
Abstract
Few existing reports have investigated the copy number variants (CNVs) in fetuses with central nervous system (CNS) anomalies. To gain further insights into the genotype–phenotype relationship, we conducted chromosomal microarray analysis (CMA) to reveal the pathogenic CNVs (pCNVs) that were associated with fetal CNS anomalies. We enrolled 5,460 pregnant women with different high-risk factors who had undergone CMA. Among them, 57 subjects with fetal CNS anomalies were recruited. Of the subjects with fetal CNS anomalies, 23 were given amniocentesis, which involved karyotype analysis and CMA to detect chromosomal abnormalities. The other 34 cases only underwent CMA detection using fetal abortive tissue. In this study, we identified five cases of chromosome aneuploid and nine cases of pCNVs in the fetuses, with a chromosomal aberration detection rate of 24.56% (14/57). In the 23 cases that were given both karyotype and CMA analysis, one case with trisomy 18 was detected by karyotyping. Moreover, CMA revealed a further three cases of pCNVs, including the 1p36.33p36.31, 7q11.23, and 1q21.1q21.2 microdeletions, with a 13.04% (3/23) increase in CMA yield over the karyotype analysis. Additionally, three cases of trisomy 13, one case of trisomy 21, and six cases of pCNVs were detected in the other 34 fetuses where only CMA was performed. Furthermore, a higher chromosomal aberration detection rate was observed in the extra CNS anomaly group than in the isolated CNS anomaly group (40.91% vs 14.29%). In conclude, several pathogenic CNVs were identified in the fetuses with CNS anomalies using CMA. Among the detected CNVs, ZIC2, GNB1, and NSUN5 may be the candidate genes that responsible for fetal CNS anomalies. Our findings provides an additional reference for genetic counseling regarding fetal CNS anomalies and offers further insight into the genotype–phenotype relationship.
Similar content being viewed by others

Introduction
Fetal central nervous system (CNS) anomalies are the most common type of congenital fetal malformation and they are generally severe. CNS anomalies include neural tube defects (NTDs), ventriculomegaly/holoprosencephaly, hydranencephaly, holoprosencephaly sequence, iniencephaly, and microcephaly1. The exact frequency of CNS anomalies is still unknown, although a previous long-term follow-up study indicated that the incidence rate may be as high as 1 in 100 births2. Generally, CNS develops between the third week and the twentieth week pregnancy. Therefore, most CNS anomalies can be diagnosed during the first or second trimester through ultrasound3. Genetic conditions are considered to be a decisive factor in central nervous system anomalies. However, in most cases of fetal CNS anomalies, the molecular etiology remains unknown.
Karyotype analysis is widely regarded as the “gold standard” method in chromosomal abnormality detection. However, its resolution is limited since it can only diagnose genetic material rearrangements larger than 5–10 Mb. Chromosomal microarray analysis (CMA) combines array-based comparative genomic hybridization (aCGH) technology and single nucleotide polymorphism (SNP) array technology. It rapidly and effectively detects chromosomal numerical abnormalities and copy number variants (CNVs) at the whole genome level4,5. Moreover, CMA has been recommended as a first-line tool for prenatal diagnosis in all pregnant women who receive amniocentesis6,7.
To date, few studies have explored the use of CMA to screen pathogenic CNVs in fetuses with CNS anomalies. In this study, we enrolled 57 subjects with fetal CNS anomalies among 5,460 pregnant women who had undergone CMA. This paper aims to provide more references for the genetic diagnosis of fetuses with CNS anomalies and counseling for parents.
Materials and methods
Subjects
For this study, we enrolled a total of 5460 pregnant women with various high-risk factors. They visited Quanzhou Women’s and Children’s Hospital between 2017 and 2023 to receive CMA for etiology diagnosis. Among them, we investigated 57 cases with fetal CNS anomalies. Only women with structural CNS anomalies were enrolled, while those with CNS soft marker anomalies were excluded. For genetics diagnosis, 23 cases underwent amniocentesis and 34 cases had abortive tissue collected. Of the 57 cases in total, 35 fetuses had isolated CNS anomalies, while 22 fetuses had extra CNS anomalies. All 57 pregnant women received CMA after providing signed written informed consent. Additionally, the 23 cases who received amniocentesis were also subject to karyotype analysis.
