
Abstract
In this study, a new nanocomposite was created by combining copper-doped nickel ferrite (NiCuFe2O4) nanoparticles with MCM-48 (Mobil Composition of Matter No. 48) on a graphene oxide (GO) substrate functionalized with poly(ρ-phenylenediamine) abbreviated as (PρPD). This nanocomposite was developed to investigate its potential for enhancing the function of a supercapacitor in energy storage. Following NiCuFe2O4@MCM-48 preparation, Hummer’s technique GO was applied. In-situ polymerization of NiCuFe2O4@MCM-48/GO nanoparticles with ρ-phenylenediamine (ρPD) in the presence of ammonium persulfate (APS) produced PρPD, a conductive polymer. Structural characterization of the nanocomposite includes FTIR, XRD, VSM, TGA-DTG, EDX, and FE-SEM. Results from BET indicate a pore size increase of up to 5 nm. Fast ion penetration and higher storage in capacitor material are explained by this. Additionally, the nanocomposite’s electrochemical performance was evaluated using GCD and CV tests. The NiCuFe2O4@MCM-48/GO/PρPD nanocomposite has a specific capacitance of 203.57 F g−1 (1 A g−1). Furthermore, cyclical stability is essential for energy storage applications. The nanocomposite retains 92.5% of its original capacitance after 3000 cycles, indicating outstanding electrochemical stability.
Similar content being viewed by others


Introduction
Excessive energy use and economic expansion have caused environmental contamination, endangering human health and natural systems. The need for alternative energy sources such as solar, wind, tidal, and nuclear power has arisen due to the conflict between fossil fuel burning and human energy needs. Efficient energy storage is crucial for successful energy management1,2,3. To choose the best energy storage option, consider variables such as durability, reliability, storage capacity, cost, and environmental effect. Supercapacitors, a potential energy storage technology, are popular for their compact size, lightweight, fast charge/discharge rates, high power density, and long cycle life4. In summary, supercapacitors store energy in two ways. The first technique, the electric double-layer capacitor (EDLC), involves ion adsorption on the electrode surface without chemical reactions. The second process, Faraday capacitance (sometimes called pseudocapacitance), involves fast redox reactions on the electrode’s surface5. Carbon electrode materials, such activated carbon, graphene oxide, and carbon nanotubes, are often utilized in supercapacitors, fuel cells, solar cells, and batteries owing to their porous structure and huge cross-sectional area. EDLC-type behavior is seen in these materials due to ion adsorption at the electrode contact6. Moreover, supercapacitor research has focused on graphene oxide (GO) due to its higher capacity of up to 189 F g−1, mechanical properties7,8, conductivity9, thermal properties10, biocompatibility11,12,13, low cost, easy processing14 and quasi-capacitive effect from oxygen-containing functional groups on baseplates15,16. However, for materials like metal oxides9,17 and conductive polymers (e.g., polyaniline18 and polypyrrole19), which exhibit quasi-capacitive behavior, charge storage is based on fast Faradic reactions20. Although the second mechanism stores more charge than the first, its cycle stability is minimized owing to lesser conductivity. To increase energy density and cycle stability without sacrificing power density, a hybrid technique combines carbon-based and quasi-capacitive materials21. Additionally, other materials may be used to enhance the characteristics of these compounds22. Porous compounds like MCM-48 are one kind23. The term “Mobil Composition of Matter” (MCM) was first used to refer to a group of mesoporous materials that were originally created by researchers at Mobil in 1992. MCM-41 and MCM-48, which stand for Mobil Composition of Matter No. 41 and Mobil Composition of Matter No. 48 respectively, are extensively researched mesoporous molecular sieves24. Porous carbon electrodes perform well due to their high electrical conductivity, charge accumulation surface area, large pore diameter, and pore connectivity enabling fast ionic movement and electrolyte wetting25. Additionally, their cheap cost, various forms, and chemical stability make them popular supercapacitor electrodes26. High specific capacitance in carbon electrodes demands increased surface area and porosity to improve composites’ surface-to-volume ratio and electrochemical reaction contact27,28. This study implanted NiCuFe2O4 nanoparticles with MCM-48 on a graphene oxide (GO) substrate functionalized with poly(p-phenylenediamine) (PρPD). This nanocomposite was researched for energy storage. Energy storage applications need cycling stability. The NiCuFe2O4@MCM-48/GO/PρPD nanocomposite maintains 92.5% of its original capacitance after 3000 cycles, demonstrating excellent electrochemical stability (Scheme 1).

Preparation of NiCuFe2O4@MCM-48/GO/PρPD nanocomposite.
Experimental
Materials and methods
The chemical materials, including reagents and solvents, used in this investigation were of high purity and purchased from international companies (Merck, Aldrich, and Fluka). Various analytical techniques were employed to study the structure of the nanocomposite. Fourier-transform infrared (FT-IR) spectroscopy (Shimadzu FT-8400 s model, Japan) was performed with a frequency range of 400–4000 cm−1 and using potassium bromide pellets for functional group identification. Field-Emission Scanning Electron Microscopy (FE-SEM) analysis (KYKY, EM8000 model) and Energy Dispersive X-Ray Analysis (EDX) analysis (BRUKER instrument) were used for morphology and elemental analysis, respectively. TGA/DTA was recorded using a BÄHR apparatus, X-ray diffraction analysis (XRD) was recorded with a BRUKER D8 ADVANCE device, and a N2 adsorption–desorption isotherm was recorded and analyzed using a MICROMERITICS ASAP 2020 instrument. Vibrating-sample magnetometer analysis (VSM) was carried out by Meghnatis Daghigh Kavir-MDKB-Kashan.
