
Abstract
Cerebral ischaemia/reperfusion (I/R) injury induces irreversible brain injury and causes functional impairment. Ubiquitination plays a crucial role in protein degradation, but its role in cerebral I/R injury remains unclear. Differentially expressed genes in stroke were identified by analysing the microarray dataset GSE119121. Cerebral I/R was simulated in vitro by treating human microglial HMC3 cells with oxygen–glucose deprivation/reperfusion (OGD/R). Cell viability was tested by Cell Counting Kit 8 (CCK-8) assays, and pyroptosis was examined by flow cytometry. Lactate dehydrogenase (LDH) and inflammatory cytokine secretion were measured by LDH cytotoxicity assays and enzyme-linked immunosorbent assay (ELISA), respectively. The cerebral I/R animal model was established by middle cerebral artery occlusion (MCAO) surgery in rats. Bioinformatic analysis indicated that tripartite motif-containing protein 59 (TRIM59) is downregulated in stroke, which was verified in cerebral I/R models. The upregulation of TRIM59 promoted viability and inhibited pyroptosis in OGD/R-treated microglia and alleviated cerebral I/R injury in vivo. TRIM59 attenuated NOD-like receptor family pyrin domain containing 3 (NLRP3) protein expression through ubiquitination, thus degrading NLRP3 and alleviating OGD/R-induced injury. TRIM59 relieves cerebral I/R injury in vivo and in vivo. Mechanistically, TRIM59 directly interacts with NLRP3 and inhibits NLRP3 through ubiquitination. Targeting the TRIM59/NLRP3 signalling axis may be an effective therapeutic strategy for cerebral I/R.
Similar content being viewed by others


Introduction
Stroke has been listed in the top 5 causes of death worldwide and is caused by a shortage of blood supply in the brain due to various causes. Stroke deprives neurons of glucose, oxygen and energy, leading to the death of neurons. In addition, the restoration of blood perfusion and reoxygenation can aggravate the injury to ischaemic cells, which is called cerebral ischaemia/reperfusion (I/R) injury1,2.
The pathological process of cerebral I/R injury involves excitatory amino acid toxicity, calcium overload, free radical damage, cytotoxic effects of nitric oxide and immune inflammation3,4,5,6. Inflammation is also related to the damage caused by cerebral I/R7,8. Microglia are considered to be the macrophages of the central nervous system and are the major resident immune cells involved in the inflammatory response9,10. Overactivated microglia lead to inflammation and aggravate tissue injury and neuronal death by inducing the excessive production of inflammatory cytokines11. Therefore, suppressing of the overactivation of microglia may be helpful to protect central nervous system.
Recently, microglial pyroptosis is a proinflammatory form of programmed cell death and has been reported to regulate stroke-induced inflammation12. Pyroptosis depends on the activation of inflammasomes. The oligomerization of NOD-like receptor family pyrin domain containing 3 (NLRP3) with the apoptosis-associated speck-like protein containing the CARD (ASC) and pro-Caspase-1 activates the NLRP3 inflammasome, leading to Caspase-1 cleavage13,14. Therefore, suppressing microglial pyroptosis might be a novel strategy for cerebral I/R.
Excessive activation of the NLRP3 inflammatory complex is significantly associated with a variety of inflammatory diseases, such as type 2 diabetes mellitus15, atherosclerosis16, Alzheimer's disease17 and cancer18. Recent studies have confirmed that activation of the NLRP3 inflammatory complex promotes the pathological development of cerebral I/R injury19,20,21. Additionally, the NLRP3 inflammasome can be activated by posttranslational modifications (PTMs), such as ubiquitination, sumoylation, alkylation, phosphorylation, and S-nitrosylation22,23,24. Ubiquitination has been proven to play an important regulatory role in NLRP3-mediated inflammation25,26. Ubiquitin is a protein composed of 76 amino acids that is typically bound to the lysine residues of substrate proteins through isopeptide bonds27. However, the role and mechanism of ubiquitination in pyroptosis and the progression of I/R injury remain unclear.
TRIM proteins participate in multiple cellular processes including cell proliferation, transcriptional regulation, immunity and cancer progression28,29. A series of studies indicated that many TRIM proteins are linked to pyroptosis30,31. TRIM59 is a member of the TRIM protein superfamily, and has a TRIM or RBCC motif consisting of a RING-finger domain (R), a B-box domain (B), and a coiled-coil domain (CC). Increasing evidence showed that TRIM59 has been identified as contributing to tumor progression32. In the current study, we aimed to analyse the role of TRIM59 in cerebral I/R injury. Whether the ubiquitination of NLRP3 mediated by TRIM59 is related to pyroptosis in cerebral I/R was also investigated. Our findings will provide new strategies for the treatment of stroke.
Materials and methods
Bioinformatic analysis
GSE119121 is an expression profile of the blood of rats with stroke. As described in Dagonnier M et al., 0-h and 2-h data were selected for analysis to obtain genes that were abnormally expressed in rats with stroke33. The criteria for gene screening were as follows: p < 0.05 and absolute value of log2 (fold change) > 1.
