
Abstract
Ab initio CASSCF/MRCI + Q calculations have been used to investigate the electronic structure and transition properties of the alkaline earth astatine molecules SrAt and BaAt. The adiabatic potential energy curves have been computed and plotted for the low-lying electronic states in the representations 2S+1Λ+/− and Ω(±) (with and without spin–orbit coupling effect). The spectroscopic and vibrational constants have been deduced for the corresponding bound states. An analysis of the Franck–Condon factors, the Einstein Coefficients, and the branching ratios among different vibrational levels has shown that both SrAt and BaAt molecules are suitable candidates for Doppler and Sysphus laser cooling. Experimental laser cooling schemes and conditions for these two molecules have been proposed. These results may pave the way for new spectroscopic and laser cooling experiments of alkaline earth astatine molecules.
Similar content being viewed by others
Introduction
Researchers have been interested in the spectroscopic studies of the alkali and alkaline earth halides1,2 because of their relevance to astrophysics. These have been detected in the interstellar medium3 and the upper atmosphere4. In this view, MgF, SrF1, and MgCl molecules are predicted to appear in S-stars2, on the sun’s surface, and in the sunspot’s spectrum. Moreover, these alkaline-earth mono-halide molecules are highly interesting for high-temperature reactions in catalysis and corrosion processes5.
From the perspective of laser cooling experiments, the compounds of alkaline-earth metals have been proposed as promising candidates for laser cooling and controlling the preparation of many-body entangled states6,7,8. SrF and YO9,10 molecules have been cooled using transverse cooling methods, while CaF has been cooled by longitudinal laser cooling11. Extensive theoretical studies have also been performed for molecules that possess similar electronic structures, such as BeF12 and MgF13
The electronic structure of the alkaline-earth halide molecules, including MgAt, has been studied in the literature. The first few low-lying excited electronic states of the molecules MgCl, MgBr, and MgI have already been investigated14,15,16,17,18,19. In 2015, Wan et al.20 presented for BeI and MgI an ab initio investigation for the effect of spin–orbit coupling on laser cooling, where they calculated the spectroscopic properties and the cooling wavelength of these molecules in the ultra-violet region. The suitability of laser cooling of alkaline earth mono halides BaX (X = F, Cl, Br, I) and MgX(X = Br, At, I) has been verified respectively by Yang et al.21 and Yang and Tao22.
We present a theoretical study by using the ab initio method (CASSCF/MRCI + Q) for the molecules SrAt and BaAt to test the candidacy of alkaline-earth astatine species for laser cooling. Section “Computational approach” includes the computational approach followed for the pursued computations. The adiabatic potential energy curves, the dipole moment curves of the low-lying doublet and quartet electronic states, and their spectroscopic constants in the 2S+1Λ+/− and Ω(±) representations are presented in Section “Potential energy curves, spectroscopic parameters, and permanent dipole moment curves”. In addition, the vibrational energy Ev, the ro-vibrational constants Bv, Dv, the abscissa of the turning points Rmin, and Rmax of the ground, and the bound excited electronic states are displayed in Section “The ro-vibrational parameters”. Section “Laser cooling study of SrAt and BaAt molecules” includes a laser cooling investigation of the molecules SrAt and BaAt, done by calculating the Franck–Condon Factors (FCF), the Einstein Coefficients, the radiative lifetime, and the branching ratio among specific vibrational levels. Experimental parameters are presented, including the minimum slowing distance, the Doppler and recoil temperatures, and the maximal deceleration of the molecules. Laser cooling schemes for the molecules SrAt and BaAt are presented with three and four lasers in the visible and near-infrared regions, respectively.
