.5–4.8 kb. The BuV is a novel virus of the family Parvoviridae, genus Protoparvovirus, which are common pathogens in many animals, including birds and mammals1. It contains 2 major open reading frames: ORF1 encoding nonstructural protein 1 (NS1) and ORF2 encoding viral structural protein 1 and 2 (VP1 and VP2)2. BuV was first reported in the fecal samples from children with diarrhea in Burkina Faso in 20123. Subsequently, BuV infections have been reported in diarrhea patients in several countries such as Bhutan, China, France, Finland, Netherlands, South Africa, Thailand and Turkey4,5,6,7,8,9,10,11. The occurrence of bufavirus infection in humans in those counties ranges from 0.3 to 4%5,12,13. These studies speculated that BuV might cause gastroenteritis in human6,7,14 BuV is not strictly found in humans but it has also been reported in several animal species such as rats, shrews, pigs, dogs, bats, and primates15,16,17,18,19.
In 2016, the first report of Canine Bufavirus (CBuV) in dogs in Italy revealed its presence in both dogs with gastroenteric and respiratory symptoms, as well as in asymptomatic dogs. The study reported that the genome of CBuV was closely related to human bufavirus (HBuV)20. However, CBuV was genetically distinct from other canine enteric viruses of the Parvoviridae family (canine bocavirus (CBoV) and canine parvovirus type 1 and 2 (CPV-1and CPV-2))21,22. After 2016, CBuV has been reported in dogs in Canada, China, India, and Italy20,23,24,25. Although there are several reports of CBuVs in dogs with symptomatic and asymptomatic dogs, the pathogenesis of CBuV infection is still unclear. The previous studies suggested that CBuV infection may be associated with gastroenteritis symptoms in dogs23,24.
In Thailand, human bufavirus (HBuV) has been reported in patients with gastroenteritis and the environment6,26. The occurrence of Thai HBuV has been reported at 0.27% from diarrhea patients6. However, CBuV and its genetic characteristics have never been reported in dogs in Thailand. This study is the first to report the detection and characterization of canine bufavirus (CBuV), a novel parvovirus, in dogs in Thailand.
Results
Canine bufavirus (CBuV) in domestic dogs
From September 2016 to October 2022, we conducted a survey of canine bufavirus in domestic dogs. Rectal swabs were collected from domestic animals (n = 531) from 10 provinces of Thailand (Ayutthaya, Bangkok, Chiang Rai, Nakhon Ratchasima, Phayao, Ratchaburi, Samutprakarn, Samutsakorn, Suphanburi, and Tak) (Supplement Fig. 1). The samples were tested for CBuV using PCR with specific primers to the VP1/VP2 gene. CBuV was detected at 9.42% (50/531). CBuVs were found in both symptomatic (9.84%; 31/315) and asymptomatic dogs (8.80%; 19/216). By age group, CBuVs were detected in dogs of varying age groups, including those younger than 1 year (12.60%; 32/254) and dogs aged 1–5 years (7.34%; 13/177), with statistical significance (p < 0.05) (Supplement Table 1). CBuVs of positivity by month, the viruses could be detected every year from 2016 to 2022, and the highest occurrence of CBuVs was observed in November 2017 (100.00%). Regarding the positivity of CBuVs and seasons, the occurrence of CBuVs was highly detected during the winter season (November to January) and summer season (February to May). However, there was no statistically significant correlation between the positivity of CBuVs and seasons with p > 0.3288–1 (Supplement Table 2). The co-infection of CBuVs with other canine enteric viruses was observed, including CBuV/CPV-2 (n = 5), CBuV/CECoV (n = 11), CBuV/RVA (n = 2), CBuV/CPV-2/CECoV (n = 5), CBuV/CPV-2/CECoV/RVA (n = 1). Out of 50 CBuVs positive samples, 20 CBuVs were selected and sequenced for complete genome sequencing (n = 15) and partial VP2 gene (n = 5) (Table 1).
Phylogenetic and genetic analysis of CBuVs
In this study, we successfully sequenced the CBuVs, and the genome size of Thai CBuVs was 4214 bp. The genome structure of the virus contains non-structural protein (NS1) and viral capsid proteins (VP1 and VP2). Comparing the genome structure to other parvoviruses (canine parvovirus type-2 (CPV-2), canine bocavirus type 1 and 2(CBoV type 1 and 2), the genome structure of Thai CBuVs were similar to reference CBuV but diverse from CPV-2 and CBoV (Supplement Fig. 2).