Karyotype analysis
Approximately 20 ml of amniotic fluid was collected from each fetus for karyotype analysis. The samples were then centrifuged at 1500 rpm for 10 min, and the supernatant was removed. The remaining amniotic fluid cells were mixed and inoculated in an amniotic fluid culture medium and cultured at 37 °C for 7–10 days. The cultured amniotic fluid cells were harvested using a Sinochrome Chromprep II automatic chromosome harvesting system (Shanghai Lechen Biotechnology Co., Ltd.), according to the standard procedures8. Subsequently, the cells in suspension were collected, followed by making sections and Giemsa banding. Finally, for each case, thirty karyotypes were counted and five karyotypes were analyzed. The procedures for classification and diagnosis of the karyotypes were conducted following the International System for Human Cytogenomic Nomenclature (ISCN 2020)9.
Genomic DNA extraction
About 10 ml of amniotic fluid was collected from each fetus and 2 ml of peripheral blood from the parents. According to the procedures described in our previous study10, genomic DNA was extracted from the enrolled subjects for chromosomal microarray analysis using the QIAamp DNA Blood Kit (QIAGEN, Germany) according to the manufacturer’s guidelines (www.qiagen.com).
Chromosomal microarray analysis
The genomic DNA was subsequently digested, ligated, PCR amplified, purified, fragmented, labeled, and hybridized in line with the Affymetrix CytoScan assay user guide, using the Affymetrix CytoScan 750 K array (Life Technologies, USA). The Genotyping Console and Chromosome Analysis Suite software were employed to assess the SNP and the CNVs. Several databases, including DGV (http://dgv.tcag.ca/dgv), OMIM (https://omim.org/), DECIPHER (https://decipher.sanger.ac.uk/), PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), and other databases, were used as reference resources10. A joint consensus of the American College of Medical Genetics (ACMG) and the Clinical Genome Resource (ClinGen) standards and guidelines was used for CNVs pathogenicity interpretation11.
Statistical analysis
Data analysis was conducted using SPSS20.0 software. The chi-square test was employed for statistical analysis among the groups. The Fisher exact probability test was applied for statistical analysis when the chi-square test results were not satisfactory. A value of P < 0.05 was considered statistically significant.
Ethics approval and consent to participate
Approval was obtained from the Institutional Ethics Committee of Quanzhou Women’s and Children’s Hospital before the commencement of the study (2023No.56). We received informed consent from the participants in the study and they agreed to the publication of the report. All procedures involving human participants followed the ethical standards of the institutional and/or national research committee and the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Results
Subject information
A total of 57 pregnancies with abnormal ultrasound examination results indicating CNS anomalies were enrolled in this study. As listed in Table 1, eight cases had holoprosencephaly and seven cases had agenesis of corpus callosum. Additionally, there were seven cases with dysplasia of vermis cerebelli and five cases harbored hydrocephalus. In this study, 23 subjects underwent amniocentesis, while 34 were given a genetic diagnosis using abortive tissue. The cases were categorized into two groups, comprising isolated CNS anomalies (n = 35) and those with extra CNS anomalies (n = 22).
Karyotype analysis results
Karyotype analysis and CMA were successfully performed on 23 of the cases with CNS anomalies. Among them, one case of chromosomal aneuploidy (trisomy 18) was detected, with a chromosomal abnormality detection rate of 4.35% (1/23).
Copy number variants detected by CMA but missed by karyotype analysis
The chromosomal aneuploidy of trisomy 18 detected by karyotype analysis was confirmed through CMA. Additionally, as Table 2 reveals, CMA detected an additional three cases of pCNVs, including the 1p36.33p36.31, 7q11.23, and 1q21.1q21.2 microdeletions. These microdeletions cover the regions of 1p36 deletion syndrome, Williams-Beuren syndrome, and 1q21.1 deletion syndrome, respectively (Fig. 1). All of these pCNVs were misdiagnosed by karyotype analysis, indicating that CMA exhibited a 13.04% (3/23) improvement over karyotype analysis. Parental verification was performed on Cases 6 and 7, and the results revealed that the 7q11.23 microdeletion in Case 6 was de novo, while the 1q21.1q21.2 microdeletion in Case 7 was inherited from the mother.