Computational approach
The Complete Active Space Self Consistent Field (CASSCF) has been used as a reference for generating the multiconfiguration wavefunctions of the considered two molecules. It is followed by the Multireference configuration interaction (MRCI) method, with Davidson correction (+ Q)23. The current calculations are done by employing the MOLPRO program package24, taking advantage of the graphical user interface GABEDIT25 to study the electronic structure of the electronic states of SrAt and BaAt in the doublet and quartet multiplicities with and without considering the spin–orbit coupling effect. For the BaAt molecule, the electronic wavefunctions of seventy-eight core electrons of At are described by the quasi-relativistic effective core potential ECP78MWB26 for s, p, d functions, while for the SrAt, they are described by ECP60MDF. Thirty-six electrons of Sr were frozen using the ECP36SDF for s, p functions, and 46 electrons of Ba were frozen using the ECP46MWB for s, p, d functions. It is worth noting that the Cꝏv group was decomposed into C2v sub-group because of the limitations of the MOLPRO software. Table 1 reports the active space orbitals for the two considered molecules. Thus, the molecular orbitals are labeled in the irreducible representation as 4a1, 1b1, 1b2, and 0a2 for SrAt denoted by [4,1,1,0], and 6a1, 3b1, 3b2, and 1a2 denoted by1,3,3,6 for BaAt. Also, the molecules SrAt and BaAt have been investigated in spin–orbit Ω(±) representation where Sr is treated as a system of 10 electrons using ECP28MDF27, Ba is treated by ECP46MDF 28, and At is treated by ECP60MDF for SrAt molecule and ECP78MDF for BaAt molecule28. Then the active space in the spin–orbit calculations of SrAt becomes 4σ (Sr: 5s; At:6p0, 6s, 7s), 1π (Sr:0; At: 6p1±), 0δ and that of BaAt is 6σ (Ba: 5d0, 5d+2,6p0, 6 s; At:7 s, 6p0), 3π (Ba:5d±1, 5p±1; At: 6p±1),1δ (Ba: 5d-2) and the molecular orbitals are labeled as [4,1,1,0] for SrAt and [1,3,36] for BaAt.
Additionally, the potential energy curves of the molecules BeAt, MgAt, and CaAt have been computed for a spectroscopic trend comparison (see Section “Potential energy curves, spectroscopic parameters, and permanent dipole moment curves”). The basis set is cc-pV5Z29 for Be and Mg atoms and ECP10MWM30 for Ca atom. For At atom, the same basis set (ECP78MDF) was used among all molecules. The corresponding potential energy curves of the doublet and quartet electronic states for the considered five molecules are given in Figs. FS1-FS9 along with their static dipole moments (Figs. FS10–FS15) in the supplementary materials.
Results and discussion
Potential energy curves, spectroscopic parameters, and permanent dipole moment curves
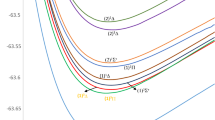
The ab initio method employed in the present work allowed the investigation of the adiabatic potential energy curves (PECs) of the electronic states of the alkaline earth astatine molecules SrAt and BaAt in their doublet and quartet multiplicities. The PECs of thirty-five electronic states (eight doublet and five quartet states of SrAt molecule) and (seven doublet and 15 quartet states of BaAt molecule), taking into account spin–orbit coupling, in the representation Ω(±) are provided in Figs. 1, 2 as a function of the internuclear distance R, while the PECs of 28 states (eight doublet and ten quartet states of SrAt) and (five doublet and five quartet states of BaAT) calculated without considering this effect are given in the supplementary material in Figs. FS5–FS8. One can notice that the two molecules have deep potential wells reflecting a dominancy of the attractive forces within the molecule’s constituents and shallower ones reflecting the dominancy of repulsive forces. Additionally, many unbound repulsive states are observed. The ground state is X2∑+, which has a deep potential well for the two molecules. The spectroscopic parameters Te, Re, \({\upomega }_{{\text{e}}}\), and Be have been calculated for the bound states upon fitting their potential energy curves into a polynomial around the equilibrium position Re. The calculated spectroscopic parameters of BaAt and SrAt molecules with and without spin–orbit coupling effects are listed in Tables 2 and 3. The data we present here has been calculated for the first time, so comparing it with the literature is not possible. Still, the validity of the spectroscopic constants can be confirmed in Table 4 through the homogeneous trend of Te, Re, and ωe of the ground and some of the low-lying electronic states of the molecules BeAt, MgAt, CaAt, SrAt, and BaAt, as in previously published work31. The correct trend of the spectroscopic constants is evident for all the investigated electronic states: an increase in the atomic mass of the alkaline earth atom corresponds to a decrease in the electronegativity, which leads to an increase in the equilibrium bond length Re, a decrease in the transition energy Te, and the harmonic frequency ωe. The spectroscopic constants are not calculated for the remainder of the excited states because they are either unbound states, have very shallow potential wells, or present an avoided crossing behavior near their minimum.