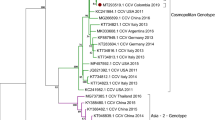
Phylogenetic analysis of the complete genome supported our observation that Thai CBuVs grouped with the canine CBuV group but in the separated cluster from BuVs from pigs, rats, and humans (Fig. 1). Pairwise comparison of the complete genome of CBuVs showed that Thai CBuVs possessed high nucleotide identity to the reference CBuVs from China and Italy with 95.20–99.70% nucleotide identities but low percentages of nucleotide identities with CPV-2 (57.90–58.00%) and CBoV (37.80–39.00%). The Thai CBuVs were closely related to Italy CBuV (ITA/2015/297, 99.70%), China CBuV (Henan38, 99.60%), and Hungary CBuV (HUN/2012/126, 99.60%) (Supplement Table 3). On the other hand, comparing Thai CBuVs and other bufaviruses from different hosts (humans, bats, rats, and pigs), the nucleotide identities ranged only from 43.20 to 65.20%.

Phylogenetic analysis of the whole genome sequences of Thai CBuV. The phylogenetic tree was constructed using MEGA v.7.0 with a neighbor-joining algorithm with a Kimura-2 parameter model with 1000 replications of bootstrap analysis. The pink circle represents Thai CBuVs.
For NS1 gene, phylogenetic analysis of the NS1 gene showed that Thai CBuVs (n = 15) were grouped into BuVs of the Protoparvovirus genus with CBuVs from Canada, China, Hungary, and Italy but separated from other canine enteric parvoviruses of Protoparvovirus genus (CPV-2) and Bocaparovirus genus (CBoV) (Fig. 2). It is noted that based on NS1 phylogenetic analysis, Parvovirinae contains 10 genera including Amdoparvovirus, Aveoparvovirus, Artiparvovirus, Bocaparvovirus, Copiparvovirus, Dependoparvovirus, Erythroparvovirus, Loliparvovirus, Protoparvovirus and Tetraparvovirus. For NS1 nucleotide comparison, Thai CBuVs were highly conserved and possessed high nucleotide identities to Italy CBuV (ITA/2015/297; 99.90%), China CBuVs (CBuV-88; 99.80%) and Hungry CBuV (HUN/2012/126; 99.80%). Moreover, the Thai CBuVs possessed low nucleotide identities with bufaviruses of other species from bats, pigs, rats, and humans (62.50–68.40% nucleotide identities) (Supplement Table 3).

Phylogenetic analysis of the NS1 gene of Thai CBuV. The maximum likelihood tree was generated by using IQ-TREE version 2.1.3 (http://www.iqtree.org/) using the TVMe + IG4 model of nucleotide substitution, default heuristic search options, and ultrafast bootstrapping with 1000 replicates. The tree was visualized by iTOL version 6.0 (https://itol.embl.de/). The color indicated parvovirus genus including dark blue (Protoparvovirus), lite blue (Amdoparvovirus), teal (Aveoparvovirus), ocean (Bocaparvovirus), lite green (copiparvovirus), medium green (Artiparvovrisus), green (Depaendoparvovirus), lite yellow (Erythroparvovirus), dark yellow (Loliparvovirus), yellow (Tetraparvovirus). The pink color indicates Thai CBuVs in this study.
For VP1 and VP2 genes, based on phylogenetic analysis, BuVs can be divided into subgroups based on host species, e.g., human, canine, swine, and bat subgroups. The CBuVs can be further divided into 2 subgroups: A and B. The Thai CBuVs (n = 18) were grouped into subgroup A, which were closely related to CBuVs from China and Italy. While 2 Thai CBuVs (CU_FS 28678 and CU_FS 28683) were grouped into subgroup B, which were like Italy CBuVs (35/ITA and 9AS/ITA) (Supplement Fig. 3 and 4). VP1 and VP2 genes of Thai CBuVs possessed the highest nucleotide identities to those of reference CBuVs (98.70–99.80%). Thai CBuVs (subgroup A) possessed the highest nucleotide identities to China CBuVs (Henan38; 99.80%) but low percentages of nucleotide identities to CPV-2 (51.40–52.70%) and CBoV (40.60–43.30%). Thai CBuVs (subgroup B; CU_FS 28678 and CU_FS 28683) possessed the highest nucleotide identities to Italy CBuVs (35/ITA (98.7–99.2%) and 9AS/ITA (98.6–99.8%).
