
Abstract
A series of new analogs of 3,5-dihydroxybenzoyl-hydrazineylidene conjugated to different methoxyphenyl triazole (11a-n) synthesized using click reaction. The structures of all synthesized compounds were characterized by FTIR, 1H, 13C-NMR spectroscopy, and CHO analysis. The tyrosinase inhibitory potential of the synthesized compounds was studied. The newly synthesized scaffolds were found to illustrate the variable degree of the inhibitory profile, and the most potent analog of this series was that one bearing 4-methoxyphenyl moiety, and exhibited an IC50 value of 55.39 ± 4.93 µM. The kinetic study of the most potent derivative reveals a competitive mode of inhibition. Next, molecular docking studies were performed to understand the potent inhibitor's binding mode within the enzyme's binding site. Molecular dynamics simulations were accomplished to further investigate the orientation and binding interaction over time and the stability of the 11m-tyrosinase complex.
Similar content being viewed by others
Introduction
Tyrosinase (EC 1.14.18.1) is a type-3 copper-containing metalloenzyme present in plants, fungi, bacteria, and mammals. Tyrosinase is a glycoprotein located in the membrane of the melanosome, a vesicle inside the melanocyte1. Tyrosinase is a key enzyme that catalyzes critical steps in melanin biosynthesis, including hydroxylation of L-tyrosine to 3,4-dihydroxyphenylalanine (L-DOPA), oxidation of L-DOPA to DOPAquinone. Also, it was reported that tyrosinase participates in the oxidation of 5,6-dihydroxyindole to indolequinone. The final products of the melanogenesis process are eumelanin (mostly a dark brown to black polymer) and pheomelanin (a yellow to red polymer)2 resulting in the formation of macromolecular pigments, melanin.
Abnormal pigment levels are linked to several problems, including pigmented patches, skin hyperpigmentation, postinflammatory hyperpigmentation, maturational dyschromia, periorbital hyperpigmentation, melasma, and Riehl melanosis3. Also, some evidence exists about the correlation between neuromelanin and CNS disease. On the other hand, the undesirable enzymatic browning of vegetables and fruit related to melanin synthesis is detrimental to the quality and color of the products4. Regarding the above considerations and the key role of tyrosine in melanogenesis, the discovery of new tyrosinase inhibitors is highly needed. Over time, different natural and synthetic tyrosinase inhibitors have been introduced. Currently, tyrosinase inhibitors, including kojic acid, arbutin, azelaic acid, and hydroquinone, possess undesirable side effects such as low clinical efficacy and carcinogenicity. Therefore, synthesizing novel inhibitors for medical and cosmetic applications is of great interest5.
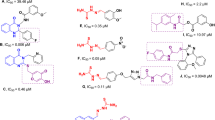
Chalcone has been introduced as a potent tyrosinase inhibitor through screening a library of compounds and different study evaluations. Vaya et al. examined non-, mono-, di-, tri-, or tetra-substituted hydroxyl derivatives as tyrosinase inhibitors, and amongst compound A exhibited the best potency6. Vanillin (Compound B), a natural flavoring agent, exhibited good tyrosinase inhibition, and vaniline–benzylidenehydrazine (Compound C) derivatives were developed7. The most potent compound in this series is exhibited in Fig. 1. The molecular docking study showed that the OH groups participate in critical interactions with two copper cofactors, plus His85 and His263, essential for tyrosinase inhibition. Also, compound D8 bearing OH moiety on the phenyl exhibited significant inhibition against tyrosinase. Compound E disclosed an IC50 near the positive control and Cu2+ chelation potential with a mole fraction of 1:2 stoichiometry9.

Potent inhibitors of mushroom tyrosinase from previous studies and newly designed compound.
Different studies also confirmed the high potency of triazole and its analogs as anticancer10,11,12, anti-microbial13,14, anti-inflammatory15,16, and anti-melanogenic agents as tyrosinase inhibitors. In this category, triazole-benzoimidazole (Compound F) with IC50 = 9.42 μM is a good example17. A new series of new triazole derivatives (Compound G) was reported, and it was shown that moieties with the ability to form hydrogen bonds with His85 and His244 improve the potency18. Recently, phenoxy methyl triazole conjugated with thiosemicarbazide (Compound H) was reported to induce the tyrosinase inhibitory effects with IC50 values of 0.11 μM and 0.17 μM in the presence of L-tyrosine and L-DOPA as substrates. The proposed mechanism of the high potency of tyrosinase inhibitor was related to its Cu chelatory potential. Compound H significantly reduced the melanin content in skin melanoma cells to 39.8% at 8 µM. The main interactions with tyrosinase active site were seen between the phenoxy group and His263 and Ala286. Residue Val283 formed an H-bond interaction with the triazole ring and two pi-sigma interactions with triazole and methoxybenzene rings19. As a result, phenoxy-triazole was chosen as a valuable starting point.
With these results in hand, 3,5-dihydroxybenzohydrazide was chosen that has similarity with native substrate L-tyrosine to explore the structural requirements of tyrosinase-inhibitory activity. The presence of such OH moiety provides the minimal structural requirements of tyrosinase inhibition. On the other hand, aryl methoxy-triazole scaffolds were connected to 3,5-dihydroxybenzohydrazide moiety through Schiff base reaction, developing and extending the structure–activity relationships (SARs), and different derivitization were conducted at the R position. It was proposed that aryl methoxy-triazole not only provides the optimum bulkiness to occupy the pocket of the enzyme but also might interact with the critical residue of the binding site to hinder the oxidation process. The designed compounds were synthesized and evaluated as possible tyrosinase inhibitors. Next, the most potent derivative was subjected to the kinetic study to determine the type of inhibition. Furthermore, molecular docking and molecular dynamic simulations were also performed.