Cell culture and oxygen–glucose deprivation/reperfusion (OGD/R) model establishment
Human microglia HMC3 were obtained from ATCC (Manassas, VA, USA) and cultured in DMEM (Gibco, Grand Island, NY,USA) with 10% FBS (Gibco), 100 U/mL penicillin (Gibco), and 100 μg/mL streptomycin (Gibco). HMC3 cells were tested for mycoplasma and authenticated by Procell Life Science & Technology Co., Ltd. (Wuhan, China) to exclude contamination with other cell lines or incorrect identification. Oxygen–glucose deprivation/reperfusion (OGD/R) was used to establish a cell model of cerebral I/R injury in vitro as previously described34. Briefly, HMC3 cells were cultured in Earle’s balanced salt solution (Gibco) at 37 °C with 95% N2 and 5% CO2. Two hours later, the medium was replaced with normal medium, and the cells were cultured normally.
Cell transfection
pcDNA3.1-TRIM59 (TRIM59), pcDNA3.1-NLRP3 (NLRP3), and corresponding controls (Vector) were purchased from GenePharma (Shanghai, China). HMC3 cells were seeded into six-well plates (3 × 105/mL) and transfected using Lipofectamine® 2000 reagent (Invitrogen, CA, USA). After 48 h, the cells were collected for OGD/R inducation and subsequent experiments.
Production of lentiviral vectors
Lentiviral particles overexpressing TRIM59 (LV-TRIM59) and the negative controls (LV-NC) were obtained from GenePharma and transfected into HEK293T cells. HEK293T culture medium was collected at 48 h after transfection.
Animals and induction of MCAO
The rat experiments were carried out in compliance with the revised Animals (Scientific Procedures) Act 1986 and the ARRIVE guidelines (http://www.nc3rs.org.uk/page.asp?id=1357). In addition, the rat experiments were approved by the Animal Ethical and Welfare Committee (approval no. MDKN-2021–058). Twenty-four SPF healthy SD rats weighing 260 ± 10 g were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, China). The production licence number was SCXK (Shanghai) 2014–0002, and the qualification licence number was 2015000518015. All animal experiments complied with ethical requirements. The rats were randomly divided into four groups, with 6 rats in each group: sham operation group (Sham), model group (MCAO), TRIM59 overexpression model group (MCAO + LV-TRIM59) and TRIM59 overexpression negative control model group (MCAO + LV-NC).
To establish MCAO in rats, the rats were anaesthetized with 2% sodium pentobarbital (2 mL/kg) after 12 h of fasting. The common carotid artery, external carotid artery and internal carotid artery were separated, a small incision was made in the common carotid artery, a thread plug was inserted, and the internal carotid artery was ligated. After 2 h of obstruction, the 4–0 (0.26 mm diameter) wire plug was removed to achieve reperfusion. The sham group underwent the same operations as the MCAO group except that the wire plug was not inserted. After the rats were awake, neurological impairment was scored according to a previous report35.
Stereotaxic injection of the lentivirus
LV-TRIM59 and LV-NC were injected into the left lateral ventricle one month before MCAO surgery to overexpress TRIM59 in rats. After anaesthesia by intraperitoneal injection of 1% pentobarbital sodium (40 mg/kg), the rats were fixed with a brain stereoscopic locator, and the left cerebral cortex was positioned using the following coordinates relative to the bregma: 1.0 mm posterior, 2.0 mm lateral, 3.0 mm below the horizontal plane of the bregma. The hole was drilled, and 2 μL lentivirus was injected at a depth of 1.2 mm with a needle. The needle was kept for 10 min after slow injection.
Measurement of brain infarct volume
After the evaluation of neurological impairment, the rats were euthanized with 400 mg/kg pentobarbital sodium. The euthanasia and anaesthesia methods were carried out in accordance with the guidelines of the American Veterinary Medical Association. Then, the brains were immediately collected and cut into six coronal slices with a thickness of 2 mm. Then, the slices were dyed using 2% 2,3,5-triphenyltetrazolium chloride (TTC; BIOPIKE, Guangzhou, China) at 37 °C for 10 min. Next, brain infarct size was calculated by the following formula: (volume of total brain tissue-volume of total ipsilateral hemispheric damage)/volume of total brain tissue × 100.
Nissl staining
The paraffin sections were dewaxed and stained with 10% methylene blue solution for 20 min. After being washed, the sections were treated with 95% alcohol, dehydrated, made transparent, sealed, and placed under an ordinary light microscope (Leica Microsystems, Wetzlar, Germany) for observation and photometry, and the number of intact neurons was counted. The total neuronal cells were divided into 10 areas and calculated, and then the degree of ischaemia-damaged neurons was calculated according to the percentage of damaged neurons in the total neurons35.