Genetic analysis of the NS1 gene showed that the NS1 gene of CBuVs contains 1917 nucleotides (639 amino acids). The conserved replication initiator motifs (GLHFHVLLQ and IVRYFLTKQP) were observed to be identical in all reference CBuVs but were not present in human bufavirus (GLHIHVLVC and IANYFLIKKP). The conserved amino acids at the walker loop ATP or GTP binding motifs of Thai CBuVs (GPASTGKS) were observed in both CBuVs and other bufaviruses (Table 2).
VP1 gene of CBuVs contains 2130 nucleotides (710 amino acids). Thai CBuVs contained three potential sites for PLA2 activity, which is required for viral entry. At one site, the calcium binding loop, Thai CBuVs contained amino acid residues (YLGPG) similar to other bufaviruses from dogs, bats, and pigs but were not present in human bufaviruses (YLGPF). The other, two catalytic sites contain amino acid residues (HDLEY and D) similar to all reference BuVs (Table 2). Interestingly, amino acid residues of VP1 related to host preference (human specific) were observed in Thai CBuVs, including PTNRP3-6AIRKA, G22F, T24Q, N71D, K86R, and K89R. This observation could suggest preference characteristics of BuVs to human hosts (Table 3).
VP2 gene of CBuVs contains 1704 nucleotides (568 amino acids). A glycine-rich motif (G-rich) was observed at the N-terminus of VP1, which was similar to other parvoviruses (Table 2). This motif is speculated to be associated with the cellular entry of the virus. Thai CBuVs subgroup B (CU_FS28678 and CU_FS28683) contained 16 unique amino acid residues, which were similar to CBuVs strain 9AS and 35 from Italy, suggesting unique subgroup B characteristics (Supplement Table 4). Moreover, amino acid residues related to host preference were observed. Amino acid insertion at 12–13 and 370 were observed in human BuVs and bat BuVs (Table 3).
Antigenic epitopes prediction, selective pressure, and recombination event of CBuVs
Based on VP2, the recommendation residues of antigenic epitopes prediction of Thai CBuV subgroup A (CU_FS20141) were at position 239–258 (KFDDIQFITVENCVPIELLR) and 99–118 (NDSYHAKVETPWSLLHANCW) which similar to CBuV strain AH001 and AH002 from China (MT542982, and MT 542983). While the residues of antigenic epitopes of Thai CBuV subgroup B (CU_FS28678) were at position 90–109 (QTLHGRDTINDSYHAKVETP) and 239–258 (KYDDIQFITVENCVPIELLR) (Supplement Table 5).
The selective pressure of CBuVs was analyzed by the alignment of all genes of CBuVs using the statistical parameters (FEL, FUBAR, and MEME). The potential positive selection sites of NS1 (n = 9), VP1 (n = 8), and VP2 (n = 7) were detected in this study (Supplement Table 6). The overall mean difference of dN/dS was 0.306 for the NS1 gene, 0.125 for the VP1 gene, and 0.118 for the VP2 gene, suggesting all gene of CBuVs was under negative selection. For the NS1 gene, only one amino acid at position 512 of NS1 was confirmed to be a positive selection (p < 0.1 by MEME and posterior probability of 0.9 by FUBAR). For the VP1 gene, the amino acid position at 256 was confirmed to be a positive selection site (p < 0.1 by MEME and FEL, posterior probability of 0.9 by FUBAR). For the VP2 gene, amino acid positions at 22 and 113 were found as positive selection (p < 0.1 by FEL and posterior probability of 0.9 by FUBAR). A potential positive selection at 113 of the VP2 gene located at the predicted B-cell epitopes of Thai CBuV (strain CU_FS20141) at the location 99–118.
Recombinant analysis of Thai CBuV was performed by using the RDP program, similarity plot, and bootScan analysis. A putative recombinant event was observed in Thai CBuV (CU_FS28678) (Fig. 3). The putative recombinant breakpoint located at the position 1684–2697 which supported by statistically significant (RDP, GENECONV, BootScan, Maxchi, Chimaera, SiScan and 3Seq with p-value of 1.067 × 10−07, 5.806 × 10−06, 4.323 × 10−05, 1.144 × 10−09, 8.612 × 10−03, 7.014 × 10−30, 2.222 × 10−21, respectively) (Supplement Table 7). Our result showed that the major and minor putative parents of Thai CBuV (CU_FS28678) were CBuV strain 9AS from Italy (MT154051; CBuV-subgroup B) and Thai CBuV (CU_FS28696) (CBuV-subgroup A).