Potential energy curves of the lowest Ω(±) doublet and quartet states of the SrAt molecule.

Potential energy curves of the lowest Ω(±) doublet and quartet states of the BaAt molecule.
Moreover, By using the basis set ECP60MDF For At atom, the comparison of our spectroscopic constants for MgAt (Table 4) with those given by Yang and Gao22 shows a very-good agreement with relative differences of 0.6%, 1.8%, and 2.5%, respectively, for ΔRe/Re, Δωe/ωe, ΔBe/Be for the ground state X2∑+. For (2)2Π states, these relative differences are 1.63%, 1.60%, 3.70%, and 3.6% for ΔTe/Te, ΔRe/Re, Δωe/ωe, and ΔBe/Be, respectively.
Given the correct trend and the very good agreement of our spectroscopic constants with those available in the literature22, we may confirm the accuracy of our results for the two molecules, SrAt and BaAt.
The permanent dipole moment curves (PDMCs) are an effective tool for understanding the polarity and the strength of the long-range dipole–dipole forces in diatomic molecules. The permanent dipole moment curves (PDMCs) of the five molecules, BeAt, MgAt, CaAt, SrAt, and BaAt (without including the spin–orbit coupling effects), are represented in Figs. FS10–FS15 of the supplementary material. The electrons’ density distribution can be understood according to the polarity of the dipole moments ranging from − µ to + µ. The dipole moment usually exhibits positive values when the electrons’ density is closer to the alkaline earth metal considered at the origin. On the contrary, flipping in the polarity occurs when the dipole moment becomes negative as the electrons’ density becomes closer to the At atom. Consequently, the positive values of dipole moments can be denoted by Srδ−Atδ+ and Baδ−Atδ+. The values of dipole moment, which tend to be zero at large internuclear distances, are evidence of the molecule’s dissociation into neutral fragments. In contrast, those with constant values indicate dissociation into ionic fragments.
The ro-vibrational parameters
The theoretical determination of a given level’s rovibrational constants is effective in the prediction process of absorption/emission line positions. These are useful in guiding experimental investigations that facilitate the detection of unknown molecules. In the conventional approach of the Rayleigh-Schrödinger perturbation theory (RSPT), the first analytical expressions of the centrifugal distortion constants (CDC) have been derived by Albritton et al.32. To overcome the complexity of the computation of such expressions, Hutson derived an algorithm33 by using the Numerov difference equation for the determination of the constants \({D}_{\upnu }\), \({H}_{\upnu }\), \({L}_{\upnu }\), and \({M}_{\upnu }\) in terms of the vibrational wave function \({\Psi }_{\upnu }\). But in this algorithm, some difficulties had appeared for some potentials (like the Lennard–Jones potential), such as the problem of treating high vibrational levels near the dissociation limit. An improvement has then been introduced to the Huston algorithm Tellinghuisen34, but it is still insufficient to reach larger orders of centrifugal distortion constants. For this purpose, the quantum mechanical canonical function method35,36,37 was developed to calculate the rotation–vibration constants for highly excited electronic states with many centrifugal distortion constants.
This approach is used in the present work to determine the rovibrational parameters of the BaAt molecules, including the vibrational energy Ev, the rotational constant Bv, the centrifugal distortion constant Dv, and the abscissas of the turning point Rmin and Rmax. These values, including the spin–orbit coupling effects, are given in (Tables 5, 6). Since most states are unbound, the spectroscopic constants and the ro-vibrational parameter of the quartet spin–orbit potential energy curves have not been calculated. There are no comparisons with other results because these constants are calculated here for the first time.
Laser cooling study of SrAt and BaAt molecules
The difference in equilibrium positions ΔRe between the ground state X2∑+ and the two excites states (1)2Π and (2)2∑+ states of SrAt and BaAt are minimal; this directed our attention to verify the laser cooling suitability for these molecules through cycles involving the aforementioned states, in the Ω(±) representation. However, an experimental confirmation of the presented electronic structure calculation is highly recommended before such step is taken.