Immunofluorescence analysis
Brain sections were fixed with 4% paraformaldehyde and then incubated in PBST containing 5% BSA (Santa Cruz, CA, USA) for 0.5 h. Next, the sections were treated with primary antibodies, including anti-IBA-1 (highly expressed in microglia) and anti-NLRP3, at 4 °C overnight, followed by incubation with the corresponding secondary antibody at 37 °C for 1 h. The nuclei were stained with DAPI (Beyotime, Shanghai, China). Finally, the sections were observed with a scanning fluorescence microscope (Leica Microsystems, Wetzlar, Germany) and analysed using ImageJ software (ver. 1.45r, National Institutes of Health, Bethesda, MD, USA).
Real-time quantitative polymerase chain reaction (RT–qPCR)
The mRNA expression of TRIM59 and NLRP3 in brain tissues and HMC3 cells was assessed by RT-qPCR. We extracted total RNA using TRIzol® reagent (Thermo Fisher, Waltham, MA, USA) and then reverse transcribed the RNA into cDNA (Hi-Fi cDNA Synthesis Kit, Abcam, Cambridge, MA, USA). RT-qPCR was performed using the SYBR Green Mix kit (Beyotime) in an Eppendorf Thermol Cycler system (Eppendorf AG, Shanghai, China). The target amplification process was performed as follows: predenaturation at 95 °C for 5 min, denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s, and then extension at 72 °C for 30 s, all for 40 cycles. Quantitative analysis was then performed using the 2-ΔΔCt method. The primer sequences are listed in Table 1.
Cell viability assay
Cell counting kit 8 (CCK-8, Beyotime) was used to evaluate the viability of HMC3 cells. In brief, the cells were seeded into 96-well plates, and 10 μL/well CCK-8 reagent was added. After 4 h of culture, cell viability was detected at an absorbance of 450 nm with a microplate reader (Bio-Rad).
Lactate dehydrogenase (LDH) cytotoxicity evaluation
To evaluate cell damage and measure cell death, LDH release by HMC3 cells was determined by an LDH Cytotoxicity Assay Kit (Beyotime). In brief, HMC3 cells were seeded into a 96-well plate and incubated with the LDH reagent for 0.5 h to measure LDH release. The absorbance values were obtained at 490 nm with a microplate reader (Bio-Rad, Hercules, CA, USA).
Enzyme-linked immunosorbent assay (ELISA)
The levels of interleukin-1β (IL-1β) and IL-18 in HMC3 cell lysates were measured by ELISA using a human IL-1β ELISA kit (ab214025, Abcam) and a human IL-18 ELISA kit (ab215539, Abcam) according to the manufacturer's instructions. IL-1β and IL-18 levels in rat serum were detected by a rat IL-1β ELISA kit (ab100768, Abcam) and a rat IL-18 ELISA kit (ab213909, Abcam), respectively.
Measurement of pyroptosis
Flow cytometry was performed to examine pyroptosis. HMC3 cells were digested with trypsin and washed with PBS. Pyroptosis was assessed with FLICA 660-YVADFMK in accordance with the manufacturer's protocol. The cells were stained with PI to identify cells with membrane pores and analysed by a BD-Verse flow cytometer (BD, NJ, USA).
Western blot analysis
HMC3 cells were collected to extract proteins (RIPA, Beyotime), and the protein concentration was determined using the BCA method. Then, the proteins were separated by 10% SDS‒PAGE and transferred to PVDF membranes. After the membrane was transferred, the proteins were blocked in 5% skim milk at room temperature for 2 h. Diluted primary antibodies (Table 2) were added and incubated overnight at 4 °C. HRP-labelled secondary antibodies were then added and incubated. Finally, the protein bands were visualized using an ECL system (Thermo Fisher).
Protein stability assessment
A cycloheximide (CHX) blocking assay was performed to determine the half-life of NLRP3 as previously reported36. HMC3 cells were incubated with CHX (100 μg/mL, Sigma-Aldrich, St. Louis, MO,USA), and the protein level of NLRP3 was detected at different time points (0, 2, 4, 8 h) after CHX treatment.
Coimmunoprecipitation (Co-IP)
HMC3 cells were lysed in RIPA lysis buffer (Beyotime). A 1/10 volume of supernatant was collected as the input, and half of the remaining supernatant was incubated with 20 µL/mL protein A/G Sepharose beads (Beyotime) at 4 °C for 1 h to remove nonspecific hybrid proteins. Afterwards, the lysates were incubated with 2 µg anti-NLRP3 antibodies (ab263899, Abcam)/anti-TRIM59 antibodies (ab69639, Abcam) or negative control IgG (Beyotime) at 4 °C overnight and then rotated at 4 °C with a mixture of protein A/G Sepharose beads (20 µL/mL) for 4 h. The bound proteins were analysed by western blotting.
Ubiquitination assay
The cells were transfected with vectors expressing Flag-NLRP3, HA-UB, TRIM59 and the control. Whole-cell extracts were immunoprecipitated with anti-HA and analysed by immunoblotting with anti-Flag antibodies.