Phylogenetic analysis and recombination analysis of Thai CBuV (CU_FS 28678). (a) Phylogenetic analysis of CBuVs was constructed using MEGA v.7.0 with a neighbor-joining algorithm with a Kimura-2 parameter model with 1000 replications of bootstrap analysis. The pink circle represents Thai CBuV strain CU_FS 28678. The blue and green triangles showed putative major and minor parents. (b) Similarity and Bootscan analysis of Thai CBuV showed the recombinant CU_FS 28678. (c) Genome organization of potential recombinant of Thai CBuV strain CU_ FS 28678.
Discussion
Bufavirus (BuV) is a novel member of the family Parvoviridae. It was first reported in humans with gastroenteric symptoms in 20128. Bufavirus infection in dogs was first described in Italy in 2016 and subsequently reported in several countries20,23,24,27. However, CBuVs have not been reported in Thailand. This study is the first to detect CBuVs in domestic dogs in Thailand. Our survey showed CBuV positivity at 9.42% (50/531), which was comparable to other previous studies (2.5–8.8% positivity)20,22,23,24. However, there was no significant difference in CBuV positivity by season. Our result showed that CBuVs was highly detected in gastroenteritis dogs, although there was no statistical significance between symptomatic and asymptomatic dogs. The previous studies showed that the CBuVs have been detected in both healthy dogs and dogs with respiratory and gastroenteric symptoms20,24,27. Some studies supported that BuVs may associated with gastroenteritis and cause systemic infection in humans and dogs10,11,23,24. However, the pathogenesis of CBuV infection is still not clear. Thai CBuVs showed a significant presence in both younger than 1 year (< 1 year) and dogs aged 1–5 years with statistical significance (p < 0.05) (Supplement Table 1). This observation agreed with the previous study, which suggested that CBuV tends to be more prevalent for dogs under 5 years of age than the older age group20. In this study, we observed co-infection of CBuVs with other enteric viruses. For example, CPV-2 and CECoV were co-infection with CBuV, which was similar to other studies23,27. However, the severity level of clinical signs of co-infection between CBuVs and other pathogens should be further investigated.
Currently, only 17 nearly complete genome sequences of CBuVs are available in the GenBank Database. Our study provided an additional 15 complete genomes of CBuVs in the database. In this study, the complete genome size of CBuVs encoding 3 ORFs, including NS1, VP1, and VP2 (Supplement Fig. 2). Based on the phylogenetic analysis of the complete genome, Thai CBuVs belong to the bufavirus of the Protoparvovirus genus (canine group) and were closely related to CBuVs from China (Henan38), Hungary (HUN/2012/126) and Italy (ITA/2015/297). It is noted that the classification of parvovirus genus was classified by NS1 gene sequencing28. The phylogenetic analysis of the NS1 gene showed that Thai CBuVs were grouped into the Protoparvovirus genus (CBuV group) but were separated from canine parvovirus type -2 (CPV-2). Thai CBuVs have high nucleotide identities to reference CBuVs at 98.70–99.90% but were distinct from CPV-2 viruses at only 57.10% nucleotide identities. The phylogenetic analysis of VP1 and VP2 genes showed that CBuVs clustered into 2 major subgroups, subgroups A and B, which agreed with the previous study23. Thai CBuVs (n = 18) were grouped into subgroup A with CBuVs from Italy and China. The other Thai CBuVs (strain CU_FS 28678 and CU_ FS 28683; n = 2) were grouped into subgroup B with CBuV from Italy. It is noted that Thai CBuVs (subgroups A and B) might share the same common ancestor with CBuVs from China and Italy.
Thai CBuVs contained conserved regions of the parvovirus-conserved replication initiator motifs. For NS1, Thai CBuVs contained a helicase motif walker (GPASTGKS), which was also observed in all reference bufaviruses8,20. For the VP1 gene, Thai CBuVs posed a unique calcium-binding loop site (YLGPG) and two catalytic sites (HDLEY and D), which were also observed in most reference bufaviruses, suggesting unique characteristics11,18. Previous studies indicated that the calcium-binding loop site and phospholipase A2 (PLA2) region may be associated with viral entry to the host cell of the viruses29. The N-terminus of the VP1/VP2 had a glycine-rich motif, which may be associated with the cellular entry of parvovirus30,31. Moreover, human-specific amino acid residues of VP1 were observed at 3-6AIRKA, 22F, 24Q, 71D, 86R, and 89R. Unique amino acid insertion at positions 12–13 and 370 of VP2 were observed in human BuVs and Bat BuVs, suggesting potential antigenic markers for human and bat BuVs. Moreover, in the comparison of the genetic analysis between CBuVs subgroup A and B, there were 16 unique amino acids of CBuVs subgroup B, suggesting the genetic characteristics of the CBuV subgroup.