Preparation of nanocomposite
Preparation of NiCuFe2O4
NiCuFe2O4 polycrystal is synthesized by a simultaneous precipitation method. In the first place, a 0.5 M solution was made separately from each of the salts, namely nickel nitrate, copper nitrate, and iron nitrate. Afterward, all three solutions were poured into the flask for stirring until all the salts were completely dissolved. In the next step, 0.2 M NaOH solution was added to the flask at a temperature of 60 °C, and then the temperature was raised to 100 °C and stirred for 6 h. Subsequently, the nanoparticles that were initially deposited were subjected to filtration and centrifugation, followed by drying in an oven at a temperature of 60 °C for a duration of 48 h. Finally, the obtained nanoparticles were ground in an agate mortar and calcined for 5 h at a temperature of 600 °C inside the furnace.
Preparation of NiCuFe2O4@MCM-48
For the in-situ synthesis of NiCuFe2O4@MCM-48, 2.4 g (6.6 mmol) of n-hexadecyltrimethylammonium bromide (CTAB) as a template was dissolved in 50 g of deionized water and 28 g (0.87 mol) of industrial ethanol. Then, after adding 0.5 g of NiCuFe2O4 and stirring the mixture for 15 min, 3.4 g (0.2 mol) of ammonia was added to the surfactant solution. After stirring the solution for 10 min, 3.4 g of TEOS were added and the mixture was stirred for an additional 2 h at ambient temperature. The solid material was then obtained by filtration, washed with distilled water through centrifugation, and dried at ambient temperature. To remove the template, the material was placed in a furnace for 6 h at a temperature of 823 K (equivalent to 550 °C).
Preparation of GO
The preparation of graphene oxide involved a modified Hummer method, as described in previous studies12. To begin, 1 g of graphite was combined with 23 ml of 98% sulfuric acid in a 1000 ml beaker. The mixture was stirred for a few minutes before adding 0.5 g of sodium nitrate (NaNO3). The stirring continued for 25 min at a temperature of 66 ℃. To ensure the complete dissolution of components, the beaker was subjected to an ultrasonic bath at room temperature for 30 min. Following that, 3 g of potassium permanganate were gradually added over a period of 1 h with continuous stirring, while the beaker was maintained in an ultrasonic ice bath. This process continued until a dark green, sludge-like substance was formed. To complete the reaction, the mixture was stirred for an additional 30 min in the ultrasonic bath at room temperature. Next, 50 ml of distilled water were added, and the mixture was stirred for 30 min at a temperature of 98 °C. To induce a significant amount of foam formation, 700 ml of distilled water and 12 ml of hydrogen peroxide were added simultaneously. Afterwards, the pH of the mixture was adjusted by adding a 2% solution of hydrochloric acid (2 ml of HCl per 100 ml of distilled water). The mixture was left to settle for 1 day, following which the water in the beaker was replaced and the elution process was repeated three times. Ultimately, the precipitate was dried in an oven at a temperature of 60 ℃ for a duration of 24 h.
Preparation of NiCuFe2O4@MCM-48/GO
To load NiCuFe2O4@MCM-48 onto GO sheets, in a 250 ml flask, 0.07 g of NiCuFe2O4@MCM-48, along with 0.4 g of GO, was stirred in 100 ml of DMF at 80 °C for 1 h.
Preparation of NiCuFe2O4@MCM-48/GO/PρPD
During the final stage, the temperature was lowered before introducing a solution consisting of 0.4 g of ρPD dissolved in 20 ml of DMF into the previously prepared mixture. While maintaining the flask in an ice bath, a solution of 2 g of APS in 40 ml of DMF was gradually added dropwise. The mixture was then stirred for 24 h at the same temperature, and the resulting product was obtained through a series of washing steps using ethanol and distilled water (repeated twice), followed by centrifugation and drying at room temperature.