Results and discussion
Synthesis
Synthesis of the target compounds 11a-n was schematically described in Fig. 2. Briefly, 3,5-dihydroxybenzoic acid (compound 1) was allowed to react with methanol under the refluxed condition to conduct an esterification reaction. After 8 h, the methanol was evaporated to obtain methyl 3,5-dihydroxybenzoate (compound 2). This methyl ester was then reacted with hydrazine hydrate to produce the intermediate 3,5-dihydroxybenzoic acid hydrazide (compound 3). In a separate reaction, chloroacetyl chloride (compound 5) was added to aniline derivatives (compound 4a-n) in DMF, yielding compound 6a-n. In another reaction vessel, propargyl bromide (compound 8) was introduced to 4-hydroxybenzaldehyde (compound 7) and potassium carbonate to produce O-propargyl benzaldehyde (compound 9). Additionally, aryl acetamide derivatives were refluxed with sodium azide and triethylamine (TEA), followed by the addition of compound 9 and catalytic CuSO4.5H2O and sodium ascorbate to produce the 10a-n. Finally, a mixture of compounds 10a-n and 3,5-dihydroxybenzohydrazide in the presence of acetic acid was refluxed in ethanol, followed by purification through diethyl ether crystallization to obtain the desired products, 11a-n20,21.

Synthesis of compounds 11a-n.
In vitro inhibition of tyrosinase
Inhibitory activities against tyrosinase were determined in the colorimetric assay for all compounds. The results are shown in Table 1. 3,5-Dihydroxybenzoyl-hydrazineylidene compounds were synthesized to assess the importance of different substituents at R positions of the terminal aryl ring.
Compound 11a, serving as the primary unsubstantiated derivative, displayed an IC50 value of 90.53 µM with a modest 53.87% inhibition observed at 100 µM concentration. This initial finding prompted us to investigate the impact of electron-withdrawing groups on the compound's potency, leading to the design and synthesis of compounds 11b-h.
Incorporating a 2-fluorophenyl group in compound 11b, reduced the inhibitory potency, with only 22.99% inhibition at 100 µM. This result suggests that the electron-withdrawing nature of the fluorine atom has a detrimental effect on the compound's activity. While shifting the substituent from the ortho to para position in compound 11c enhanced the inhibitory activity, with a measured inhibition of 32.28% at 100 µM. This suggests that the para position is more favorable for improving the compound's potency.
A more detailed evaluation of para-position substitutions revealed that 4-fluorophenyl (11c) had the highest potency, surpassing 4-chlorophenyl (11d) > 4-bromophenyl (11e). This ranking indicates that increasing the bulkiness of the para-position substituents negatively correlates with the inhibitory potency. The observed trend underscores the importance of understanding the impact of both electron-withdrawing and steric effects in optimizing the compound's activity.
Furthermore, introducing multi-halogen substitutions at different positions of the phenyl ring was attempted to improve inhibition potentially. However, it was observed that no improvement in potency was achieved in compounds 11f (R = 2,4-dichlorophenyl) and 11g (R = 2,6-dichlorophenyl) when compared to the unsubstituted analog 11a.
Given that the halogen-substituted group did not significantly improve the potency compared to the unsubstituted moiety, introducing nitro groups was considered an alternative approach to assess their electron-donating potential and the potential for hydrogen-bond interactions. It was observed that compounds 11h (R = 4-nitrophenyl) and 11i (R = 2-methyl-4-nitrophenyl), as nitro-substituted derivatives, exhibited lower inhibitory potencies when compared to 11a as the unsubstituted derivative.
In light of the lack of activity observed in the electron-withdrawing substituted derivative, the synthesis of electron-donating groups was pursued. Introducing a para-methyl group resulted in a significant activity loss, with only 15.22% inhibition observed at 100 µM. However, the inhibitory activity of compound 11k, which featured 2,6-dimethyl substitutions, improved when compared to the monomethyl substituted group, with an inhibition rate of 38.59% at 100 µM. Additionally, introducing a larger electron-donating group like 4-ethyl substitution (11l) further enhanced the compound's potency, resulting in an IC50 value of 87.54 µM. This data underscores the importance of considering the electron-donating nature and steric effects when optimizing the inhibitory potential of these derivatives.
Remarkably, the para-methoxy group emerged as the most effective substitution, with compound 11m displaying the highest potency, featuring an IC50 value of 58.88 μM. This finding underscores the potential of electron-donating properties, particularly when coupled with heteroatoms, to facilitate hydrogen-bond interactions, positively influencing the compound's inhibitory activity.
Additionally, introducing a benzyl group (11n), which elongates the linker, resulting in an inhibition of 17.48% at 100 µM. However, it is apparent that extending the linker is not well-tolerated and may not be conducive to improving the compound's inhibitory potency.
Enzyme inhibitory kinetics
The enzyme inhibitory interaction mechanism of 11m with the binding site of tyrosinase was determined using Michaelis–Menten kinetic studies. The inhibitor exhibits a dose-dependent inhibition of the enzyme tyrosinase. Inhibition kinetics was analyzed by the Lineweaver–Burk plot with 1/Vmaxvs. 1/[S] at different doses of 11m (Fig. 3a). Results exhibited that the Michaelis–Menten constant (Km) changes while that of 1/Vmax remained the same, representing the competitive nature of the most potent inhibitor (11m) (Fig. 3a). The dissociation constant Ki for 11m was 52.77 μM as shown in Fig. 3b.

(a) Line weaver-burk plot of 11m against tyrosinase; (b) The secondary plot between the Km and various concentrations of 11m.
Docking study
The molecular docking study was conducted to provide insights into the binding mode of 11m, the most potent derivative against the tyrosinase enzyme.