Our study showed that the VP1 and VP2 genes of Thai CBuVs have higher variations than the NS1 gene, which were similar to the previous study32. The VP2 gene of parvoviruses may have functions relating to viral entering, receptor binding, and immunogenicity and contains many major epitopes32,33,34. Moreover, B-cell epitopes are recognized as an immunogenicity classification. Thus, the mutations of this epitope may affect immunogenicity or generate a novel serotype of the virus35. The top list of antigenic epitope predictions on the VP2 gene of Thai CBuVs (CU_FS 20141 and CU_FS 28678) were observed in this study at 239–258 and 90–109, respectively (Supplement Table 5). In the selective pressure analysis based on the VP2 gene, two positive selections (22, 113) were observed in this study. A residue at 113 of VP2 was located at predicted B-cell epitopes of Thai CBuV (CU_FS20141) (location 99–118). Notably, the possibility of an antigenic shift to escape host immune responses of Thai CBuVs should be considered. However, due to the limited number of isolates in this study, further investigation on an extensive sample scale is necessary.
The positive selection pressure and genetic recombination are the factors that affect to high evolution rate of parvovirus evolution36,37. Previous study showed that natural recombination events of protoparvovirus between bufaviruses strains WUHARV and MgBuV1 can occur, suggesting cross-species transmission or sharing a common ancestor between bat, swine, and non-human primate bufavirus15. In this study, the recombinant Thai CBuV (CU_FS28678) was observed between Thai CBuV (CU_FS28696) and Italy CBuVs (9AS), suggesting a possible common ancestor of Thai and Italy CBuVs and inducing genetic diversity of CBuVs.
In conclusion, this is the first to report and genetically characterize the complete genome of CBuVs in domestic dogs in Thailand. Our result showed that Thai CBuVs were detected in both healthy and dogs with gastroenteric signs. The phylogenetic analysis showed that Thai CBuVs might share a common ancestor with CBuVs from Italy and China. However, the genetic database of CBuVs is still limited. Thus, surveillance and genetic characterization of CBuVs in domestic animals should be further investigated on a larger scale to elucidate the dynamic, evolution, and distribution of CBuVs.
Materials and methods
Canine samples
In this study, we conducted a cross-sectional sample collection from Chulalongkorn University’s Veterinary Teaching Hospital and private small animal hospitals in Thailand from September 2016 to October 2022. A total of 531 rectal swab samples were collected from dogs with asymptomatic (n = 216) and gastroenteritis (n = 315) symptoms, including vomiting, diarrhea, and dehydration. The samples were collected from dogs of young age (< 1 year) (n = 254), adults (1–5 years) (n = 177), and seniors (> 5 years) (n = 100). The animal demographic data, including age, sex, breed, and vaccination history, were recorded. This study was performed in accordance with the Chulalongkorn University, Animal Care and Uses Protocol (CU-VET IACUC#2031050, CU-VET IACUC#2031035) guidelines and regulations.
Canine bufavirus identification
All rectal swab samples were subjected to DNA extraction using DNA/RNA GENTi Automated Nucleic Acid Extraction System (GeneAll® Seoul, Korea) following the manufacturer’s recommendations. For canine bufavirus identification, DNA samples were screened using PCR with specific primers for the VP1/VP2 gene. The primers used in this study were previously described, including CPPV 165F (5′-CTGGTTTAATCCAGCAGACT3′), CPPV 371R (5′-TGAAGACCAAGGTAGTAGGT3′) corresponded to the position 2872–2891 and 3060–3079 of CBuV, respectively22. The PCR was performed in a final volume of 50 μl comprising 4 μl of template DNA, 25 μl of 2 × reaction buffer of the HotStarTaq® Master Mix (Qiagen, Hilden, Germany), 0.2 μM of each forward and reverse primers and distilled water to a final volume of 50 μl. The PCR condition included an initial denaturation step at 95 °C for 15 min, following 40 cycles of denaturation at 94 °C for 30 s, annealing at 52 °C for 30 s, and extension at 72 °C for 1 min, as well as a final extension step at 72 °C for 10 min. PCR products were run on a 1.5% agarose gel, which was mixed with RedSafe™ (iNtRON Biotechnology, Inc., Korea) at 100 V for 45 min. The expected size of the CBuV-positive sample was 208 bp. In this study, other canine viral enteric pathogens, including Canine Parvovirus, Canine Coronavirus, and Canine Rotavirus, were also tested in all samples38,39,40.