The main criterion for keeping a molecule in a closed-loop cycle is a highly diagonal Franck–Condon factor (FCF) among the lowest vibrational levels of a bound excited state and those of the ground state 38. The vibrational FCF of the transition X2∑+1/2—(1)2\(\sum_{1/2}^{ + }\) of the molecule SrAt (calculated by using the LEVEL 11 program39) is plotted in Fig. 3. One can notice that the transition among the vibrational levels v′ = v = 0 has a higher probability than the remaining ones. At the same time, the deexcitation of the vibrational level v′ = 0 takes place mainly through the channel v′0v1, v′0v2, and v′0v3 with the following FCF, respectively f0′0 = 0.812067, f0′1 = 0.161978, f0′2 = 0.022776 and f0′3 = 0.002822. The deexcitation through the remaining channels is minimal.

Franck–Condon factor for the transitions X2\(\sum_{1/2}^{ + }\) − (2)2∑1/2 and X2\(\sum_{1/2}^{ + }\) − (1)2Π1/2 of the molecules SrAt and BaAt, respectively.
A short radiative lifetime among vibrational levels involved in the cooling cycle is the second criterion for a successful laser cooling process, as it maximizes the cooling rate and produces a strong Doppler force. This can be done by calculating the vibrational Einstein coefficient Aν′ν given by40
where M(r) is the electronic transition dipole moment (in Debye), and ΔE is the energy difference between the two studied electronic states. The computed X2\(\sum_{1/2}^{ + }\) − (1)2\(\sum_{1/2}^{ + }\) transition dipole moment is represented in Fig. 4. The radiative lifetimes (given by \(\tau_{{v^{\prime}}} = \frac{1}{{\mathop \sum \nolimits_{v} A_{{v^{\prime}v}} }}\)) of six considered vibrational levels (v′), and the vibrational branching ratio (given by Rv′v = \(\frac{{A_{{v^{\prime}v}} }}{{\mathop \sum \nolimits_{v} A_{{v^{\prime}v}} }}\)41,42) among the vibrational transitions between different levels (v′) and (v) are displayed in Table 7. The transition X2\(\sum_{1/2}^{ + }\) − (1)2\(\sum_{1/2}^{ + }\) of SrAt molecule satisfies this condition, given the short radiative lifetimes that vary as 92.50 ns ≤ τ ≤ 101.9 ns among different values of v′.

Transition dipole moments for the transitions X2\(\sum_{1/2}^{ + }\) − (2)2\(\sum_{1/2}^{ + }\) and X2\(\sum_{1/2}^{ + }\) − (1)2Π1/2 of the molecules SrAt and BaAt, respectively.
Finally, the number of cycles (N) for photon absorption/emission should be maximized to decelerate the molecule sufficiently43,44. One can define N in terms of total decay channels involved (ɳ) as the following:
In our case, we propose ɳ = R0′0 + R0′1 + R0′2 + R0′3, for which N = 1786. The corresponding laser cooling scheme is given in Fig. 5. The solid red lines represent the cycling lasers, while the dotted lines represent the spontaneous decay. The values of the vibrational transitions FCF (fν′ν) and the vibrational branching ratios Rν′ν are annotated under the ground state vibrational level involved in the corresponding transition. The proposed laser wavelengths are in the visible domain, with the primary pumping laser at λ0′0 = 666.8 nm, and the three repumping lasers used to close the leaks from higher vibrational levels at wavelengths λ0′1 = 673.3 nm, λ0′2 = 679.8 nm, λ0′3 = 686.4 nm.

Laser cooling scheme with the transition X2\(\sum_{1/2}^{ + }\) − (2)2\(\sum_{1/2}^{ + }\) of the molecule SrAt.
The lowest SrAt temperature that can be reached through the Doppler and Sysphus laser cooling processes are in the order of the μK, as shown with the following corresponding experimental parameters needed below43,45:
where amax and Tini are the molecule’s maximum deceleration and initial temperature, respectively, and Vrms is the rms velocity. The parameters m and Lmin are the molecule’s mass and minimum slowing distance, respectively. Ne is the number of excited states in the main cycling transition, and Ntot is the number of the excited states connected to the ground state plus Ne. According to the SrAt laser cooling scheme, the ratio Ne/Ntot equals 1/5, considering the vibrational ground and excited states. T.D. and Tr are, respectively, the Doppler and recoil temperatures. The slowing distance Lmin is relatively small; however, a close scale (12 mm) stopping length has been proposed to slow down hydrogen atoms44.