The validation of the molecular docking process was initially conducted through the redocking of a co-crystallized ligand, tropolone, within the binding site of the enzyme. The procedure resulted in an RMSD (root mean square deviation) value of less than 2 Å, confirming that the docking approach was reliable. This low RMSD value implies a strong correlation between the docked conformation and the crystalographic conformation, suggesting that the docking protocol accurately predicts the ligand's orientation and position within the enzymatic binding site. This validation step is critical, as it establishes the credibility of the docking method for further exploration and analysis of potential ligand-enzyme interactions in subsequent studies.
The in silico studies conducted on the designed analogs were examined. As determined by the molecular docking analysis results presented in Table 2, the docking scores of the derivatives against tyrosinase ranged from – 7.570 to – 4.157 kcal/mol. Notably, there was a positive correlation between these docking scores and the biological results, strengthening the validity of the approach.
Particularly, compounds 11m and 11l were identified as the most potent inhibitors. Compound 11m, with an IC50 value of 55.39 ± 4.93 µM, demonstrated a remarkable docking score of – 7.57 kcal/mol. In contrast, compound 11l, exhibiting an IC50 value of 87.54 ± 7.99 µM, was associated with a docking score of – 6.254 kcal/mol. These findings underscore their prominence as active inhibitors in the series. Conversely, the compounds considered as less active, including 11i, 11j, 11e, and 11f, were noted for their relatively minimal docking scores against tyrosinase, recorded – 4.157, – 4.416, – 4.351, and – 4.905 kcal/mol, respectively. This denotes a clear separation between active and less active molecules regarding their potency.
Further examination into the binding interactions has revealed that the most potent inhibitors in our dataset primarily interact with the Cu+2 cofactor through a metal coordination bond. This type of interaction is critical for inhibiting tyrosinase activity, suggesting that the metal binding is a key pathway for inhibitory action amongst the active compounds studied.
The docking results of 11m as the most potent compound, characterized by its chalcone structure, displayed five noteworthy interactions. These interactions encompassed associations with both Cu2+ ions, H-bond interactions with Glu98, and three π-π stacking interactions with His61, Tyr97, and Phe292.On the other side of the molecule, the phenolic linker exhibited another π-π stacking interaction with His263, and the amide group formed an H-bond with the crucial His85, along with another H-bond with Asn81. These results confirm that 11m interacts critically with Cu ions and His residues (Fig. 4).

Representation of compound 11m within the active site of tyrosinase.
Molecular dynamics simulation
The molecular dynamics (MD) simulation was performed to confirm the 11m stability over the enzyme active site. First, the root mean square deviation (RMSD) of the enzyme's backbone was analyzed over during 100 ns MD simulation to study the perturbation of the protein–ligand complex. As shown in Fig. 5, atoms of the protein alone fluctuated at around 1.75 Å. Specifically, within the first 8 ns of the simulation, the RMSD experienced a pronounced increase. Subsequently, between 8–18 ns, it stabilized at an RMSD value of 1.75 Å. Notably, for the remaining duration of the simulation, the complex exhibited a consistently lower RMSD compared to the protein alone, which maintained an RMSD of around 1.4 Å, confirming the stability of the complex.

RMSD plot of the tyrosinase in complex with compound 11m in the MD simulation time. RMSD values of the apo-enzyme are depicted in blue, and the 11m-enzyme is exhibited in orange.
Next, The root mean square fluctuation (RMSF) is also evaluated. RMSF recorded the local changes along the protein chain during the MD run. As exhibited, the secondary structure elements like α-helical and β-strand are usually rigid. The high fluctuations observed in the RMSD values can be attributed to the unstructured region of the enzyme (Fig. 6). As indicated, the reduction in movement in the region spanning residues 68–81 (the red dashed line), residues 245 to 251 (the yellow dashed line), along with residues 187–195 (the green dashed line), compared to the apoenzyme in these areas, played a significant role in stabilizing the complex.

RMSF plot of the tyrosinase residues in complexed with 11m. RMSF values of the apo-enzyme are depicted in blue, and the 11m-enzyme is exhibited in orange.
In Fig. 7 depicts the Ligand Root Mean Square Fluctuation (L-RMSF) values of the heavy atoms of ligand 11m when bound to tyrosinase. With the exception of the 4-methoxyphenyl regions, all atoms in 11m exhibit RMSF values below 2 Å. This minimal fluctuation signifies a stable complex formation with tyrosinase, primarily due to robust intermolecular interactions that restrict their mobility during the molecular dynamics simulation. This persistent binding interaction strongly suggests that ligand 11m holds promising potential as an effective tyrosinase inhibitor.

L-RMSF graph of the heavy atoms of 11m in complex with tyrosinase.
Interaction types with the ligand throughout the simulation run are exhibited in Fig. 8a, and the interaction with each residue is exhibited in Fig. 8b. 3,5-dihydroxybenzoyl displayed multiple interactions within the system. It engaged in two hydrogen-bond interactions with Glu98 and Phe292 and two pi-pi stacking interactions with Phe292 and His296. Notably, the C=O group of the benzoylhydrazone moiety consistently participated in metal coordination with the copper cofactor throughout the entire simulation period (100%). Moreover, the hydrazine group formed an essential metal interaction with another copper ion involved in the oxidation process. This hydrazine linker was observed to effectively engage with the critical copper cofactor throughout the total simulation time (100%), ultimately impeding the oxidation process in the melanogenesis pathway. Within the molecular structure, the phenoxy linker in the middle of the molecule demonstrated a hydrogen bond interaction with Val283, facilitated by water molecules, occurring during 52% of the MD simulation run. This phenoxy linker recorded another H-bound interaction with Asn260 mediated with water (52%). Additionally, the 4-methoxyphenyl group at the terminal of the molecule exhibited a hydrogen bond interaction with Tyr65. These critical interactions confirm the high potency of 11m, which properly occupies the binding site.