Canine bufavirus characterization
Representatives of positive CBuV were selected for whole genome sequencing (n = 15) and VP1/VP2 sequencing (n = 5). The CBuVs were selected based on epidemiological and demographic data such as age, collection date, breed, and vaccination history. Whole genome sequencing was conducted by amplification of each gene of the viruses by using modified oligonucleotide primer sets as previously described and new primer sets designed by using the Primer 3 plus program (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi) (Supplement Table 8)22,23,25,41. Nucleotide amplification was conducted in 50 μl PCR reaction comprising 4 μl of template DNA, 25 μl of 2 × reaction buffer of the HotStarTaq® Master Mix (Qiagen, Hilden, Germany), 0.2 μM of each forward and reverse primer and distilled water to a final volume of 50 μl. The PCR condition was set as initial denaturation step at 95 °C for 15 min; 40 cycles of denaturation at 94 °C for 30 s, annealing at 50–55 °C for 30 s and extension at 72 °C for 1–1.30 min, and final extension step at 72 °C for 10 min. PCR products were then purified and sequenced at 1st Base Laboratories Sdn Bhd, Malaysia. The nucleotide sequences were edited, validated, and assembled using SeqMan software v.5.03 (DNASTAR Inc.; Wisconsin, USA). In this study, whole genome and VP2 sequences of Thai CBuVs were deposited into the GenBank database under the accession numbers OQ730240- OQ730259 (Table 1).
Phylogenetic and genetic analyses of canine bufavirus
Phylogenetic and genetic analyses of CBuV were conducted by comparing nucleotide sequences of Thai CBuVs with those of reference viruses of the Parvoviridae family available from the GenBank database, including canine parvovirus, canine bocavirus, human bufavirus, bat bufavirus, rat bufavirus, and swine bufavirus. The reference nucleotide sequences of CBuV were also included. It should be noted that only 17 complete genome sequences of domestic dog and canid wildlife BuVs have been reported and were available from the GenBank database. Phylogenetic trees of WGS and VP1 gene of CBuVs were constructed using MEGA v.7.0 (Tempe, AZ, USA) with the neighbor-joining method applied with the Kimura 2-parameter and 1000 bootstrap replicates. For NS1 and VP2, phylogenetic tree and best models were generated by using IQ-TREE version 2.1.3 (http://www.iqtree.org/) with the TVMe + IG4 model of nucleotide substitution, default heuristic search options, and ultrafast bootstrapping with 1000 replicates. The tree was visualized by iTOL version 6.0 (https://itol.embl.de/) and Figtree V1.3.1 (http://tree.bio.ed.ac.uk › software).
For genetic analysis, the nucleotide sequences and deduced amino acids of CBuVs were aligned and compared with those of reference viruses using MegAlign software v.5.03 (DNASTAR Inc.; Wisconsin, USA). A pairwise comparison of nucleotides and amino acids of Thai CBuV, and those of reference CBuV was conducted. The variable and unique amino acids related to receptor binding of the viruses and host preferences were evaluated. For analysis of the selective pressure of CBuVs, the ratio of non-synonymous (dN) to synonymous (dS) substitutions was estimated using static methods on the online software (http://www.datamonkey.org/). The values dN/dS > 1, dN/dS = 1, and dN/dS < 1 were used to define positive selection, neutral mutations, and negative selection, respectively. The positive selection position site was identified by at least 2 algorithms. The significance levels were set at p = 0.1. Antigenic epitope prediction of Thai CBuVs was identified using the online software (http://sysbio.unl.edu/SVMTRiP/). Recombination analysis was performed using the Recombination Detection Program (RDP) package version 4.0 with a statistical method including RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, and 3Seq. The potentially positive recombination was analyzed using a potential breakpoint signal of at least four methods with p-values < 0.01. The related phylogenetic tree with potential recombinant and its putative major and minor parents were generated using RDP 4 package software.
Statistical analysis
Correlation among CBuVs positivity and the sample collection date, age of dogs, and clinical signs were analyzed using Fisher's exact test (https://www.socscistatistics.com/tests/fisher). A p-value of < 0.05 was considered as statistical significance.
Ethics statement
This study was conducted under the approval of the Institute for Animal Care and Use Protocol of the CU-VET, Chulalongkorn University (CU-VET IACUC#2031050, CU-VET IACUC#2031035).
References
Cotmore, S. F. et al. ICTV virus taxonomy profile: Parvoviridae. J. Gen. Virol. 100, 367–368 (2019).
Väisänen, E. et al. Epidemiology of two human protoparvoviruses, bufavirus and tusavirus. Sci. Rep. 6, 39267 (2016).