Ultrasound examination and chromosomal microarray analysis results in the enrolled families. (A) Ultrasound examination results showed a ventriculomegaly in Case 3. (B) A 5.0-Mb microdeletion was observed in the fetus of Case 3 using chromosomal microarray analysis (arr[GRCh37] 1p36.33p36.31(849,467_5,866,441) × 1). (C) Dysplasia of corpus callosum was observed in the fetus of Case 6 using ultrasound examination. (D) A 1.4-Mb microdeletion in the 7q11.23 region was detected in the fetus of Case 6 by chromosomal microarray analysis.
Chromosomal microarray analysis results in fetuses using abortive tissue
In this study, 34 samples of fetal abortive tissue were collected for genetics diagnosis. Among them, four cases of chromosomal aneuploidies were diagnosed, including three cases of trisomy 13 and one case of trisomy 21. Moreover, six cases of pCNVs were identified, including 13q31.1q34 deletion, 7q32.1q36.3 duplication, 10q26.2q26.3 deletion, 13q31.3q34 duplication, 4p16.3p16.2 deletion, 4p16.2p15.1 duplication, 15q11.2 deletion, and 2p25.3p24.1 duplication. These CNVs covered Wolf-Hirschhorn Syndrome, 13q32 deletion syndrome, and 15q11.2 deletion syndrome (Table 2 and Fig. 2). Additionally, four cases of variants of unknown significance (VOUS) were detected (Table 3). In Case 10, we performed parental verification, which indicated that the 13q31.1q31.3 microduplication was inherited from the father.

Other detected pathogenic copy number variants associated with CNS anomalies. (A) A 28.6 Mb deletion in the 13q31.1q34 region was detected in Case 1 by chromosomal microarray analysis. (B,C) In Case 2, a 7q32.1q36.3 duplication combined with a 10q26.2q26.3 microdeletion was identified using chromosomal microarray analysis. (D) In Case 5, a 4p16.3p16.2 microdeletion and a 4p16.2p15.1 duplication were detected using chromosomal microarray analysis. (E) A 20.2 Mb duplication in the 2p25.3p24.1 region was observed in Case 9 using chromosomal microarray analysis.
Detection rates of chromosomal abnormalities between the groups
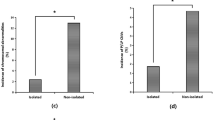
In this study, five cases of chromosome aneuploid and nine cases of pCNVs were identified in the fetuses, with a pCNVs detection rate of 15.79% (9/57) and a total chromosomal aberration detection rate of 24.56% (14/57). We further investigated the chromosomal aberration detection rates between the groups (Table 4). All five cases with chromosome aneuploid abnormalities had extra CNS anomalies, exhibiting a chromosome aneuploid detection rate of 22.73%. A significant difference in the total chromosomal aberration detection rate was observed in the extra CNS anomaly group compare with the isolated CNS anomaly group (40.91% vs 14.29%, χ2 = 5.168, P = 0.023). However, no obvious difference in pCNV detection rate was observed between the groups (18.18% vs 14.29%, χ2 = 0.000, P = 0.984).
Pregnancy outcome follow-up
Of the 23 cases that received amniocentesis, one did not attend a follow-up. Of those who did follow up, three cases with pCNVs, one case with VOUS, and three cases with normal CMA results chose to terminate their pregnancies. The remaining 15 cases with normal CMA results decided to continue their pregnancies. One year after birth, follow-up revealed that 14 newborns had reached their normal development milestones, while one case exhibited developmental delays.
Discussion
Fetal central nervous system anomalies are primarily caused by genetic material changes. Although CNS anomalies can have devastating effects, most cases are effectively diagnosed through ultrasound during the first and early second trimesters3,12. Despite occurring frequently, the etiology and mechanism of fetal CNS anomalies are still poorly understood. Moreover, there are disputes regarding the inclusion of certain indicators in the study of CNS anomalies. Some studies incorporate ultrasound soft indicators for etiological analysis, such as widening of the lateral ventricle, widening of the posterior fossa cistern, and Blake cysts13,14. In contrast, other studies exclude such soft indicator anomalies3,15,16. In this study, we recorded 57 cases of fetuses with prenatal structural CNS anomalies from 5,460 pregnant women who received prenatal etiological diagnoses. The CNS anomaly incidence rate was 1.04%, which was similar to previous reports2,17.