Following the same investigation type, we considered the transition X2\(\sum_{1/2}^{ + }\) − (1)2Π1/2 for the molecule BaAt. The FCF and the transition dipole moment for this transition are represented respectively in Figs. 3 and 4. This system shows a more evident FCF scheme diagonal feature compared to that of SrAt, where (v′,v) transitions among (0,0), (1,1) and (2,2) vibrational states have a higher probability compared to non-diagonal ones. The corresponding branching ratio values and radiative lifetime are given in Table 8. Several laser cooling loops can be built up for this molecule, with different numbers of cycles (N) for photon absorption/emission (Eq. 3). The number of cycles (N) and the corresponding schemes are given in Table 9, along with the corresponding experimental parameters (L, Vrms, amax, and Ntot). Ne was considered equal to one for all schemes.
The slowing distances of the three schemes are within the experimental conditions for the cooling of a molecule, as they range between 2.52 mm and 1.36 m. The laser cooling scheme (A) is represented in Fig. 6a, where the solid red lines represent the driven lasers, and the dotted lines represent the spontaneous decays. Their main pumping and repumping laser wavelengths, in addition to the FCF (fν′ν) and the vibrational branching ratios Rν′ν among different transitions, are also represented. The wavelength of the primary pumping laser is λ0′0 = 1041.2 nm, and those of the repumping laser are λ0′1 = 1053.7 nm, λ0′2 = 1068.2 nm, and λ0′3 = 1083.3 nm in the near-infrared region. The graphical representation of the scheme (B) (by using three lasers) is given in Fig. 6b. Scheme (C) represents another suggested scheme with four lasers for the molecule BaAt, given in Fig. 6c. This last scheme presents new pumping lasers whose wavelengths are λ1′2 = 1055.3 nm and λ1′3 = 1070.0 nm. The lowest attainable Doppler and recoil temperatures for BaAt are TD = 104.0 μK, and Tr = 51.9 nK among all three schemes as they only depend on the value of τ and λ00 for a given molecule.

Laser cooling schemes with the transition X2\(\sum_{1/2}^{ + }\) − (1)2Π1/2 of the molecule BaAt.
Conclusion
The MRCI + Q technique allowed the investigation of 63 electronic states with and without considering the spin–orbit coupling effect of the doublet and quartet electronic states of SrAt and BaAt molecules. The adiabatic potential energy curves and the static dipole moment curves have been plotted for these electronic states. The spectroscopic constants \({{\text{T}}}_{{\text{e}}}\), Re, \({\upomega }_{{\text{e}}}\), Be were deduced here for the first time to the best of our knowledge. The results are compatible with our previously published work of molecules containing alkaline earth metals and halogens, obtained using the same calculation method25,26. Based on the canonical function approach, the values of the ro-vibrational constants Ev, Bv, Dv, with the abscissas of turning points Rmin and Rmax, have been calculated for the ground and some low-lying excited states of the BaAt molecule. Transition parameters such as the FCFs, the radiative lifetime, the branching ratio, and the experimental parameters for the molecules SrAt and BaAt confirm their candidacy for Doppler and Sysphus laser cooling. The proposed laser cooling schemes may open the way for new laser cooling experiments.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Herzberg, G. Molecular Spectra and Molecular Structure I. Spectra of Diatomic Molecules (Van Nostrand Reinhold, 1950).
Sauval, A. J. predicted presence and tentative identification of new molecules in the pure S star R CYG. Astron. Astrophys. 62, 295–298 (1978).
Anderson, M. A., Allen, M. D. & Ziurys, L. M. Millimeter-wave spectroscopy of MgF: Structure and bonding in alkaline-earth monofluoride radicals. J. Chem. Phys. 100, 824 (1994).
Cernicharo, J. & Guélin, M. Metals in IRC+10216: Detection of NaCl, AlCl, and KCl, and tentative detection of AlF. Astron. Astrophys. 183, L10 (1987).
Alkemade, C. T. J., Hollander, T., Snelleman, W. & Zeegers, P. J. T. Metal Vapors in Flames (Pergamon Press, 1982).