Phan, T. G. et al. A new protoparvovirus in human fecal samples and cutaneous T cell lymphomas (mycosis fungoides). Virology 496, 299–305. https://doi.org/10.1016/j.virol.2016.06.013 (2016).
Simo-Fouda, F. et al. Investigation of bufavirus and parvovirus 4 in patients with gastro-enteritis from the south-east of France. Pathogens 10, 1151 (2021).
Vaisanen, E. et al. Bufavirus in feces of patients with gastroenteritis, Finland. Emerg. Infect. Dis. 20, 1077–1079. https://doi.org/10.3201/eid2006.131674 (2014).
Chieochansin, T., Vutithanachot, V., Theamboonlers, A. & Poovorawan, Y. Bufavirus in fecal specimens of patients with and without diarrhea in Thailand. Arch. Virol. 160, 1781–1784. https://doi.org/10.1007/s00705-015-2441-z (2015).
Yahiro, T. et al. Novel human bufavirus genotype 3 in children with severe diarrhea, Bhutan. Emerg. Infect. Dis. 20, 1037–1039. https://doi.org/10.3201/eid2006.131430 (2014).
Phan, T. G. et al. New parvovirus in child with unexplained diarrhea, Tunisia. Emerg. Infect. Dis. 20, 1911 (2014).
Smits, S. L. et al. New viruses in idiopathic human diarrhea cases, the Netherlands. Emerg. Infect. Dis. 20, 1218 (2014).
Altay, A. et al. Bufavirus genotype 3 in Turkish children with severe diarrhoea. Clin. Microbiol. Infect. 21(965), e961-964. https://doi.org/10.1016/j.cmi.2015.06.006 (2015).
Huang, D. D. et al. Identification of bufavirus-1 and bufavirus-3 in feces of patients with acute Diarrhea, China. Sci. Rep. 5, 13272. https://doi.org/10.1038/srep13272 (2015).
Ayouni, S. et al. Cosavirus, salivirus and bufavirus in diarrheal Tunisian infants. PLoS One 11, e0162255 (2016).
Razizadeh, M. H., Khatami, A. & Zarei, M. Global status of bufavirus, cosavirus, and saffold virus in gastroenteritis: A systematic review and meta-analysis. Front. Med. https://doi.org/10.3389/fmed.2021.775698 (2022).
Daprà, V. et al. Bufavirus, cosavirus, and salivirus in diarrheal Italian infants. Intervirology 64, 165–168 (2021).
Kemenesi, G. et al. Genetic diversity and recombination within bufaviruses: Detection of a novel strain in Hungarian bats. Infect. Genet. Evol. 33, 288–292. https://doi.org/10.1016/j.meegid.2015.05.017 (2015).
Liu, L. et al. Identification of a novel bufavirus in domestic pigs by a viral metagenomic approach. J. Gen. Virol. 97, 1592–1596. https://doi.org/10.1099/jgv.0.000476 (2016).
Sasaki, M. et al. Distinct Lineages of bufavirus in wild shrews and nonhuman primates. Emerg. Infect. Dis. 21, 1230–1233. https://doi.org/10.3201/eid2107.141969 (2015).
Yang, S. et al. Bufavirus Protoparvovirus in feces of wild rats in China. Virus Genes 52, 130–133. https://doi.org/10.1007/s11262-015-1262-1 (2016).
Zhou, L. et al. Viral communities associated with porcine diarrhoeal disease and genetic characterization of a bufavirus in Tibetan pigs in China. Arch. Virol. 166, 613–617. https://doi.org/10.1007/s00705-020-04932-9 (2021).
Martella, V. et al. Novel parvovirus related to primate bufaviruses in dogs. Emerg. Infect. Dis. 24, 1061 (2018).
Decaro, N. & Buonavoglia, C. Canine parvovirus—a review of epidemiological and diagnostic aspects, with emphasis on type 2c. Vet. Microbiol. 155, 1–12 (2012).
Wang, Y. et al. Genetic and phylogenetic analysis of canine bufavirus from Anhui Province, Eastern China. Infect. Genet. Evol. 86, 104600 (2020).
Di Martino, B. et al. Genetic heterogeneity of canine bufaviruses. Transbound. Emerg. Dis. 68, 802–812. https://doi.org/10.1111/tbed.13746 (2021).
Li, J. et al. Canine bufavirus in faeces and plasma of dogs with diarrhoea, China. Emerg. Microbes Infect. 8, 245–247. https://doi.org/10.1080/22221751.2018.1563457 (2019).