Statistical analysis
Each experiment was repeated three times, and all data are expressed as the mean ± SD and were calculated by GraphPad Prism 8.3. Student’s t test was used to analyse the differences between two groups, and one-way analysis of variance (ANOVA) was performed for comparisons between multiple groups. All results were considered significantly different when p < 0.05.
Ethics approval
The rat experiments were approved by the Animal Ethical and Welfare Committee (approval no. MDKN-2021-058). All animal experiments complied with ethical requirements.
Results
TRIM59 is expressed at low levels in cerebral I/R and OGD/R models
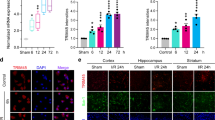
Differentially expressed genes in the blood samples of 8 spontaneously hypertensive rats at 0 and 2 h post stroke induction by MCAO were identified (Fig. 1A). As indicated in Fig. 1B, 344 aberrantly elevated genes and 267 downregulated genes were identified. Ten ubiquitination-related genes that were significantly downregulated in MCAO rats were screened (Fig. 1C–L), among which TRIM59 was selected as the most downregulated gene (Fig. 1I). Then, the low protein expression of TRIM59 was verified in the brain tissues of MCAO rats, which were established to mimic cerebral I/R in vivo (Fig. 1M, N). The OGD/R model in HMC3 cells was established, and TRIM59 mRNA and protein expression was shown to be dramatically downregulated (Fig. 1O, Q).

TRIM59 is expressed at low levels in a cerebral I/R animal model and cell model. (A) Heatmap analysis of differentially expressed genes in stroke from the microarray expression profiling dataset GSE119121. (B) A total of 344 aberrantly upregulated genes and 267 downregulated genes were identified. (C–L) The mRNA levels of ten ubiquitination-related genes in MCAO rats were detected by RT-qPCR, n = 6. (M, N) The protein expression of TRIM59 in the brain tissues of rats was detected by western blotting, n = 6. The mRNA (O) and protein expression (P, Q) of TRIM59 in HMC3 cells were evaluated by RT-qPCR and western blot analysis, respectively. n = 3. **p < 0.01, ***p < 0.001.
TRIM59 protects microglia from OGD/R injury
Then, the role of TRIM59 was studied using the cerebral OGD/R model. TRIM59 mRNA expression was significantly upregulated in HMC3 cells after transfection (Fig. 2A). A two-hour OGD/R treatment significantly reduced the viability of HMC3 cells (Fig. 2B). The pyroptosis rate of HMC3 cells was detected by flow cytometry (Fig. 2C, D). OGD/R treatment accelerated pyroptosis in HMC3 cells, which was further confirmed by the increases in LDH, IL-1β and IL-18 (Fig. 2E–G). Moreover, NLRP3, cleaved Caspase-1 p20, and GSDMD-N levels were upregulated in OGD/R-treated HMC3 cells (Fig. 2H–K). The upregulation of TRIM59 significantly alleviated OGD/R-induced injury by promoting cell growth and inhibiting pyroptosis (Fig. 2B–K).

(A) TRIM59 protects microglia from OGD/R injury. TRIM59 expression levels were detected by RT-qPCR after transfection with TRIM59 overexpression vectors, n = 3. (B) Cell viability was detected by CCK-8 assays two houres after OGD/R performace. (C, D) Pyroptosis in HMC3 cells was measured by flow cytometry. (E) The release of LDH was examined 48 h after transfection. (F, G) ELISA was used to evaluate the levels of IL-1β and IL-18. (H–K) Western blotting was used to detect the protein levels of NLRP3, cleaved Caspase-1 p20 and GSDMD-N in HMC3 cells. n = 3, **p < 0.01, ***p < 0.001, versus Vector or control group. ###p < 0.001, versus OGD/R + Vector group.
TRIM59 attenuates NLRP3 protein expression through ubiquitination
As an E3 ubiquitin ligase, TRIM59 has ubiquitin protein transferase activity. Therefore, TRIM59 was thought to modulate NLRP3 stability through the ubiquitin‒proteasome pathway. The co-IP results revealed the interaction between TRIM59 and NLRP3 (Fig. 3A). Then, a ubiquitination assay was performed on HMC3 cells after transfection. As shown in Fig. 3B, OGD/R knocked down the protein expression of TRIM59 and decreased the ubiquitination of NLRP3. However, the upregulation of TRIM59 enhanced the ubiquitination of NLRP3 and inhibited NLRP3 protein levels (Fig. 3B), suggesting that TRIM59 might affect the stability of NLRP3. To substantiate this conclusion, we treated HMC3 cells with OGD/R and the protein synthesis inhibitor CHX. The results suggested that OGD/R extended the half-life of the NLRP3 protein, while overexpression of TRIM59 shortened the half-life of the NLRP3 protein in HMC3 cells after CHX treatment (Fig. 3C, D).