Pérez-Ríos, J., Herrera, F. & Krems, R. V. External field control of ́ collective spin excitations in an optical lattice of 2Σ molecules. New J. Phys. 12, 103007 (2010).
Otto, R., Mikosch, J., Trippel, S., Weidemüller, M. & Wester, R. Nonstandard behavior of a negative ion reaction at very low temperatures. Phys. Rev. Lett. 101, 063201 (2008).
Biennier, L. et al. Low temperature reaction kinetics of C.N.− + HC3N and implications for the growth of anions in Titan’s atmosphere. Icarus 227, 123 (2014).
Shuman, E. S., Barry, J. F. & DeMille, D. Laser cooling of a diatomic molecule. Nature (London) 467, 820 (2010).
Hummon, M. T. et al. 2D magneto-optical trapping of diatomic molecules. Phys. Rev. Lett. 110, 143001 (2013).
Zhelyazkova, V. et al. Laser cooling and slowing of CaF molecules. Phys. Rev. A 89, 053416 (2014).
Lane, I. C. Ultracold fluorine production via Doppler cooled BeF. Phys. Chem. Chem. Phys. 14, 15078–15087 (2012).
Yan, K. et al. A new route for laser cooling and trapping of cold molecules: Intensity-gradient cooling of MgF molecules using localized hollow beams. New J. Phys. 22, 033003 (2020).
Rao, V. S. & Rao, P. T. Emission spectrum of MgCl : A new doublet system. Indian J. Phys. 37, 640–644 (1963).
Morgan, E. & Barrow, R. F. Rotational analysis of the A2Π - X2Σ+ system of MgCl. Nature 192, 1182–1185 (1961).
Patel, M. M. & Patel, P. D. Rotational analysis of the A2Π - X2Σ+ system of MgCl molecule. Indian J. Phys. 42, 254–259 (1968).
Walker, K. A. & Gerry, M. C. L. Investigation of the pure rotational spectrum of magnesium monobromide by Fourier transform microwave spectroscopy. J. Chem. Phys. 107, 9835 (1997).
Kuz’menko, N. E. & Chumak, L. V. Comparative analysis and search for regularities in the behavior of Franck-Condon factors for diatomic molecules. J. Quant. Spectros. Radiat. Transfer 35, 419–429 (1986).
Wu, D. et al. Ab initio calculations on potential energy curves and radiative lifetimes for the band systems A2Π - X2Σ+ of magnesium monohalides MgX (X= F, Cl, Br, I). Spectrochim. Acta, Part A 150, 499–503 (2015).
Wan, M. et al. Effects of spin-orbit coupling on laser cooling of BeI and MgI. J. Chem. Phys. 143, 164312. https://doi.org/10.1063/1.4934719 (2015).
Yang, R., Tang, B. & Yu Han, X. Laser cooling of InF, InCl and InH with an ab initio study. RSC Adv. 9, 31543 (2019).
Yang, Q.-S. & Gao, T. The feasibility of laser cooling: An investigation of ab initio of MgBr, MgI and MgAt molecular. Spectrochim. Acta, Part A: Mol. Biomol. Spectrosc. 231, 118107 (2020).
Werner, H. J. & Knowles, P. J. An efficient internally contracted multiconfiguration-reference configuration interaction method. J. Chem. Phys. 89, 5803 (1988).
Werner, H. J., et al. version, 2010.1, a package of ab initio programs, 2010, see http://www.molpro.net/info/users
Allouche, A. R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 32, 174 (2011).
Küchle, W., Dolg, M., Stoll, H. & Preuss, H. Ab initio pseudopotentials for Hg through Rn. Mol. Phys. 74, 1245 (1991).
Lim, I. S., Stoll, H. & Schwerdtfeger, P. Relativistic small-core energy-consistent pseudopotentials for the alkaline-earth elements from Ca to Ra. J. Chem. Phys. 124, 034107 (2006).
Stoll, H., Metz, B. & Dolg, M. Relativistic energy-consistent pseudopotentials–recent developments. J. Comput. Chem. 23, 767 (2002).
Prascher, B. P., Woon, D. E., Peterson, K. A., Dunning, T. H. Jr. & Wilson, A. K. Gaussian basis sets for use in correlated molecular calculations. VII. Valence, core-valence, and scalar relativistic basis sets for Li, Be, Na, and Mg. Theo. Chem. Acc. 128, 69–82. https://doi.org/10.1007/s00214-010-0764-0 (2011).