Results and discussion
Characterization
XRD pattern
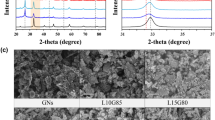
The XRD pattern of NiCuFe2O4@MCM-48/GO/PρPD is shown in Fig. 1. The peaks at 2θ are characteristic of the ferrite material, with the highest peak at 35.676° (311), confirming the formation of the cubic spinel structure29. The cubic phase of spinel ferrites (NiCuFe2O4) is characterized by the presence of peaks indexed as 30.16° (220), 35.64° (311), 43.03° (400), 57.05° (511), 62.77° (440), and 74.54° (533) according to the JCPDS Card No: 00-025-028330,31,32. The identified peaks in 2θ correspond to NiFeO (35.676° (131), 30.287° (022), 37.320° (222), 57.361° (151), 62.997° (044)), Cu (43.621° (111), 50.810° (020), 74.706° (022)), CuO (38.854° (111), 35.571° (− 111), 48.708° (-202), 35.543° (002)), NiCu (43.96 (111), 51.22 (020), 75.37 (022)), NiCuF (35° (311), 31° (220)), and Fe2O3 (24.13° (012), 33.153° (104), 35.612° (110), 49.480° (024), 54.091° (116)). The identified peaks match with the ICDD (International Diffraction Data Center) cards (96-591-0065), (96-431-3208), (96-410-5686), (96-152-4233)33, and (00-033-0664), respectively. The peaks at 2θ = 22.51°, 29.09°, 32.34°, 36.83°, and 43.6° are attributed to the tetragonal SiO2 phase of MCM-48 (JCPDF: 01-082-1235)34. Two distinct XRD peaks at approximately 2θ = 17° and 21° were observed for PρPD35,36, while the main and well-defined diffraction peak of GO was identified at 2θ = 10° (001)37.

XRD pattern of NiCuFe2O4@MCM-48/GO/PρPD.
Fourier-transform infrared spectra
Fourier transform infrared (FTIR) spectroscopy was performed on NiCuFe2O4 (Fig. 2A), MCM-48 (Fig. 2B), NiCuFe2O4@MCM-48 (Fig. 2C), and NiCuFe2O4@MCM-48/GO/PρPD (Fig. 2D) in the range of 4400–440 cm−1. The sharp peaks associated with MCM-48 were consistent with previous reports. A wide absorption band that appears within the range of 3400–3900 cm−1 is typically linked to the presence of Si (OH), Si (OH)2, or Si (OH)3 groups on the surface of the pores. The peak located at 2188 cm−1 corresponds to the stretching vibration of Si–OH, while the band situated at 1314 cm−1 is associated with an asymmetric stretching vibration of Si–O–Si. The band located around 870 cm−1 is attributed to the symmetric stretching vibration, and the peak situated at 460 cm−1 corresponds to the bending vibration of Si–O–Si38,39,40,41. The NiCuFe2O4 spectrum exhibited a band centered around 475 cm−1, which can be attributed to the stretching vibration of a metal–metal (Ni–Cu) band42,43. In the case of spinel ferrites, FTIR bands resulting from ion vibrations within the crystal lattice usually appear in the range of 1000–100 cm−1. The band at a higher frequency (555–600 cm−1) and the band at a lower frequency (v2) (480–495 cm−1) are attributed to the tetrahedral and octahedral complexes of spinel ferrites, respectively. The obtained spectrum indicated a band at approximately 528 cm−1. These intense absorption bands are characteristic of inverted spinel ferrites44. Bands with wavenumbers below 400 cm−1 were not clearly observed due to limitations of our FTIR instrument. The band situated at 3414 cm−1 could be related to the stretching vibration of the –OH group, potentially present as a result of adsorbed water molecules. Additional bands were identified at 2918, 2849, 2356, and 1384 cm−1, which may correspond to C–H stretching modes linked to surfactant molecules attached to the NiCu alloy nanoparticles45,46. The absorption band located at 600 cm−1 may be assigned to the presence of copper ferrite CuFe2O4 within the sample47. The spectrum of NiCuFe2O4@MCM-48 displayed all the peaks that were present in the spectra of NiCuFe2O4 and MCM-48. In the FTIR spectrum of NiCuFe2O4@MCM-48/GO/PρPD, the broad peak observed in the 3400–3750 cm−1 region is assigned to the stretching vibration mode of hydroxyl groups in GO and the stretching mode of the N–H of the secondary amine group in the polymer chain of PρPD. Two peaks are also visible at approximately 1314 and 1228 cm−1, which correspond to the stretching vibrations of the quinone imine units (–C–N– and C–N))10,20,48,49,50,51.

Fourier-transform infrared (FTIR) spectra of samples.
Field-emission scanning electron microscopy
Figure 3A depicts the morphology and structure of MCM-48 quasi-spherical particles, with a dominant particle size of approximately 100–500 nm52. The NiCuFe2O4 nanoparticles are well-covered by the MCM-48 silicate structure, although some parts of the image show the nanoparticles uncovered or inside cracked MCM-48 spheres. Further, Fig. 3B provides a zoomed-in view of Fig. 3A. The morphology of the final product can be observed in Fig. 3C, which shows NiCuFe2O4-covered nanoparticles with MCM-48, composite GO sheets, and PρPD polymer strands forming a matrix. Furthermore, Fig. 3D is a zoomed-in image of Fig. 3C.

FE-SEM image of (A) NiCuFe2O4@MCM-48, (B) zoomed image of (A), (C) NiCuFe2O4@MCM-48/GO/PρPD and (D) zoomed image of (C).
Energy-dispersive X-ray spectroscopy
Based on the results obtained from the EDX spectrum of this nanocomposite in Fig. 4A, the existence of NiCuFe2O4 nanoparticles covered by MCM-48 was confirmed by observing peaks for Nickel, Copper, Iron, oxygen, and Silicon. Besides, the presence of GO sheets and PρPD polymer was confirmed by observing three peaks of carbon, nitrogen, and a sharpened peak of oxygen.