(A) TRIM59 attenuates NLRP3 protein expression through ubiquitination. The interaction between TRIM59 and NLRP3 was detected by Co-IP. (B) After transfection, IP using anti-Flag was performed. The ubiquitination and expression of HA in the pull-down product was detected by western blotting. (C, D) Western blot analysis of the half-life of NLRP3 protein in HMC3 cells under OGD/R conditions and with TRIM59 overexpression. n = 3, ***p < 0.001, versus control group. ##p < 0.01, versus OGD/R + Vector group.
NLRP3 reverses the TRIM59-mediated protection against OGD/R injury
To further assess whether TRIM59 protects HMC3 cells from OGD/R injury via NLRP3, NLRP3 was elevated in TRIM59-overexpressing cells (Fig. 4A). CCK-8 and caspase-1/PI apoptosis assays demonstrated that the upregulation of NLRP3 prominently neutralized the effects of TRIM59 overexpression on decreasing viability and increasing pyroptosis in HMC3 cells treated with OGD/R (Fig. 4B, D). Moreover, LDH, IL-1β and IL-18 concentrations that were decreased by TRIM59 overexpression were dramatically decreased by the upregulation of NLRP3 (Fig. 4E–G). The protein levels of NLRP3, cleaved Caspase-1 p20, and GSDMD-N were markedly enhanced by NLRP3 overexpression due to TRIM59 overexpression (Fig. 4H–K).

NLRP3 is required for TRIM59-mediated suppression of OGD/R injury. (A) NLRP3 expression levels were detected by RT-qPCR after transfection with NLRP3 vectors. (B) Cell viability was detected by CCK-8 assays two houres after OGD/R performace. (C, D) Pyroptosis in HMC3 cells was measured by flow cytometry. (E) The release of LDH was examined 48 h after transfection. (F, G) ELISA was used to evaluate the levels of IL-1β and IL-18. (H–K) Western blotting was used to detect the protein levels of NLRP3, cleaved Caspase-1 p20 and GSDMD-N in HMC3 cells. ***p < 0.001, versus Vector or control group. n = 3, ###p < 0.001, versus OGD/R group. &&p < 0.01, versus OGD/R + TRIM59 + Vector group.
TRIM59 alleviates cerebral I/R injury in vivo
The neurological score and infarct volume were evaluated 7 days after MCAO. The infarct volume was prominently elevated in the MCAO group, but there was almost no infarct volume in the sham-operated group. The increase in TRIM59 notably decreased the infarct volume in MCAO rats (Fig. 5A, B). Moreover, the neurological score was prominently enhanced in MCAO-treated rats, while these effects were suppressed after TRIM59 upregulation (Fig. 5C). The Nissl staining results confirmed that MCAO dramatically aggravated neuronal injury in rats, and shrunken nuclei and disorderly arrays were observed in model rats (Fig. 5D, E). Additionally, MCAO prominently promoted the levels of IL-1β and IL-18 in rat serum, while TRIM59 dramatically inhibited this increase (Fig. 5F, G). Furthermore, the immunofluorescence results demonstrated that NLRP3 expression was elevated in the MCAO group, and TRIM59 overexpression significantly downregulated NLRP3 levels (Fig. 5H).

TRIM59 alleviates cerebral I/R injury in vivo. Images of TTC-stained complete coronal sections in the MCAO and sham groups. (B) The infarct volume was determined. (C) Neurologic scores in rats of the MCAO and Sham groups were evaluated. (D) Quantitative evaluation of the neuronal damage rate of the hippocampal CA3 region and (E) cerebral cortex was performed. (F, G) The expression of IL-18 and IL-1β in the serum of rats was detected by ELISA. (H) Immunofluorescence analysis was performed to evaluate the localization and expression of IBA1 and NLRP3. n = 6, ***p < 0.001, versus Sham group. ##p < 0.01, ###p < 0.001, versus MCAO + LV-NC group.
Discussion
TRIM59 was downregulated in the brain tissues of MCAO rats and OGD/R-treated HMC3 cells. Our data suggested that the upregulation of TRIM59 promoted cell viability and inhibited pyroptosis in microglia after OGD/R treatment. Moreover, TRIM59 directly interacted with NLRP3 and inhibited NLRP3 through ubiquitination. These findings indicated that TRIM59 could alleviate cerebral I/R injury by inhibiting NLRP3 expression.
Cerebral I/R injury refers to injury caused by insufficient blood supply to the brain tissue and the restoration of blood perfusion. This process triggers an innate immune response and leads to an inflammatory cascade19. Inhibiting NLRP3 inflammasome activation has been used as one of the therapeutic methods for cerebral I/R21,37. For example, ibrutinib, a Bruton's tyrosine kinase inhibitor, can inhibit NLRP3 inflammasome, caspase-1 and IL-1β activation, thereby alleviating cerebral I/R injury38.