In this study, all five cases with chromosome aneuploid abnormalities had extra CNS anomalies. Among them, trisomy 13 was the most common chromosome aneuploid abnormality. However, in other studies13,14, trisomy 18 and trisomy 21 were more prevalent, which may have been due to the inclusion of ultrasound soft indicator CNS anomalies. In this study, nine cases had pCNVs, with a pCNV detection rate of 15.79% (9/57). Moreover, CMA appeared in an additional three pCNV cases, with a 13.04% (3/23) incremental yield compared to the karyotype analysis. These findings were similar to those of existing studies13,14,16.
In our study, several pCNVs associate with CNS anomalies were identified in the fetuses. The most common structural anomaly of the human brain is holoprosencephaly, which is associated with deletions and duplications of chromosome 13. Moreover, haploinsufficiency of the ZIC2 gene is associated with autosomal dominant holoprosencephaly-5 disease18,19. In Case 1 of our study, a 28.6 Mb deletion in the 13q31.1q34 region was identified in a fetus with the isolated CNS anomaly of holoprosencephaly. This indicated that the ZIC2 gene was the principal cause of holoprosencephaly in the patient. In Case 4, a 13q31.3q34 duplication was identified in a fetus with brain malformations without holoprosencephaly. However, other candidate genes may have been responsible for the fetal brain malformations in this case. Partial trisomy 7q32.1q36.3 and 10q26.2q26.3 microdeletions were observed in Case 2 with the isolated ultrasound anomaly of hydrocephalus. Existing studies have suggested that partial trisomy or tetrasomy 7q may result in hydrocephalus20,21. Thus, we believe that the 7q32.1q36.3 duplication in this case may be the prime reason for the clinical feature of fetal hydrocephalus.
The 1p36 deletion is the most common terminal deletion syndrome in humans, with a prevalence of approximately 1 in 5000 newborns22. It is commonly characterized by developmental delays, intellectual disability, seizures, short stature, distinctive facial features, brain anomalies, congenital heart defects, and other organ defects23. Cai et al.13 identified the 1p36 deletion syndrome in a fetus with mild ventriculomegaly combined with renal cysts. Additionally, Guterman et al.24 described prenatal findings in ten cases of 1p36 deletion syndrome, suggesting that a prenatal observation of ventriculomegaly, congenital heart defects, or facial dysmorphism may be associated with 1p36 deletion syndrome. In Case 3 of this study, the 1p36.33p36.31 deletion was identified in the fetus with isolated ventriculomegaly. This aided the prenatal detection of ventriculomegaly with 1p36 deletion syndrome. The heterozygous pathogenic GNB1 variant in the 1p36.33 region leads to GNB1 encephalopathy, which manifests as moderate-to-severe developmental delays, intellectual disability, or structural brain abnormalities25. Also, it may be the candidate gene that causes brain malformation in patients with 1p36 deletion syndrome.
In Case 5, a 4p16.3p16.2 microdeletion covering Wolf-Hirschhorn syndrome with multiple structural anomalies was identified in the fetus. In this case, CNS anomalies were also detected, which was consistent with previous reports26. In Cases 6 and 7, both of the fetuses had isolated dysplasia of corpus callosum. Moreover, CMA revealed 7q11.23 and 1q21.1q21.2 microdeletions in the respective fetuses. The 7q11.23 microdeletion was associated with Williams-Beuren syndrome (WBS; MIM 194,050), which is typically expressed in the cardiovascular, central nervous, gastrointestinal, and endocrine systems27. Several studies have revealed morphologic abnormalities and volumetric reductions of corpus callosum in patients with WBS28,29, which was further verified in this study. Additionally, an Nsun5-knockout mouse model in a previous study revealed that the Nsun5 deletion suppressed the proliferation of callosal oligodendrocyte precursor cells. This signified the involvement of the Nsun5 deletion in the agenesis of corpus callosum in WBS30. However, to our knowledge, no reports have described the relationship between 1q21.1 deletion syndrome and dysplasia of corpus callosum.
Two fetuses (Cases 8 and 9) with spina bifida were identified as harboring the 15q11.2 microdeletion and the 2p25.3p24.1 duplication in this present study. A previous study established the relationship between partial trisomy 2p24 and neural tube defects (NTDs), including anencephaly, occipital encephalocele, and spina bifida31. Furthermore, another study stated that approximately 23% of patients with trisomy 2p24 exhibited NTDs32. To date, no reports have elicited the relationship between the 15q11.2 microdeletion and NTDs. Therefore, we presumed that the 15q11.2 microdeletion may not be the main reason for fetal NTDs.