EDX spectrum of NiCuFe2O4@MCM-48/GO/PρPD (A), N2 adsorption–desorption isotherm of MCM-48, NiCuFe2O4@MCM-48 and NiCuFe2O4@MCM-48/GO/PρPD (B), The TGA, DTG curve (C (A), (B)) and VSM (D) of NiCuFe2O4@MCM-48/GO/PρPD.
BET analysis
Figure 4B shows the Nitrogen adsorption–desorption isotherms for MCM-48, NiCuFe2O4@MCM-48, and NiCuFe2O4@MCM-48/GO/PρPD. The isotherms display a type IV adsorption–desorption behavior with a clearly distinguishable hysteresis loop, which is characteristic of mesoporous materials53. The information in Table 1 shows that adding GO sheets and PρPD polymer to the NiCuFe2O4@MCM-48 composite material that was made on-site led to a gradual decrease in both surface area and pore volume compared to pure MCM-48. The presence of organic groups led to pore blockages, reducing the surface area of the mesoporous product52. Meanwhile, the pore size gradually increased due to the continuous decrease in micropores, and rich mesoporous structures formed. Over time, the pore size increased gradually as a result of a continuous decrease in micropores and the development of rich mesoporous structures. It is worth noting that high surface area does not necessarily translate to high capacitance values, and this holds true for all pore types54. The microporous structure provides channels for ion transfer, lowers ion-transport resistance, and, in turn, facilitates charge storage55,56.
Thermogravimetric analysis (TGA)
Thermogravimetric analysis (TGA) was performed under an argon atmosphere over the temperature range of 50–900 °C, as depicted in Fig. 4C(A). The weight loss observed during the heating process can be divided into three stages. In the first stage, there was a minor weight loss of around 3% at temperatures below 100 °C, which is likely due to the physical adsorption of water on the external surface or the removal of water from the mesoporous channels57. The second stage exhibited a sharp drop in weight, related to the removal of organic components at a temperature below 400 °C (with a decomposition rate of approximately 50% by weight)58, which was also confirmed by DTG curve Fig. 4C(B). The reduction in mass between 200 and 300 °C can be attributed to the thermal decomposition of oxygen-containing functional groups present in GO, such as carboxyl, epoxide, and hydroxyl groups12. Furthermore, the thermal degradation of PρPD polymer chains likely begins at a temperature range of 250 to 340 °C59. The final stage is observed in the temperature range of 400–600 °C and corresponds to the elimination of carbonaceous residues58.
Vibrating‑sample magnetometer (VSM)
A vibrating-sample magnetometer (VSM), shown in Fig. 4D, was used to check the magnetic properties of the NiCuFe2O4@MCM-48/GO/PρPD that was synthesized. The magnetization of the synthesized composite was measured at approximately 1.56 emu g−1. Compared to other research on NiCuFe2O4 nanoparticles, the composite has a relatively low magnetic saturation60. This may be because the nanoparticles are confined within the MCM-48 porous material and are surrounded by polymer and GO sheets.
Electrochemical measurements
The electrochemical experiments were carried out using a Autolab PGST200 electrochemical analyzer in a routine three-electrode configuration, with an Ag/AgCl reference electrode and a Platinum wire counter electrode. The working electrode was prepared by mixing the electroactive material (NiCuFe2O4@MCM-48/GO/PρPD), acetylene black, and polytetrafluoroethylene (PTFE) (80:10:10 wt%) in NMP under ultrasonic conditions. The resulting ink was then sprayed onto a stainless-steel mesh, followed by thermal treatment at 120°C. The capacitive behavior of the synthesized materials was studied in 1.0 M H2SO4 solutions. In the three-electrode system, cyclic voltammetry tests were carried out over a potential range of − 0.2 V to 0.8 V at various scan rates from 1 to 10 mV s−1. Additionally, galvanostatic charge–discharge measurements were conducted at different current densities (1–5 A g−1) with a potential window of − 0.2 to 0.8 V.
Electrochemical application of the NiCuFe2O4@MCM-48/GO/PρPD
The electrochemical behavior of the NiCuFe2O4@MCM-48/GO/PρPD electrode was analyzed by performing cyclic voltammetry (CV) measurements in 1.0 M H2SO4 electrolyte within the potential window of − 0.2 to 0.8 V (vs. AG/AgCl) at varying scan rates ranging from 1.0 to 10 (mV s−1). As illustrated in Fig. 5A, the resulting CV curves display anodic and cathodic peaks in a quasi-rectangular shape, suggesting the existence of both electrical double-layer capacitors (EDLC) and pseudocapacitive behavior in the synthesized material. The distinctive redox peaks observed in the cyclic voltammogram can be attributed to the Faradaic redox reactions taking place at the surface of the electrode material. As the scan rate increases, it becomes apparent that the position of the anodic and cathodic peaks shifts towards more positive and negative potentials, respectively. This polarization phenomenon implies that the transfer of electrolyte ions onto the surface of the material is restricted at higher scan rates. The shape of the cyclic voltammogram remains relatively unchanged at different scan rates within the measured potential window, indicating the excellent rate capability of the electrode materials61,62.