(a) Protein interaction types with the 11m throughout the simulation, (b) 2D ligand interactions with the protein residues.
In addition to the interaction analysis, the Prime/MM-GBSA module was used to estimate the strengths of interactions between the ligand–protein complex generated by the clustering method. ΔGbind of tyrosinase/compound 11m complex was estimated to be – 43.46 kcal/mol.
ADMET properties and in silico toxicity
Table 3 demonstrates drug-likeness prediction for 11a-n. Drug-likeness refers to the set of properties that make a molecule suitable for use as a drug. Lipinski's Rule of Five evaluates a compound's drug-likeness based on its physicochemical properties. This rule includes the molecular weight (MW ≤ 500), the number of hydrogen bonding acceptors (≤ 10), the number of hydrogen bonding acceptors (≤ 5), the lipophilicity index (logP ≤ 5), rotatable bond count (≤ 10) and polar surface area (≤ 140) of compounds22 . Regardless of molecular weight, most compounds have successfully met Lipinski's rule23.
ADMET stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity of pharmaceutical compounds within a biological system. These properties are crucial component of drug development that can significantly improve the safety, efficacy, and efficiency of bringing new medications. Table 4 presents the ADMET predictions for all compounds, calculated using the pkCSM online servers23. The estimated Human Intestinal Absorption (HIA) of the compounds ranged from 60.123 to 67.123, indicating a relatively high absorption rate within the gastrointestinal tract. This suggests that the compounds have favorable pharmacokinetic profiles for oral administration, an essential characteristic for effective systemic drug delivery. The compounds under investigation are estimated to have a low steady-state volume of distribution, ranging from – 2.735 to – 0.386 log L/kg, which indicates limited distribution primarily to the plasma and extracellular spaces rather than extensive tissue penetration. This profile potentially minimizes adverse effects associated with tissue accumulation, making these compounds particularly suitable for treatments necessitating rapid systemic availability and clear dose–response relationships. Regarding metabolism, all compounds were predicted not to be inhibitors of CYP2D6 and CYP2C19 while expected to be CYP3A4 and CYP2C9 inhibitors. This profile may exhibit an advantage by minimizing the risk of adverse drug interactions mediated via CYP2D6 and CYP2C19 enzymes involved in the metabolism of a wide range of pharmaceuticals. The total clearance of derivatives is in the range of 0.267 to – 0.437. A higher clearance rate may be desired to achieve a rapid onset of action and shorter duration. In contrast, slower clearance may be necessary to maintain therapeutic levels over an extended period. In silico models have quantified the maximum tolerated dose for humans between 0.270 and 0.319 mg/kg/day for the compounds studied, denoting a closely defined dosing range advantageous for further refinement of dosage regimens in clinical trials.
Meanwhile, the oral rat acute toxicity studies showed a narrow range from 2.415 to 2.487, suggesting a consistent toxicity profile in this model, which rationalizes the extrapolation to potential human toxicology outcome. hERG1 inhibition is a valuable parameter in ADME-T prediction, particularly for assessing the risk of drug-induced long QT syndrome (LQTS) and associated cardiac arrhythmias. As can be seen, none of the derivitives predicted to be hERG1 inhibitors.
Conclusion
Regarding the important role of tyrosinase in the melanogenesis process, a series of 3,5-dihydroxybenzoyl-hydrazineylidene derivatives, 11a-n, was designed and synthesized as tyrosinase inhibitor. The structure of all derivatives was comprehensively characterized using various spectroscopic data and elemental analysis. The in vitro tyrosinase inhibitory activity of these compounds revealed promising results, with most derivatives displaying moderate to good potency. Remarkable inhibitory potential was exhibited by compound 11m, with an IC50 value of 55.39 ± 4.93 µM. A competitive mode of inhibition was indicated by kinetic analysis of the most potent analogs, with a Ki of 52.77 μM calculated for 11m.
Furthermore, the molecular docking study exhibited significant interactions of 11m with critical and highly conserved His amino acid and Cu2+ cofactors. Stability and consistent binding throughout a 100 ns simulation period were revealed by MD simulations of 11m with tyrosinase. Notably, reduced fluctuations in critical residue regions compared to tyrosinase alone were indicated by the RMSD values of 11m-tyrosinase complexes. Importantly, consistent interaction with Cu2 + cofactors was observed throughout the entire simulation period (100%). The enhanced potency of these compounds, particularly compound 11m, was attributed to their chalcone-based structure with an amide linker and a terminal 4-methoxyphenyl tail moiety. The designed structure provides a promising foundation for developing potent tyrosinase inhibitors, which can be utilized as primary frameworks for forthcoming tyrosinase inhibitor development.
Method and materials
Chemistry
1H and 13C NMR spectra were recorded using a Bruker spectrometer 400 MHz instrument. Chemical shifts were reported in parts per million (ppm). Multiplicities were indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and coupling constant J was reported in hertz (Hz). IR spectra were obtained with a Nicolet, FR -IR Magna 550. Melting points were also recorded using Kofler hot-stage apparatus. All the chemicals were purchased from Merck and Sigma.
Synthesis of methyl 3,5-dihydroxybenzoate (2)
3,5-Dihydroxybenzoic acid (10.00 g, 64.9 mmol) was dissolved in methanol (50 mL concentrated sulfuric acid (750 μL in 10 mL methanol) ) and the solution was allowed to reflux for 8 h. After cooling to ambient temperature, methanol was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate (50 mL), which was then washed with saturated NaHCO3 solution and water. The organic layer was dried over anhydrous Na2SO4, and ethyl acetate was removed in vacuo to afford methyl 3,5-dihydroxylbenzoate (10.74 g, 98%) as a white powder.