Kaupp, M., Schleyer, P. V. R., Stoll, H. & Preuss, H. Pseudopotential approaches to Ca, Sr, and Ba hydrides. Why are some alkaline earth MX2 compounds bent?. J. Chem. Phys. 94, 1360 (1991).
Elmoussaoui, S., El-Kork, N. & Korek, M. Electronic structure of the ZnCl molecule with rovibrational and ionicity studies of the ZnX (X=F, Cl, Br, I) compounds. Comput. Theor. Chem. 1090, 94–104 (2016).
Albritton, D. L., Harrop, W. J., Schmeltekopf, A. L. & Zare, R. N. Calculation of centrifugal distortion constants for diatomic molecules from RKR potentials. J. Mol. Spectrosc. 46, 25–36 (1973).
Hutson, J. M. Centrifugal distortion constants for diatomic molecules: an improved computational method. J. Phys. B 14, 851–857 (1981).
Tellinghuisen, T. An improved method for the direct computation of diatomic centrifugal distortion constants. J. Mol. Spectrosc. 122, 455–461 (1987).
Korek, M. & El-Kork, N. Solution of the rovibrational Schrödinger equation of a molecule using the volterra integral equation. Adv. Phys. Chem. https://doi.org/10.1155/2018/1487982 (2018).
El-Kork, N. et al. Electronic structure with dipole moment calculations of the high-lying electronic states of BeH, MgH and SrH molecules. J. Phys. Commun. 2(5), 055030 (2019).
Zeid, I., Al Abdallah, R., El-Kork, N. & Korek, M. Ab-initio calculations of the electronic structure of the alkaline earth hydride anions XH−(X= Mg, Ca, Sr, and Ba) toward laser cooling experiment. Spectrochim. Acta A Mol. Biomol. Spectrosc. 224, 117461 (2020).
Di Rosa, M. D. Laser-cooling molecule. Eur. Phys. J. D At. Mol. Opt. Plas. Phys. 31(2), 395–402 (2004).
Le Roy, R. J. LEVEL: A computer program for solving the radial schrödinger equation for bound and quasibound levels. J. Quant. Spectrosc. Radiat. Transf. 186, 167–178. https://doi.org/10.1016/j.jqsrt.2016.05.028 (2017).
Bernath, P. F. Spectra of Atoms and Molecules (Oxford University Press, 2020).
Kang, S.-Y. et al. Ab initio study of laser cooling of AlF+ and AlCl+ molecular ions. J. Phys. B At. Mol. Opt. Phys. 50(10), 105103 (2017).
Lane, C. Production of ultracold hydrogen and deuterium via Doppler-cooled Feshbach molecules. Phys. Rev. A 92, 022511 (2015).
Li, R. et al. Laser cooling of the SiO+ molecular ion: A theoretical contribution. Chem. Phys. 525, 110412. https://doi.org/10.1016/j.chemphys.2019.110412 (2019).
Metcalf, H. J. & van der Straten, P. Laser cooling and trapping of atoms. J. Opt. Soc. Am. B 20, 887–908 (2003).
Moussa, A., El-Kork, N. & Korek, M. Laser cooling and electronic structure studies of CaK and its ions CaK ±. New J. Phys. 23(1), 013017. https://doi.org/10.1088/1367-2630/abd50d (2021).
Acknowledgements
This publication is based upon work supported by the Khalifa University of Science and Technology under Award No. CIRA-2019-054. Khalifa University, a high-power computer was used to complete this work. Faculty: N.E.K(initials) is partly supported by the internal grant (8474000336-KU-SPSC).
Author information
Authors and Affiliations
Contributions
A.M. and I.Z. contributed in data collection, data analysis and interpretation, and drafting the article. N.E.K. and M.K. contributed in the conception, supervision and critical review of this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Madi, A., El-Kork, N., Zeid, I. et al. Theoretical electronic structure with spin–orbit coupling effect of the molecules SrAt and BaAt for laser cooling studies. Sci Rep 14, 6289 (2024). https://doi.org/10.1038/s41598-024-53564-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53564-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