In this study, we analyzed the CMA results of 57 fetuses with prenatal structural CNS anomalies from 5460 pregnant women. Several pathogenic CNVs that were potentially associated with CNS anomalies were identified, including the 13q31.1q34 deletion, 7q32.1q36.3 duplication, 1p36.33q36.31 microdeletion, 4p16.3p16.2 microdeletion, 7q11.23 microdeletion, and 2p25.3p24.1 duplication. Among them, our study indicated that ZIC2, GNB1, and NSUN5 were the possible candidate genes. Additionally, several microdeletion syndromes were identified in fetuses with isolated CNS anomalies, which provided a further reference for the genetic diagnosis and counseling of fetuses with isolated CNS anomalies.
Data availability
The datasets used and analyzed in this study are available from the corresponding author upon reasonable request.
References
Csabay, L., Szabó, I., Papp, C., Tóth-Pál, E. & Papp, Z. Central nervous system anomalies. Ann. N Y Acad. Sci. 847, 21–45 (1998).
Chitty, L. S. & Pilu, G. The challenge of imaging the fetal central nervous system: An aid to prenatal diagnosis, management and prognosis. Prenat. Diagn. 29(4), 301–302 (2009).
Onkar, D., Onkar, P. & Mitra, K. Evaluation of fetal central nervous system anomalies by ultrasound and its anatomical co-relation. J. Clin. Diagn. Res. 8(6), 5–7 (2014).
Dugoff, L., Norton, M. E. & Kuller, J. A. The use of chromosomal microarray for prenatal diagnosis. Am. J. Obstet. Gynecol. 215(4), B2-9 (2016).
Breman, A. et al. Prenatal chromosomal microarray analysis in a diagnostic laboratory; experience with >1000 cases and review of the literature. Prenat. Diagn. 32(4), 351–361 (2012).
Stosic, M., Levy, B. & Wapner, R. The use of chromosomal microarray analysis in prenatal diagnosis. Obstet. Gynecol. Clin. N. Am. 45(1), 55–68 (2018).
Prenatal Screening And Diagnosis Group Birth Defect Prevention And Control Professional Committee Chinese Preventive Medical Association, Prenatal Diagnosis Group Society Of Medical Genetics Chinese Medical Association. Liu J. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 40(9), 1051–1061 (2023).
Zhuang, J. et al. A prenatal diagnosis and genetics study of five pedigrees in the Chinese population with Xp22.31 microduplication. Mol Cytogenet. 12, 50 (2019).
Wang, H. Introduction and interpretation of the updated contents of the international system for human cytogenomic nomenclature. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 38(12), 1165–1170 (2021).
Zhuang, J. et al. Cytogenetic and molecular analysis of distal 4q duplication with distinctive phenotype using single-nucleotide polymorphism array. Mol. Cytogenet. 14(1), 46 (2021).
Riggs, E. R., Andersen, E. F., Cherry, A. M., et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) [published correction appears in Genet Med. 2021 Nov;23(11):2230]. Genet. Med. 22(2), 245–257 (2020).
Noronha Neto, C., Souza, A. S., Moraes Filho, O. B. & Noronha, A. M. Validação do diagnóstico ultrassonográfico de anomalias fetais em centro de referência [Validation of ultrasound diagnosis of fetal anomalies at a reference center in Pernambuco]. Rev. Assoc. Med. Bras. 55(5), 541–546 (2009).
Cai, M., Huang, H., Xu, L. & Lin, N. Clinical utility and the yield of single nucleotide polymorphism array in prenatal diagnosis of fetal central nervous system abnormalities. Front. Mol. Biosci. 8, 666115 (2021).
Tao, H., Wu, J., Han, Y., Zhang, B. & Zhai, J. Genetic etiology and pregnancy outcomes of fetuses with central nervous system anomalies. Arch. Gynecol. Obstet. https://doi.org/10.1007/s00404-023-07152-z (2023).
Schumann, M. et al. Array-based molecular karyotyping in fetuses with isolated brain malformations identifies disease-causing CNVs. J. Neurodev. Disord. 8, 11 (2016).
Krutzke, S. K. et al. Array-based molecular karyotyping in fetal brain malformations: Identification of novel candidate genes and chromosomal regions. Birth Defects Res. A Clin. Mol. Teratol. 106(1), 16–26 (2016).