Synthesis of 3,5-dihydroxybenzohydrazide (3)
Methyl 3,5-dihydroxybenzoate (compound 2, 1.060 mmol) was dissolved in absolute ethanol (5.0 mL) and hydrazine hydrate (0.5 mg, 15.625 mmol) was added. The reaction was allowed to react at room temperature for 16 h, concentrated, filtered, and dried. The intermediate 3,5-dihydroxybenzoic acid hydrazide was obtained.
Synthesis of Substituted 2-chloro-N-phenylacetamide (6a-n)
On an ice bath, chloroacetyl chloride (6.78 g, 0.06 mol) was added to a solution containing 0.050 mol of aniline derivatives dissolved in 40 mL of DMF. The resulting mixture was stirred at room temperature for 2 h, then poured into water and subsequently filtered to isolate compound 6a-n24,25.
Synthesis of 4-(prop-2-yn-1-yloxy)benzaldehyde (9)
Propargyl bromide (23 mmol) was added to a solution containing 4-hydroxybenzaldehyde (compound 7, 23 mmol) and potassium carbonate (22 mmol) in 10 mL of dry DMF. The reaction mixture was stirred at room temperature for 2 h and then extracted with ethyl acetate to obtain crude O-propargyl benzaldehyde.
Synthesis of 2-(4-((4-formylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (10a-n)
At first, 2 mmol of different aryl acetamide derivatives (6a-m) and 2 mmol sodium azide in the presence of TEA were dissolved in DMF/H2O, and the mixture was refluxed for 3 h. The resulting solution was then refluxed for 3 h. Subsequently, 2 mmol of compound 9, the catalytic quantity of CuSO4·5H2O, and sodium ascorbate were introduced. After 24 h, the products were synthesized, and the reaction's progress was monitored using TLC to ensure completion.
Synthesis of 11a-n derivatives
A mixture containing 0.020 mol of compounds 10a-n and 0.021 mol of 3,5-dihydroxybenzoyl-hydrazineylidene, along with approximately 2 mL of acetic acid as the acid catalyst, was subjected to reflux in 96% ethanol (50 mL) for 8 h. Upon the completion of the reaction, the solvent was removed under reduced pressure, and the crude product was subsequently purified through crystallization in diethyl ether. After filtration and washing with diethyl ether, the product was left to dry at room temperature.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (11a)
IR (KBr): 3521, 1679, 1364, 1196 cm−1. 1H NMR (400 MHz, ) δ 11.59 (s, 1H), 10.51 (s, 1H), 9.59 (s, 2H), 8.38 (s, 1H), 8.30 (s, 1H), 7.67 (dd, J = 8.8, 2.6 Hz, 2H), 7.33 (dt, J = 17.7, 7.3 Hz, 4H), 7.12 (dt, J = 23.1, 7.9 Hz, 3H), 6.74 (d, J = 2.1 Hz, 2H), 6.42 (d, J = 2.2 Hz, 1H), 5.28 (s, 2H), 5.21 (s, 2H).
13C NMR (101 MHz, DMSO) δ 164.64, 163.61, 160.02, 158.83, 147.75, 142.67, 138.87, 130.46, 129.40, 129.08, 128.84, 127.86, 126.91, 119.66, 115.52, 106.17, 105.97, 61.59, 52.69 ppm; Anal. Calcd for C25H22N6O5: C, 61.72; H, 4.56; N, 17.28; Found: C 61.66; H 4.35; N 17.49.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(2-fluorophenyl)acetamide (11b)
IR (KBr): 3545, 1688, 1361, 1167, 768 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.60 (s, 1H), 9.84 (s, 1H), 9.59 (s, 2H), 8.37 (s, 1H), 8.29 (s, 1H), 7.66 (d, J = 8.6 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 7.12 – 7.06 (m, 3H), 6.73 (d, J = 1.8 Hz, 2H), 6.41 (s, 1H), 5.41 (s, 2H), 5.24 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.49, 163.65, 160.03, 158.95, 153.78, 149.95, 147.73, 142.65, 136.08, 135.55, 134.67, 129.08, 128.24, 127.76, 127.24, 126.86, 115.53, 106.15, 61.58, 52.09 ppm; Anal. Calcd for C25H22N6O5: C, 59.52; H, 4.20; N, 16.66; Found: C, 59.71; H, 4.34; N, 16.81.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-fluorophenyl)acetamide (11c)
IR (KBr): 3541, 1681, 1358, 1171, 761 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.70 (s, 1H), 9.62 (s, 2H), 8.38 (s, 1H), 8.30 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 6.74 (d, J = 1.6 Hz, 2H), 6.45 (s, 1H), 5.38 (s, 2H), 5.25 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.87, 163.62, 161.22, 160.02, 158.84, 147.76, 142.72, 137.84, 136.11, 129.34, 129.09, 127.84, 126.92, 121.25, 115.53, 106.18, 61.58, 52.67 ppm; Anal. Calcd for C25H21FN6O5: C, 59.52; H, 4.20; N, 16.66; Found: C, 59.46; H, 4.02; N, 16.43.