Ubiquitination can affect the biological functions of cells by regulating the stability and expression level of intracellular proteins, including cell proliferation, cell cycle, apoptosis, tumour development, metastasis, and invasion39. Changes in protein stability and abnormal expression are related to the development of diseases, and so the study of protein ubiquitination mechanisms can better explain the molecular mechanisms of diseases40,41. Ubiquitination was shown to modulate the stability of the NLRP3 inflammasome42,43. At present, only E3 ubiquitin ligases have been shown to negatively regulate the inflammasome by targeting NLRP344. One of the main mechanisms involves controlling NLRP3 levels through the proteasome degradation pathway. Recently, TRIM59 has been identified as a novel molecular biomarker in various tumours45. TRIM59 promotes lung cancer progression by regulating autophagy or the NLRP3 inflammasome signalling pathway28,46. TRIM59 participates in tumour disease progression by regulating the ubiquitination of genes47,48,49. The presence of the unique N-terminal RING domain enables most TRIM family members to have extensive ubiquitination enzymatic activities and induce target proteins to enter the proteasome-dependent ubiquitination degradation process by binding them to ubiquitin. TRIM59 has the conserved RBCC domain of typical TRIM family proteins28,46. In addition to the three typical RBCC domains (RING finger domain, B-box domain and coiled-coil domain), the C segment of TRIM59 also has a TM domain that may be involved in the transmembrane localization of TRIM59, which is involved in regulating the intracellular localization of TRIM59. In the current work, bioinformatic analysis was carried out to screen genes in stroke related to ubiquitination, and TRIM59 was shown to be expressed at low levels in cerebral I/R animal and cell models. An increase in TRIM59 dramatically protected HMC3 cells from OGD/R injury. Afterwards, TRIM59 was found to regulate the ubiquitination of NLRP3.
Microglia, the resident immune cells tiling the whole CNS, exhibit an amazing capacity for regeneration50,51. Even when depleted in large quantities in the uninflamed central nervous system, microglia are able to quickly refill and return to normal density within a few days through the proliferation of residual cells. After neurodegeneration, they proliferate and accumulate in the diseased central nervous system52. Therefore, if the microglia are protected well, it may be possible to convert them into neurons to repair the damaged central nervous system where neurons are lost. In this study, TRIM59 promotes microglial survival and reduces infarction size and neuronal damage. Whether the protective mechanism benifits neurons should be investigated in the future. Meanwhile, which amino acid residue of NLRP3 is ubiquitination modified has not been elucidated.
Therefore, it is suggested that TRIM59 may act as a ubiquitin ligase to induce the ubiquitination-mediated degradation of NLRP3. Our data revealed that TRIM59 binds endogenously to NLRP3 and attenuates NLRP3 protein expression through ubiquitination. Furthermore, the half-life of the NLRP3 protein was decreased in TRIM59-overexpressing cells. These results suggest that the overexpression of TRIM59 contributes to NLRP3 protein degradation in microglia.
Conclusion
Overall, TRIM59 relieves cerebral I/R injury in vivo and in vitro. Mechanistically, TRIM59 directly interacts with NLRP3 and inhibits NLRP3 through ubiquitination. The TRIM59/NLRP3 signalling axis may be a promising prognostic factor and a valuable therapeutic target for cerebral I/R (Supplementary Information).
Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
References
Li, C. et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol. Res. 174, 105933. https://doi.org/10.1016/j.phrs.2021.105933 (2021).
Przykaza, L. Understanding the connection between common stroke comorbidities, their associated inflammation, and the course of the cerebral ischemia/reperfusion cascade. Front. Immunol. 12, 782569. https://doi.org/10.3389/fimmu.2021.782569 (2021).
Amantea, D. & Bagetta, G. Excitatory and inhibitory amino acid neurotransmitters in stroke: from neurotoxicity to ischemic tolerance. Curr. Opin. Pharmacol. 35, 111–119. https://doi.org/10.1016/j.coph.2017.07.014 (2017).
Zhang, T. et al. Pseudoginsenoside-F11 protects against transient cerebral ischemia injury in rats involving repressing calcium overload. Neuroscience 411, 86–104. https://doi.org/10.1016/j.neuroscience.2019.05.030 (2019).
Sun, M. S. et al. Free radical damage in ischemia-reperfusion injury: An obstacle in acute ischemic stroke after revascularization therapy. Oxid. Med. Cell. Longev. 2018, 3804979. https://doi.org/10.1155/2018/3804979 (2018).
Narne, P., Pandey, V. & Phanithi, P. B. Role of nitric oxide and hydrogen sulfide in ischemic stroke and the emergent epigenetic underpinnings. Mol. Neurobiol. 56, 1749–1769. https://doi.org/10.1007/s12035-018-1141-6 (2019).
Franke, M. et al. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav. Immun. 92, 221–231. https://doi.org/10.1016/j.bbi.2020.12.009 (2021).