Paladini, D., Malinger, G., Birnbaum, R., et al. ISUOG Practice Guidelines (updated): sonographic examination of the fetal central nervous system. Part 2: performance of targeted neurosonography [published correction appears in Ultrasound Obstet Gynecol. 2022 Oct;60(4):591]. Ultrasound Obstet Gynecol. 57(4), 661–671 (2021).
Brown, S. A. et al. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat. Genet. 20(2), 180–183 (1998).
Orioli, I. M. et al. Identification of novel mutations in SHH and ZIC2 in a South American (ECLAMC) population with holoprosencephaly. Hum. Genet. 109(1), 1–6 (2001).
Stetten, G. et al. A paternally derived inverted duplication of 7q with evidence of a telomeric deletion. Am. J. Med. Genet. 68(1), 76–81 (1997).
Lehnen, H. et al. Severe phenotype in a girl with partial tetrasomy 7, karyotype 46, XX, trp(7)(q35q36). Cytogenet. Genome Res. 125(3), 248–252 (2009).
Shaffer, L. G. & Lupski, J. R. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu. Rev. Genet. 34, 297–329 (2000).
Jordan, V. K., Zaveri, H. P. & Scott, D. A. 1p36 deletion syndrome: An update. Appl. Clin. Genet. 8, 189–200 (2015).
Guterman, S. et al. Prenatal findings in 1p36 deletion syndrome: New cases and a literature review. Prenat. Diagn. 39(10), 871–882 (2019).
Revah-Politi, A., Sands, T. T., Colombo, S., Goldstein, D. B., & Anyane-Yeboa, K. GNB1 Encephalopathy. In: Adam MP, Mirzaa GM, Pagon RA, et al., eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; March 5, 2020.
De Keersmaecker, B., Albert, M., Hillion, Y. & Ville, Y. Prenatal diagnosis of brain abnormalities in Wolf-Hirschhorn (4p-) syndrome. Prenat. Diagn. 22(5), 366–370 (2002).
Kozel, B. A. et al. Williams syndrome. Nat. Rev. Dis. Primers. 7(1), 42 (2021).
Luders, E. et al. Callosal morphology in Williams syndrome: A new evaluation of shape and thickness. Neuroreport. 18(3), 203–207 (2007).
Schmitt, J. E., Eliez, S., Warsofsky, I. S., Bellugi, U. & Reiss, A. L. Corpus callosum morphology of Williams syndrome: Relation to genetics and behavior. Dev. Med. Child Neurol. 43(3), 155–159 (2001).
Yuan, Z., Chen, P., Zhang, T., Shen, B. & Chen, L. Agenesis and hypomyelination of corpus callosum in mice lacking Nsun5, an RNA methyltransferase. Cells. 8(6), 552 (2019).
Lurie, I. W. et al. Trisomy 2p: Analysis of unusual phenotypic findings. Am. J. Med. Genet. 55(2), 229–236 (1995).
Doray, B. et al. Recurrent neural tube defects associated with partial trisomy 2p22-pter: Report of two siblings and review of the literature. GenetCouns. 14(2), 165–172 (2003).
Acknowledgements
We would like to express our appreciation to the patients and the other subjects who participated in this study. We would also like to thank the Fujian Provincial Commission and Quanzhou Science and Technology Bureau for funding this work.
Funding
This study was supported by the Fujian Provincial Science and Technology Project (2020QNB045), the Quanzhou Science and Technology Program of China (2023NS068), and the Natural Science Foundation of Fujian Province (2023J01104).
Author information
Authors and Affiliations
Contributions
J.Z. designed and wrote the article; X.C., Y.C., and Y.J. recruited the participants and analyzed the data; W.C. performed the karyotype analysis; N.Z., C.C., and J.Z. revised and polished the paper. All authors have approved the final article. We confirm that written informed consent was signed by the parents for the publication of their own and their children’s genetic data and relevant information. The written informed consent is available on request.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhuang, J., Zhang, N., Chen, Y. et al. Prenatal diagnosis and molecular cytogenetic characterization of fetuses with central nervous system anomalies using chromosomal microarray analysis: a seven-year single-center retrospective study. Sci Rep 14, 2271 (2024). https://doi.org/10.1038/s41598-024-52831-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52831-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