N-(4-chlorophenyl)-2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)Acetamide (11d)
IR (KBr): 3537, 1687, 1364, 1165, 758 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.65 (s, 1H), 9.58 (s, 2H), 8.38 (s, 1H), 8.30 (s, 1H), 7.64 (dd, J = 18.2, 8.6 Hz, 4H), 7.46 – 7.34 (m, 2H), 7.15 (d, J = 8.4 Hz, 2H), 6.74 (d, J = 2.2 Hz, 2H), 6.42 (d, J = 2.2 Hz, 1H), 5.38 (s, 2H), 5.25 (s, 2H). 13C NMR (101 MHz, DMSO) δ 164.85, 163.61, 160.01, 158.83, 147.75, 142.71, 137.82, 136.09, 129.33, 129.08, 127.83, 127.75, 126.91, 121.24, 115.52, 106.17, 105.96, 61.58, 52.67. Anal. Calcd for C25H21ClN6O5: C, 57.64; H, 4.06; N, 16.13; Found: C 57.37; H 3.89; N 16.31.
N-(4-bromophenyl)-2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11e)
IR (KBr): 3537, 1658, 1556, 1253, 786 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.61 (s, 1H), 9.58 (s, 2H), 8.38 (s, 1H), 8.30 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 6.76 – 6.72 (m, 2H), 6.42 (s, 1H), 5.38 (s, 2H), 5.25 (s, 2H) ppm; 13C NMR (101 MHz, DMSO-d6) δ 166.09, 163.62, 161.68, 158.84, 147.76, 142.72, 137.84, 136.11, 129.34, 129.09, 127.84, 126.92, 121.25, 115.53, 106.18, 61.58, 52.67 ppm; Anal. Calcd for C25H21BrN6O5: C, 53.11; H, 3.74; N, 14.86; Found: C, 52.97; H, 3.56; N, 14.62.
N-(2,4-dichlorophenyl)-2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11f.)
IR (KBr): 3532, 1651, 1552, 1249, 792 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 9.80 (s, 1H), 9.58 (s, 2H), 8.37 (s, 1H), 8.29 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 1.1 Hz, 3H), 6.73 (d, J = 2.1 Hz, 2H), 6.41 (d, J = 2.3 Hz, 1H), 5.41 (s, 2H), 5.24 (s, 2H), 2.16 (s, 6H). 13C NMR (101 MHz, DMSO) δ 164.47, 163.58, 160.03, 158.83, 147.71, 142.63, 136.09, 133.59, 131.20, 130.32, 129.07, 128.23, 127.74, 127.23, 126.84, 122.58, 115.51, 106.16, 105.70, 61.57, 52.09 ppm; Anal. Calcd for C25H20Cl2N6O6: C, 54.07; H, 3.63; N, 15.13; Found: C 53.89; H 3.47; N 15.02.
N-(2,6-dichlorophenyl)-2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)Acetamide (11g)
IR (KBr): 3533, 1655, 1325, 1270, 802 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 9.80 (s, 1H), 9.61 (s, 1H), 8.37 (s, 1H), 8.29 (s, 1H), 7.66 (d, J = 8.3 Hz, 2H), 7.12 (d, J = 28.2 Hz, 5H), 6.73 (d, J = 2.3 Hz, 2H), 6.41 (d, J = 2.2 Hz, 1H), 5.41 (s, 2H), 5.24 (s, 2H). 13C NMR (101 MHz, DMSO) δ 164.47, 163.63, 160.01, 158.93, 147.72, 142.64, 136.06, 135.54, 134.65, 129.07, 128.23, 127.75, 127.23, 126.85, 115.51, 106.12, 105.99, 61.57, 52.09 ppm; Anal. Calcd for C25H20Cl2N6O5: C, 54.07; H, 3.63; N, 15.13; Found: C 54.16; H 3.36; N 15.35.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-nitrophenyl)Acetamide (11h)
IR (KBr): 3539, 1696, 1551, 1347, 1228 cm−1. 1H NMR (400 MHz, ) δ 11.58 (s, 1H), 11.02 (s, 1H), 9.58 (s, 2H), 8.61 (d, J = 2.2 Hz, 2H), 8.33 (d, J = 8.4 Hz, 2H), 7.97 (dd, J = 8.2, 2.3 Hz, 2H), 7.90 (d, J = 8.6 Hz, 3H), 7.67 (d, J = 8.3 Hz, 3H), 7.28 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 2.2 Hz, 1H), 6.42 (d, J = 2.2 Hz, 1H), 5.34 (s, 2H), 5.26 (s, 2H); 13C NMR (101 MHz, DMSO) δ 164.43, 163.47, 160.55, 158.83, 148.46, 147.53, 144.81, 142.36, 139.93, 129.09, 127.48, 126.43, 125.69, 118.84, 115.67, 106.03, 105.41, 61.88, 52.69 ppm; Anal. Calcd for C25H21N7O7: C, 56.50; H, 3.98; N, 18.45; Found: C 56.27; H 4.21; N 18.62.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(2-methyl-4-nitrophenyl)acetamide (11i)
IR (KBr): 3527, 1690, 1551, 1352, 1205 cm−1. 1H NMR (400 MHz, ) δ 11.58 (s, 1H), 9.89 (s, 1H), 9.58 (s, 2H), 8.38 (s, 1H), 8.32 (s, 1H), 8.18 (d, J = 2.6 Hz, 1H), 7.96 (dd, J = 8.9, 3.1 Hz, 1H), 7.90 (d, J = 8.7 Hz, 1H), 7.67 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 2.1 Hz, 2H), 6.42 (d, J = 2.3 Hz, 1H), 5.33 (s, 2H), 5.26 (s, 2H), 2.42 (s, 3H). 13C NMR (101 MHz, DMSO) δ 164.39, 163.85, 160.88, 158.17, 147.06, 143.95, 142.21, 139.83, 136.35, 132.28, 129.08, 127.35, 126.04, 123.72, 120.44, 115.52, 109.84, 106.27, 105.87, 61.12, 52.63, 18.32. Anal. Calcd for C26H23N7O7: C, 57.25; H, 4.25; N, 17.97; Found: C 57.10; H 4.14; N 18.09.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (11j)