Tian, T. et al. Targeted delivery of neural progenitor cell-derived extracellular vesicles for anti-inflammation after cerebral ischemia. Theranostics 11, 6507–6521. https://doi.org/10.7150/thno.56367 (2021).
Liu, X. et al. Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 phenotypes. Exp. Neurol. 341, 113700. https://doi.org/10.1016/j.expneurol.2021.113700 (2021).
Xu, X. et al. Annexin A1 protects against cerebral ischemia-reperfusion injury by modulating microglia/macrophage polarization via FPR2/ALX-dependent AMPK-mTOR pathway. J. Neuroinflamm. 18, 119. https://doi.org/10.1186/s12974-021-02174-3 (2021).
Surinkaew, P., Sawaddiruk, P., Apaijai, N., Chattipakorn, N. & Chattipakorn, S. C. Role of microglia under cardiac and cerebral ischemia/reperfusion (I/R) injury. Metab. Brain Dis. 33, 1019–1030. https://doi.org/10.1007/s11011-018-0232-4 (2018).
Xu, P. et al. Microglial TREM-1 receptor mediates neuroinflammatory injury via interaction with SYK in experimental ischemic stroke. Cell Death Dis. 10, 555. https://doi.org/10.1038/s41419-019-1777-9 (2019).
Shao, B. Z., Xu, Z. Q., Han, B. Z., Su, D. F. & Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 6, 262. https://doi.org/10.3389/fphar.2015.00262 (2015).
Kelley, N., Jeltema, D., Duan, Y. & He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20133328 (2019).
Jo, E. K., Kim, J. K., Shin, D. M. & Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 13, 148–159. https://doi.org/10.1038/cmi.2015.95 (2016).
Grebe, A., Hoss, F. & Latz, E. NLRP3 Inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 122, 1722–1740. https://doi.org/10.1161/CIRCRESAHA.118.311362 (2018).
Heneka, M. T. et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. https://doi.org/10.1038/nature11729 (2013).
Moossavi, M., Parsamanesh, N., Bahrami, A., Atkin, S. L. & Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 17, 158. https://doi.org/10.1186/s12943-018-0900-3 (2018).
Gong, Z., Pan, J., Shen, Q., Li, M. & Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 15, 242. https://doi.org/10.1186/s12974-018-1282-6 (2018).
Ma, C. et al. Qingkailing injection ameliorates cerebral ischemia-reperfusion injury and modulates the AMPK/NLRP3 inflammasome signalling pathway. BMC Complement. Altern. Med. 19, 320. https://doi.org/10.1186/s12906-019-2703-5 (2019).
Liu, H. et al. Adiponectin peptide alleviates oxidative stress and NLRP3 inflammasome activation after cerebral ischemia-reperfusion injury by regulating AMPK/GSK-3beta. Exp. Neurol. 329, 113302. https://doi.org/10.1016/j.expneurol.2020.113302 (2020).
Qin, Y. et al. TRIM28 SUMOylates and stabilizes NLRP3 to facilitate inflammasome activation. Nat. Commun. 12, 4794. https://doi.org/10.1038/s41467-021-25033-4 (2021).
Shim, D. et al. BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci. Rep. 7, 15012–15020. https://doi.org/10.1038/s41598-017-15314-8 (2017).
Tang, X. et al. SNO-MLP (S-nitrosylation of muscle LIM protein) facilitates myocardial hypertrophy through TLR3 (toll-like receptor 3)-mediated RIP3 (receptor-interacting protein kinase 3) and NLRP3 (NOD-like receptor pyrin domain containing 3) inflammasome activation. Circulation 141, 984–1000. https://doi.org/10.1161/CIRCULATIONAHA.119.042336 (2020).
Bednash, J. S. & Mallampalli, R. K. Regulation of inflammasomes by ubiquitination. Cell Mol Immunol 13, 722–728. https://doi.org/10.1038/cmi.2016.15 (2016).
Shim, D. W. & Lee, K. H. Posttranslational regulation of the NLR family pyrin domain-containing 3 inflammasome. Front. Immunol. 9, 1054. https://doi.org/10.3389/fimmu.2018.01054 (2018).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem. 81, 203–229. https://doi.org/10.1146/annurev-biochem-060310-170328 (2012).
Han, T. et al. TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy 14, 2035–2048. https://doi.org/10.1080/15548627.2018.1491493 (2018).
Tsuchida, T. et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776. https://doi.org/10.1016/j.immuni.2010.10.013 (2010).
Wan, T., Li, X. & Li, Y. The role of TRIM family proteins in autophagy, pyroptosis, and diabetes mellitus. Cell Biol. Int. 45, 913–926. https://doi.org/10.1002/cbin.11550 (2021).
Guo, M. et al. TRIM21 regulates virus-induced cell pyroptosis through polyubiquitination of ISG12a. J. Immunol. 209, 1987–1998. https://doi.org/10.4049/jimmunol.2200163 (2022).