CV curves of NiCuFe2O4@MCM-48/GO/PρPD at various scan rates in 1.0 M H2SO4 electrolyte solutions (A), GCD curves (second cycle) of NiCuFe2O4@MCM-48/GO/PρPD at different current densities in 1.0 M H2SO4 electrolyte solutions (B); variation of the specific capacitance values versus the current density (C) and cycling stabilities of the NiCuFe2O4@MCM-48/GO/PρPD over 3000 GCD 1.0 M H2SO4 electrolyte solutions (D).
To assess the storage capacitance of the nanocomposite, galvanostatic charge–discharge (GCD) test was conducted. The resulting GCD curves (second cycle), shown in Fig. 5B, were obtained at different current densities (1–5 A g−1) in 1.0 M H2SO4 electrolyte within the potential range of − 0.2 to 0.8 V. The GCD cycles exhibited nonlinear characteristics, suggesting that the capacitive behavior of the electrode was due to a combination of electrical double layer and pseudocapacitive processes63. As expected, the nonlinear shape of the charge/discharge curves is due to the quasi-capacitive behavior of metal oxides caused by the electrochemical reaction of adsorption–desorption or redox at the electrode–electrolyte interface. As is clear from the curves, discharge/charge times are almost parallel, indicative of good coulombic efficiency and electrochemical reversibility of NiCuFe2O4@MCM-48/GO/PρPD nanocomposite. It is also worth noting that the IR drop remains unchanged as the current density increases, indicating the exceptional capacitor performance and the reversible nature of the electrode materials, the results being in perfect agreement with the CV curves.
Specific capacitance values were calculated using Eq. (1) for various current densities.
The following symbols are used: Cs (F g−1) for specific capacitance, I (A) for discharging current, \(\Delta t (s)\) for discharge time, m (g) for the mass of the electrode material, and \(\Delta v\) (V) for potential difference. Figure 5C illustrates the specific capacitance of NiCuFe2O4@MCM-48/GO/PρPD electrode materials at various current densities. As the current density increases, the specific capacitance decreases due to restricted ion diffusion into the active sites. Specifically, the specific capacitance declined from 203.57 F g−1 at 1 A g−1 to 64.85 F g−1 at 5 A g−1.
The longevity of an electrode material is crucial for energy storage applications. To assess the cycling stability of the synthesized electrode materials in 1.0 M H2SO4, cyclic tests were performed at a current density of 3 A g−1, as illustrated in Fig. 5D. The results demonstrate that the electrode material’s capacitance retention after 3000 cycles is 92.5% of its original capacitance, indicating the exceptional electrochemical stability of the NiCuFe2O4@MCM-48/GO/PρPD electrode materials.
Table 2 presents a comparison between the capacitor ability of the NiCuFe2O4@MCM-48/GO/PρPD nanocomposite and other materials reported in recent research. The results reveal the high specific capacitance of the nanocomposite compared to each of its components in other compounds, along with its exceptional ability to maintain the initial capacity for reuse. Therefore, this makes it a potential candidate for use in supercapacitor applications.
Conclusion
To summarize, a composite material consisting of NiCuFe2O4 coated with MCM-48, GO nanosheets, and PρPD (produced by an in-situ technique) has been created and used as an electrode substance in supercapacitor applications. Using NiCuFe2O4 as a transition metal oxide along with MCM-48 porous carbon material improves the electrochemical efficiency of composite electrodes by adding redox centers that make it easier for charge to be stored through redox reactions. GO demonstrates a significant capacitance in capacitive applications because to the supplementary quasi-capacitive impact of oxygen-containing functional groups bonded to its base plates. Moreover, PρPD is an electrically conductive polymer capable of undergoing redox reactions with an electrolyte. These qualities provide them advantageous options for electrode components in supercapacitors. Analytical methods including FT-IR spectroscopy, XRD, VSM, TGA-DTG, EDX, and FE-SEM were used to assess the physical and chemical characteristics of the NiCuFe2O4@MCM-48/GO/PρPD nanocomposite. According to the findings from BET analysis, the produced nanocomposite has a maximum pore size of 5 nm. This supports the need for establishing channels that enable rapid ion transport with little resistance, hence enhancing the capacity for charge storage. The nanocomposite materials were analyzed for their electrical supercapacitive performance using cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) experiments, and a cyclic stability investigation of the electrode. The composite exhibits a specific capacitance of 203.57 F g−1 (at 1 A g−1) and retains 92.5% of its original capacitance after 3000 cycles, indicating its suitability for supercapacitor applications.
Data availability
The whole of the data produced or examined during this investigation is included within this published publication.
References
Dubey, R. & Guruviah, V. Review of carbon-based electrode materials for supercapacitor energy storage. Ionics 25, 1419–1445 (2019).
Sharma, K., Arora, A. & Tripathi, S. K. Review of supercapacitors: Materials and devices. J. Energy Storage 21, 801–825 (2019).
Wang, Y. et al. Recent progress in carbon-based materials for supercapacitor electrodes: A review. J. Mater. Sci. 56, 173–200 (2021).
Saini, S., Chand, P. & Joshi, A. Biomass derived carbon for supercapacitor applications. J. Energy Storage 39, 102646 (2021).