IR (KBr): 3448, 1671, 1389, 1231 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.44 (s, 1H), 9.61 (s, 1H), 8.38 (s, 1H), 8.29 (s, 1H), 7.67 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.3 Hz, 2H), 7.16 (t, J = 7.9 Hz, 4H), 6.87 – 6.61 (m, 2H), 6.42 (s, 1H), 5.35 (s, 2H), 5.25 (s, 2H) ), 2.31 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.10, 161.35, 160.03, 158.84, 156.00, 147.75, 142.66, 136.10, 131.97, 129.09, 127.75, 126.90, 121.22, 115.53, 114.49, 106.17, 61.59, 52.61, 20.80 ppm; Anal. Calcd for C26H24N6O5: C, 62.39; H, 4.83; N, 16.79; Found: C, 62.25; H, 4.63; N, 16.64.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(2,6-dimethylphenyl)Acetamide (11k)
IR (KBr): 3562, 1678, 1341, 1210 cm−1. 1H NMR (400 MHz, ) δ 11.58 (s, 1H), 10.25 (s, 0H), 9.58 (s, 2H), 8.37 (s, 1H), 8.29 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.09 (s, 8H), 6.73 (d, J = 2.2 Hz, 2H), 6.42 (t, J = 2.2 Hz, 1H), 5.41 (s, 3H), 5.24 (s, 2H), 2.16 (s, 6H) ppm; 13C NMR (101 MHz, DMSO) δ 164.47, 163.59, 160.01, 158.82, 147.73, 142.63, 137.87, 136.09, 130.50, 129.07, 128.23, 127.23, 126.85, 126.76, 115.51, 106.16, 105.59, 61.46, 52.08, 18.50 ppm; Anal. Calcd for C27H26N6O5: C, 63.03; H, 5.09; N, 16.33; Found: C 63.28; H 5.14; N 16.19.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-ethylphenyl)acetamide (11l)
IR (KBr): 3544, 1677, 1330, 1171 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.44 (s, 1H), 9.61 (s, 2H), 8.38 (s, 1H), 8.29 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.56 – 7.45 (m, 2H), 7.16 (t, J = 7.9 Hz, 4H), 6.74 (d, J = 2.1 Hz, 2H), 6.42 (d, J = 2.3 Hz, 1H), 5.35 (s, 2H), 5.25 (s, 2H), 2.56 (q, J = 7.6 Hz, 2H), 1.16 (t, J = 7.6 Hz, 3H) ppm; 13C NMR (101 MHz, DMSO) δ 164.37, 163.61, 160.02, 158.85, 147.75, 142.65, 139.66, 136.54, 129.08, 128.57, 127.75, 126.90, 122.87, 119.75, 115.52, 106.17, 105.97, 61.58, 52.66, 28.05, 16.11 ppm; Anal. Calcd for C27H26N6O5: C, 63.03; H, 5.09; N, 16.33; Found: C 62.84; H 5.26; N 16.52.
2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-methoxyphenyl)acetamide (11m)
IR (KBr): 3442, 1678, 1383, 1235 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 10.37 (s, 1H), 9.59 (s, 2H), 8.38 (s, 1H), 8.29 (s, 1H), 7.67 (d, J = 8.4 Hz, 2H), 7.54 – 7.46 (m, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.97 – 6.86 (m, 2H), 6.74 (d, J = 2.1 Hz, 2H), 6.41 (d, J = 2.2 Hz, 1H), 5.33 (s, 2H), 5.25 (s, 2H), 3.73 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.09, 163.60, 160.02, 158.83, 155.98, 147.74, 142.64, 136.09, 131.96, 129.08, 127.74, 126.89, 121.21, 115.52, 114.48, 106.16, 105.95, 61.58, 55.62, 52.60 ppm; Anal. Calcd for C26H24N6O6: C, 60.46; H, 4.68; N, 16.27; Found: C 60.63; H 4.47; N 16.01.
N-benzyl-2-(4-((4-((2-(3,5-dihydroxybenzoyl)hydrazineylidene)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (11n)
IR (KBr): 3445, 1668, 1328, 1212 cm−1. 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 9.58 (s, 2H), 8.87 (t, J = 5.9 Hz, 1H), 8.38 (s, 1H), 8.24 (s, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.38 – 7.23 (m, 5H), 7.15 (t, J = 6.8 Hz, 3H), 6.73 (d, J = 2.1 Hz, 2H), 6.42 (t, J = 2.2 Hz, 1H), 5.23 (s, 2H), 5.21 (s, 2H), 4.34 (d, J = 5.8 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 165.87, 163.59, 160.02, 158.82, 147.73, 142.58, 139.16, 136.09, 129.37, 129.07, 128.84, 127.86, 127.48, 126.79, 115.51, 106.16, 105.94, 61.58, 52.07, 42.84. Anal. Calcd for C26H24N6O5: C, 62.39; H, 4.83; N, 16.79; Found: C 62.11; H 4.62; N 16.57.
Tyrosinase inhibitory assay
The tyrosinase inhibitory activities of derivatives were performed according to the previously reported procedures26,27. All the test samples were first dissolved in DMSO at dilution to the required final concentrations. Initially, in a 96-well microplate, 10 µl of test samples were added to 160 µl of phosphate buffer (pH = 6.8), and then 10 µl tyrosinase was added. After the mixture was pre-incubated at 28 °C for 20 min, 20 µl of L-DOPA solution was added. DMSO without test compounds was used as the control, and kojic acid was used as a positive control. After 8 min incubation absorbance of samples was measured at 490 nm. Each assay was conducted as three separate replicates.