Tan, P., He, L. & Zhou, Y. TRIM59 deficiency curtails breast cancer metastasis through SQSTM1-selective autophagic degradation of PDCD10. Autophagy 15, 747–749. https://doi.org/10.1080/15548627.2019.1569951 (2019).
Dagonnier, M. et al. Hyperacute changes in blood mRNA expression profiles of rats after middle cerebral artery occlusion: Towards a stroke time signature. Plos One 13, e206321. https://doi.org/10.1371/journal.pone.0206321 (2018).
Cao, X., Wang, Y. & Gao, L. CHRFAM7A overexpression attenuates cerebral ischemia-reperfusion injury via inhibiting microglia pyroptosis mediated by the NLRP3/Caspase-1 pathway. Inflammation 44, 1023–1034. https://doi.org/10.1007/s10753-020-01398-4 (2021).
Zhang, H. S., Ouyang, B., Ji, X. Y. & Liu, M. F. Gastrodin alleviates cerebral ischaemia/reperfusion injury by inhibiting pyroptosis by regulating the lncRNA NEAT1/miR-22-3p axis. Neurochem. Res. 46, 1747–1758. https://doi.org/10.1007/s11064-021-03285-2 (2021).
Cai, B. et al. USP5 attenuates NLRP3 inflammasome activation by promoting autophagic degradation of NLRP3. Autophagy 18, 990–1004. https://doi.org/10.1080/15548627.2021.1965426 (2022).
Ye, Y. et al. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by Inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-kappaB signaling pathway. Front. Cell. Neurosci. 13, 553. https://doi.org/10.3389/fncel.2019.00553 (2019).
Ito, M. et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6, 7360. https://doi.org/10.1038/ncomms8360 (2015).
Popovic, D., Vucic, D. & Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 20, 1242–1253. https://doi.org/10.1038/nm.3739 (2014).
van Wijk, S. J., Fulda, S., Dikic, I. & Heilemann, M. Visualizing ubiquitination in mammalian cells. EMBO Rep. https://doi.org/10.15252/embr.201846520 (2019).
Carroll, E. C. & Marqusee, S. Site-specific ubiquitination: Deconstructing the degradation tag. Curr. Opin. Struct. Biol. 73, 102345. https://doi.org/10.1016/j.sbi.2022.102345 (2022).
Akther, M., Haque, M. E., Park, J., Kang, T. B. & Lee, K. H. NLRP3 ubiquitination—A new approach to target NLRP3 inflammasome activation. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22168780 (2021).
Tang, J. et al. Sequential ubiquitination of NLRP3 by RNF125 and Cbl-b limits inflammasome activation and endotoxemia. J. Exp. Med. https://doi.org/10.1084/jem.20182091 (2020).
Song, H. et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 7, 13727. https://doi.org/10.1038/ncomms13727 (2016).
Jin, Z. et al. TRIM59: A potential diagnostic and prognostic biomarker in human tumors. Plos One 16, e257445. https://doi.org/10.1371/journal.pone.0257445 (2021).
Liang, M. et al. Cancer-derived exosomal TRIM59 regulates macrophage NLRP3 inflammasome activation to promote lung cancer progression. J. Exp. Clin. Cancer Res. 39, 176. https://doi.org/10.1186/s13046-020-01688-7 (2020).
Wang, F., Wang, H., Sun, L., Niu, C. & Xu, J. TRIM59 inhibits PPM1A through ubiquitination and activates TGF-beta/Smad signaling to promote the invasion of ectopic endometrial stromal cells in endometriosis. Am. J. Physiol. Cell Physiol. 319, C392-401. https://doi.org/10.1152/ajpcell.00127.2019 (2020).
Sang, Y. et al. CDK5-dependent phosphorylation and nuclear translocation of TRIM59 promotes macroH2A1 ubiquitination and tumorigenicity. Nat. Commun. 10, 4013. https://doi.org/10.1038/s41467-019-12001-2 (2019).
Zhou, Z. et al. TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 147, 1043–1054. https://doi.org/10.1053/j.gastro.2014.07.021 (2014).
Xu, Z. et al. Efficient strategies for microglia replacement in the central nervous system. Cell Rep. 32, 108041. https://doi.org/10.1016/j.celrep.2020.108041 (2020).
Zhan, L. et al. Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. Plos Biol. 17, e3000134. https://doi.org/10.1371/journal.pbio.3000134 (2019).
Prinz, M., Erny, D. & Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 18, 385–392. https://doi.org/10.1038/ni.3703 (2017).
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors participated in the design, interpretation of the studies, analysis of the data and review of the manuscript. L.Z. drafted the work and revised it critically for important intellectual content. G.L. was responsible for the acquisition, analysis and interpretation of data for the work. Y.L. made substantial contributions to the conception or design of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Li, G. & Li, Y. TRIM59 suppresses the brain ischaemia/reperfusion injury and pyroptosis of microglial through mediating the ubiquitination of NLRP3. Sci Rep 14, 2511 (2024). https://doi.org/10.1038/s41598-024-52914-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52914-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