Vickers, N. J. Animal communication: When I’m calling you, will you answer too? Curr. Biol. 27, R713–R715 (2017).
Sadat, Z. et al. A comprehensive review about applications of carbon-based nanostructures in wound healing: From antibacterial aspects to cell growth stimulation. Biomater. Sci. 10, 6911 (2022).
Eivazzadeh-Keihan, R. et al. Magnetic chitosan-silk fibroin hydrogel/graphene oxide nanobiocomposite for biological and hyperthermia applications. Carbohydr. Polym. 300, 120246 (2023).
Eivazzadeh-Keihan, R. et al. Biological investigation of a novel nanocomposite based on functionalized graphene oxide nanosheets with pectin, silk fibroin and zinc chromite nanoparticles. J. Biotechnol. 358, 55–63 (2022).
Eivazzadeh-Keihan, R. et al. Fe3O4/GO@ melamine-ZnO nanocomposite: A promising versatile tool for organic catalysis and electrical capacitance. Colloids Surf. A Physicochem. Eng. Asp. 587, 124335 (2020).
Eivazzadeh-Keihan, R. et al. Magnetic graphene oxide–lignin nanobiocomposite: A novel, eco-friendly and stable nanostructure suitable for hyperthermia in cancer therapy. RSC Adv. 12, 3593–3601 (2022).
Eivazzadeh-Keihan, R. et al. Functionalized graphene oxide nanosheets with folic acid and silk fibroin as a novel nanobiocomposite for biomedical applications. Sci. Rep. 12, 1–12 (2022).
Eivazzadeh-Keihan, R. et al. Graphene oxide-lignin/silk fibroin/ZnO nanobiocomposite: A novel bioactive scaffold with antibacterial activity. Sci. Rep. 1, 1 (2021).
Eivazzadeh-Keihan, R. et al. A novel, bioactive and antibacterial scaffold based on functionalized graphene oxide with lignin, silk fibroin and ZnO nanoparticles. Sci. Rep. 12, 1–12 (2022).
Gorab, M. G. et al. Decoration of graphene oxide nanosheets with carboxymethylcellulose hydrogel, silk fibroin and magnetic nanoparticles for biomedical and hyperthermia applications. Nanoscale Adv. 4, 2826 (2022).
Xu, B. et al. What is the choice for supercapacitors: Graphene or graphene oxide? Energy Environ. Sci. 4, 2826–2830 (2011).
Wang, R., Li, X., Nie, Z., Zhao, Y. & Wang, H. Metal/metal oxide nanoparticles-composited porous carbon for high-performance supercapacitors. J. Energy Storage 38, 102479 (2021).
Wang, M., Wang, Y., Dou, H., Wei, G. & Wang, X. Enhanced rate capability of nanostructured three-dimensional graphene/Ni3S2 composite for supercapacitor electrode. Ceram. Int. 42, 9858–9865 (2016).
Cong, H.-P., Ren, X.-C., Wang, P. & Yu, S.-H. Flexible graphene–polyaniline composite paper for high-performance supercapacitor. Energy Environ. Sci. 6, 1185–1191 (2013).
Zhang, H., Wang, J., Shan, Q., Wang, Z. & Wang, S. Tunable electrode morphology used for high performance supercapacitor: Polypyrrole nanomaterials as model materials. Electrochim. Acta 90, 535–541 (2013).
Eivazzadeh-Keihan, R. et al. Effective combination of rGO and CuO nanomaterials through poly (p-phenylenediamine) texture: Utilizing it as an excellent supercapacitor. Energy Fuels 35, 10869–10877 (2021).
Zhang, J. et al. Hierarchical MnO2/activated carbon cloth electrode prepared by synchronized electrochemical activation and oxidation for flexible asymmetric supercapacitors. J. Chem. Eng. 372, 1047–1055 (2019).
Liu, Q., Ma, C., Qiao, W., Ling, L. & Wang, J. Nanoarchitectured MnO2 confined to mesoporous carbon microspheres as bifunctional electrodes for high-performance supercapacitors and lithium-ion capacitors. Ind. Eng. Chem. Res. 61, 1748–1760 (2022).
Vix-Guterl, C. et al. Supercapacitor electrodes from new ordered porous carbon materials obtained by a templating procedure. J. Mater. Sci. Eng. B 108, 148–155 (2004).
Singh, P. K. & Sharma, K. Mesoporous silica nanomaterial: An over view. Mater. Today Proc. 45, 3458–3461 (2021).
Frackowiak, E. & Beguin, F. Carbon materials for the electrochemical storage of energy in capacitors. Carbon 39, 937–950 (2001).
Xia, K., Gao, Q., Jiang, J. & Hu, J. Hierarchical porous carbons with controlled micropores and mesopores for supercapacitor electrode materials. Carbon 46, 1718–1726 (2008).
Wang, Y. et al. Ball milling-assisted synthesis and electrochemical performance of porous carbon with controlled morphology and graphitization degree for supercapacitors. J. Energy Storage 38, 102496 (2021).
Zhan, Y. et al. Preparation of highly porous activated carbons from peanut shells as low-cost electrode materials for supercapacitors. J. Energy Storage 34, 102180 (2021).