Sun, W. et al. First identification of a novel parvovirus distantly related to human bufavirus from diarrheal dogs in China. Virus Res. 265, 127–131. https://doi.org/10.1016/j.virusres.2019.03.020 (2019).
Nantachit, N., Khamrin, P., Kumthip, K., Malasao, R. & Maneekarn, N. Molecular surveillance and genetic analyses of bufavirus in environmental water in Thailand. Infect. Genet. Evol. 75, 104013. https://doi.org/10.1016/j.meegid.2019.104013 (2019).
Ganji, V. K., Buddala, B., Yella, N. R. & Putty, K. First report of canine bufavirus in India. Arch. Virol. 167, 1145–1149. https://doi.org/10.1007/s00705-022-05398-7 (2022).
Cotmore, S. F. et al. The family parvoviridae. Arch. Virol. 159, 1239–1247 (2014).
Farr, G. A., Zhang, L.-G. & Tattersall, P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. 102, 17148–17153 (2005).
Tu, M., Liu, F., Chen, S., Wang, M. & Cheng, A. Role of capsid proteins in parvoviruses infection. Virol. J. 12, 114. https://doi.org/10.1186/s12985-015-0344-y (2015).
Castellanos, M. et al. A slender tract of glycine residues is required for translocation of the VP2 protein N-terminal domain through the parvovirus MVM capsid channel to initiate infection. Biochem. J. 455, 87–94 (2013).
Allison, A. B. et al. Role of multiple hosts in the cross-species transmission and emergence of a pandemic parvovirus. J. Virol. 86, 865–872 (2012).
Allison, A. B. et al. Single mutations in the VP2 300 loop region of the three-fold spike of the carnivore parvovirus capsid can determine host range. J. Virol. 90, 753–767 (2016).
De Souza, A. R. et al. Porcine parvovirus VP1/VP2 on a time series epitope mapping: Exploring the effects of high hydrostatic pressure on the immune recognition of antigens. Virol. J. 16, 1–11 (2019).
Zhang, C. et al. The relationship between B-cell epitope and mimotope sequences. Protein Pept. Lett. 23, 132–141 (2016).
Ohshima, T. & Mochizuki, M. Evidence for recombination between feline panleukopenia virus and canine parvovirus type 2. J. Vet. Med. Sci. 71, 403–408 (2009).
Shackelton, L. A., Parrish, C. R., Truyen, U. & Holmes, E. C. High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc. Natl. Acad. Sci. 102, 379–384 (2005).
Buonavoglia, C. et al. Evidence for evolution of canine parvovirus type 2 in Italy. J. Gen. Virol. 82, 3021–3025 (2001).
Ortega, A. F., Martínez-Castañeda, J. S., Bautista-Gómez, L. G., Muñoz, R. F. & Hernández, I. Q. Identification of co-infection by rotavirus and parvovirus in dogs with gastroenteritis in Mexico. Braz. J. Microbiol. 48, 769–773 (2017).
Pratelli, A., Tempesta, M., Greco, G., Martella, V. & Buonavoglia, C. Development of a nested PCR assay for the detection of canine coronavirus. J. Virol. Methods 80, 11–15 (1999).
Rozen, S. & Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Bioinform. Methods Protoc. https://doi.org/10.1385/1-59259-192-2:365 (1999).
Acknowledgements
We would like to thank the Faculty of Veterinary Science, Chulalongkorn University, for financial support (Veterinary Science Research Fund, (RI 1/2566). This work was supported by Thailand Science Research and Innovation Fund Chulalongkorn University [FOODF67310016]. We are grateful for the financial support from Chulalongkorn University for the research fund under the Chulalongkorn University’s TSRI Fundamental Fund (HEA663100105), the Center of Excellence for Emerging and Re-emerging Infectious Diseases in Animals (CUEIDAs), and the One Health Research Cluster. This project was partially funded by the National Research Council of Thailand (NRCT): NRCT Senior scholar 2022 #N42A650553.
Author information
Authors and Affiliations
Contributions
K.C., Y.T., E.P. and W.J. performed sample collection, molecular detection, whole genome characterization and analysis. C.N., E.C., S.P. and T.J. participated in whole genome sequencing and phylogenetic analysis. K.C. drafted the manuscript. A.A. (PI) designed the study, performed data analysis, drafted, revised and approved the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Charoenkul, K., Thaw, Y.N., Phyu, E.M. et al. First detection and genetic characterization of canine bufavirus in domestic dogs, Thailand. Sci Rep 14, 4773 (2024). https://doi.org/10.1038/s41598-024-54914-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54914-z
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