Singh, G. et al. Effect of mixed ternary transition metal ferrite nanocrystallites on thermal decomposition of ammmonium perchlorate. Thermochim. Acta 477, 42–47 (2008).
Salavati, H., Taei, M., Rasouli, N., Zohoor, A. & Ashian, G. Application of Spinel-structured NiCuFe2O4 nanoparticles for determination of naproxen in the presence of acetaminophen. Appl. Chem. 14, 119–134 (2019).
Kazempour, S. & Naeimi, H. Design, fabrication and characterization of multi-yolk@ shell NiCuFe2O4@ mSiO2 mesoporous nanocomposite spheres for the synthesis of pyrimido-quinolines under solvent-free conditions. ChemistryOpen 12, e202300053 (2023).
Audi, D. M. Synthesis of doped nickel ferrite Wi elucidation and photocatalytic study interdiscipline. Res. J. Chem. 7, 6–12 (2017).
Murthy, Y., Viswanath, I. K., Rao, T. K. & Singh, R. Synthesis and characterization of nickel copper ferrite. Int. J. Chemtech Res. 1, 1308–1311 (2009).
Qin, H. et al. Photoluminescence and phosphorescence from MCM-48 nanoparticle-embedded composite nanofibers prepared by electrospinning. RSC Adv. 2, 11207–11210 (2012).
Dong, Y. Z. & Choi, H. J. Electrorheological characteristics of poly (diphenylamine)/magnetite composite-based suspension. Materials 12, 2911 (2019).
Min, Y.-L., Wang, T., Zhang, Y.-G. & Chen, Y.-C. The synthesis of poly (p-phenylenediamine) microstructures without oxidant and their effective adsorption of lead ions. J. Mater. Chem. 21, 6683–6689 (2011).
Ameer, S. & Gul, I. H. Influence of reduced graphene oxide on effective absorption bandwidth shift of hybrid absorbers. PLoS ONE 11, e0153544 (2016).
Benhamou, A., Baudu, M., Derriche, Z. & Basly, J.-P. Aqueous heavy metals removal on amine-functionalized Si-MCM-41 and Si-MCM-48. J. Hazard. Mater. 171, 1001–1008 (2009).
Gu, X. et al. Adsorption of methyl violet onto mesoporous MCM-48 from aqueous solution. J. Nanosci. Nanotechnol. 14, 4655–4663 (2014).
Benhamou, A.-F., Basly, J., Baudu, M., Derriche, Z. & Hamacha, R. Amino-functionalized MCM-41 and MCM-48 for the removal of chromate and arsenate. J. Colloid Interface Sci. 404, 135–139 (2013).
Hu, X. N. et al. Adv. Mater. Res. 148–153 (Trans Tech Publ).
Mohamed Saeed, G., Radiman, S., Gasaymeh, S. S., Lim, H. & Huang, N. M. Mild hydrothermal synthesis of Ni–Cu nanoparticles. J. Nanomater. 2010, 1 (2010).
Taqui, S. N. et al. Bioremediation of textile industrial effluents using nutraceutical industrial spent: Laboratory-scale demonstration of circular economy. Nanomaterials 12, 1684 (2022).
Bhujun, B., Tan, M. T. & Shanmugam, A. S. Study of mixed ternary transition metal ferrites as potential electrodes for supercapacitor applications. Results Phys. 7, 345–353 (2017).
Ahmed, J. et al. Bimetallic Cu–Ni nanoparticles of varying composition (CuNi3, CuNi, Cu3Ni). Colloids Surf. A Physicochem. Eng. Asp. 331, 206–212 (2008).
MacPhail, R., Strauss, H., Snyder, R. & Elliger, C. Carbon-hydrogen stretching modes and the structure of n-alkyl chains. 2. Long, all-trans chains. J. Phys. Chem. 88, 334–341 (1984).
Elfalaky, A. Structural and IR studies on Cu–Ti mixed ferrite. Appl. Phys. A 59, 389–393 (1994).
Amiri, A., Baghayeri, M. & Nori, S. Magnetic solid-phase extraction using poly (para-phenylenediamine) modified with magnetic nanoparticles as adsorbent for analysis of monocyclic aromatic amines in water and urine samples. J. Chromatogr. A 1415, 20–26 (2015).
Cao, Y. et al. Eco-friendly mass production of poly (p-phenylenediamine)/graphene oxide nanoplatelet composites and their electrorheological characteristics. Compos. Sci. Technol. 122, 36–41 (2016).
Eivazzadeh-Keihan, R., Radinekiyan, F., Madanchi, H., Aliabadi, H. A. M. & Maleki, A. Graphene oxide/alginate/silk fibroin composite as a novel bionanostructure with improved blood compatibility, less toxicity and enhanced mechanical properties. Carbohydr. Polym. 248, 116802 (2020).
Eivazzadeh-Keihan, R. et al. Functionalization of chitosan by metformin, nickel metal ions and magnetic nanoparticles as a nanobiocomposite for purification of alkaline phosphatase from hen’s egg yolk. J. Chromatogr. A 1679, 463376 (2022).
Ghomari, K. et al. TPD and DSC insights in the basicity of MCM-48-like silica and modified counterparts. Thermochim. Acta 600, 52–61 (2015).
Li, Y. et al. Gradient adsorption of methylene blue and crystal violet onto compound microporous silica from aqueous medium. ACS Omega 5, 28382–28392 (2020).
Kang, Y. et al. Three-dimensionally macroporous nitrogen and boron co-doped graphene aerogels derived from polyaspartamide for supercapacitor electrodes. Mater. Today Commun. 25, 101495 (2020).
Liu, M., Li, W., Ruan, S. & Fei, Y. N-Doped hierarchical mesoporous carbon from mesophase pitch and polypyrrole for supercapacitors. Energy Fuels 34, 5044–5051 (2020).
Liu, B., Liu, L., Yu, Y., Zhang, Y. & Chen, A. Synthesis of mesoporous carbon with tunable pore size for supercapacitors. New J. Chem. 44, 1036–1044 (2020).
Pena, M. et al. Elucidating the local environment of Ti(IV) active sites in Ti-MCM-48: A comparison between silylated and calcined catalysts. Microporous Mesoporous Mater. 44, 345–356 (2001).
Longloilert, R., Chaisuwan, T., Luengnaruemitchai, A. & Wongkasemjit, S. Synthesis and characterization of M-MCM-48 (M= Cr, Ce) from silatrane via sol–gel process. J. Solgel. Sci. Technol. 61, 133–143 (2012).
Amer, I. & Brandt, S. Synthesis and characterization of poly (p-phenylenediamine) and its derivatives using aluminium triflate as a co-catalyst. Cogent Eng. 5, 1499701 (2018).
Begum, S. S. & Bellad, S. Magnetic and magnetoelectric properties of NixCu1–xFe2O4–PbZr0.52Ti0.48O3 single and multilayered thick films. Bull. Mater. Sci. 45, 1–7 (2022).
Hussain, I., Mak, T. & Zhang, K. Boron-doped trimetallic Cu-Ni-Co oxide nanoneedles for supercapacitor application. ACS Appl. Nano Mater. 4, 129–141. https://doi.org/10.1021/acsanm.0c02411 (2021).
Asen, P. & Shahrokhian, S. A high performance supercapacitor based on graphene/polypyrrole/Cu2O–Cu(OH)2 ternary nanocomposite coated on nickel foam. J. Phys. Chem. C 121, 6508–6519. https://doi.org/10.1021/acs.jpcc.7b00534 (2017).
Kumbhar, V. S., Jagadale, A. D., Shinde, N. M. & Lokhande, C. D. Chemical synthesis of spinel cobalt ferrite (CoFe2O4) nano-flakes for supercapacitor application. Appl. Surf. Sci. 259, 39–43. https://doi.org/10.1016/j.apsusc.2012.06.034 (2012).
Benzigar, M. R. et al. Ordered mesoporous C70 with highly crystalline pore walls for energy applications. Adv. Funct. Mater. 28, 1803701 (2018).
Wang, G. et al. Fabrication of monodisperse hollow mesoporous carbon spheres by using “confined nanospace deposition” method for supercapacitor. J. Alloys Compd. 736, 35–41 (2018).
Baro, M. & Ramaprabhu, S. Conductive and nitrogen-enriched porous carbon nanostructure derived from poly (para-phenylenediamine) for energy conversion and storage applications. Appl. Surf. Sci. 503, 144069 (2020).
Aboutalebi, S. H. et al. Comparison of GO, GO/MWCNTs composite and MWCNTs as potential electrode materials for supercapacitors. Energy Environ. Sci. 4, 1855–1865 (2011).
Acknowledgements
The authors express their gratitude for the partial funding provided by the Research Council of Iran University of Science and Technology.
Author information
Authors and Affiliations
Contributions
R.E.-K.: The corresponding author of current study Substantial contributions to the conception, Design of the work, have drafted the work, Writing—Review & Editing, Analysis and interpretation of data and wrote the main manuscript. Z.S.: Have drafted the work, Analysis and interpretation of data, substantively revised it. Wrote the main manuscript and prepared figures. V.D.-E.: Analysis and interpretation of data, substantively revised it, wrote the main manuscript and prepared figures. S.D.: Analysis and interpretation of data, substantively revised it, wrote the main manuscript and prepared figures. A.K.: Analysis and interpretation of data, substantively revised it. A.M.: The corresponding (submitting) author of current study, Substantial contributions to the conception, have drafted the work, Analysis and interpretation of data and wrote the main manuscript, Writing—Review & Editing, substantively revised it.
Corresponding authors
Ethics declarations
Competing interests
The authors have stated that they do not have any financial or personal relationships that could have affected the work presented in this paper, and therefore they do not have any competing interests to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sadat, Z., Eivazzadeh-Keihan, R., Daneshvari-Esfahlan, V. et al. Fabrication of a novel porous nanostructure based on NiCuFe2O4@MCM-48, embedded with graphene oxide/poly (p-phenylenediamine) to construct an efficient supercapacitor. Sci Rep 14, 3137 (2024). https://doi.org/10.1038/s41598-024-53241-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53241-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