Enzyme kinetic studies
The kinetic study for tyrosinase inhibition by 11m as the most potent analog was carried out using four different concentrations of inhibitor (10, 20, 30, 50 and 70 µM) against tyrosinase with different concentrations of L-DOPA (0.25, 0.5, 0.75, and 1 mM) as the substrate. The Lineweaver–Burk reciprocal plot was provided by plotting 1/V against 1/[S] at variable concentrations of the L-DOPA.
Molecular docking
The molecular docking studies were performed using the Maestro molecular modeling platform (version 10.5) by Schrödinger, LLC (Maestro, Schrödinger, LLC, New York, NY, 2021). The 3D crystal structure of tyrosinase was retrieved from the Protein Data Bank (PDB code: 2Y9X). Protein was prepared in which the water molecules and the cognate ligand (tropolone) were removed from the receptor and the hydrogen atoms were added and non-polar hydrogens were merged into related atoms of the receptor via protein preparation. To prepare the ligand, the 2D structures of the ligands were drawn in ChemDraw version 12, converted into SDF files, and subjected to the LigPrep module. Ligands were prepared by OPLS_2005 force field using EPIK. The derivative was docked on binding sites using induced-fit docking with a box size of 20 Å, reporting 10 poses per ligand to form the final complex. The other parameters were as follows: confirmation sampling with an energy window of 2.5 kcal/mol, receptor and ligand van der Waals scaling 0.5, and Glide redocking into receptor structures within 30 kcal/mol of the best structure, and all parameters were set as default28,29.
MD simulation
The molecular simulation was conducted utilizing the Desmond of Schrödinger package (version 10.5, Maestro, Schrödinger, LLC, New York, NY, 2021). To prepare the system for MD simulation, the protein–ligand complexes were immersed in an orthorhombic box of suitable dimensions with periodic boundary conditions and solvated using explicit water molecules of the SPC type. Additionally, the system was neutralized by incorporating an appropriate number of counter-ions, and a 0.15 M solution of NaCl was employed to mimic realistic cellular ionic concentrations20. The MD protocol involved minimization, pre-production, and production MD simulation steps. Finally, the system was subjected to produce MD simulations for 100 ns for ao enzyme and protein–ligand complex. The systems' dynamic behavior and structural changes were analyzed by calculating the RMSD and RMSF30.
Data availability
The datasets generated and/or analysed during the current study are available in the Worldwide Protein Data Bank (wwPDB) repository. (https://www.rcsb.org/structure/2y9x).
References
Li, J. et al. Recent advances in the design and discovery of synthetic tyrosinase inhibitors. Eur. J. Med. Chem. 224, 113744 (2021).
Carradori, S., Melfi, F., Rešetar, J., Şimşek, R. Tyrosinase enzyme and its inhibitors: An update of the literature, Metalloenzymes (2024) 533–546.
Taylor, S.C. Diagnosing Skin Disease in Skin of Color, An Issue of Dermatologic Clinics, E-Book, Elsevier Health Sciences2023.
Moon, K. M., Kwon, E.-B., Lee, B. & Kim, C. Y. Recent trends in controlling the enzymatic browning of fruit and vegetable products. Molecules 25(12), 2754 (2020).
Obaid, R. J. et al. Natural and synthetic flavonoid derivatives as new potential tyrosinase inhibitors: A systematic review. RSC Adv. 11(36), 22159–22198 (2021).
Nerya, O., Musa, R., Khatib, S., Tamir, S. & Vaya, J. Chalcones as potent tyrosinase inhibitors: the effect of hydroxyl positions and numbers. Phytochemistry 65(10), 1389–1395 (2004).
Iraji, A. et al. Synthesis, biological evaluation and molecular docking analysis of vaniline–benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem. 14(1), 1–11 (2020).
Nazir, Y. et al. Molecular docking synthesis, and tyrosinase inhibition activity of acetophenone amide: Potential inhibitor of melanogenesis. BioMed Res. Int. 2022, 1040693 (2022).
Iraji, A., Panahi, Z., Edraki, N., Khoshneviszadeh, M. & Khoshneviszadeh, M. Design, synthesis, in vitro and in silico studies of novel Schiff base derivatives of 2-hydroxy-4-methoxybenzamide as tyrosinase inhibitors. Drug Dev. Res. 82(4), 533–542 (2021).
Othman, E. M., Fayed, E. A., Husseiny, E. M. & Abulkhair, H. S. Rationale design, synthesis, cytotoxicity evaluation, and in silico mechanistic studies of novel 1,2,3-triazoles with potential anticancer activity. New J. Chem. 46(25), 12206–12216 (2022).
Othman, E. M., Fayed, E. A., Husseiny, E. M. & Abulkhair, H. S. The effect of novel synthetic semicarbazone- and thiosemicarbazone-linked 1,2,3-triazoles on the apoptotic markers, VEGFR-2, and cell cycle of myeloid leukemia. Bioorganic Chem. 127, 105968 (2022).
Othman, E. M., Fayed, E. A., Husseiny, E. M. & Abulkhair, H. S. Apoptosis induction, PARP-1 inhibition, and cell cycle analysis of leukemia cancer cells treated with novel synthetic 1,2,3-triazole-chalcone conjugates. Bioorganic Chem. 123, 105762 (2022).
Marinescu, M. Benzimidazole-triazole hybrids as antimicrobial and antiviral agents: A systematic review. Antibiotics 12(7), 1220 (2023).
Tian, G. et al. Recent advances in 1, 2, 3-and 1, 2, 4-triazole hybrids as antimicrobials and their SAR: A critical review. Eur. J. Med. Chem. 259, 115603 (2023).
Mlakić, M. et al. New naphtho/thienobenzo-triazoles with interconnected anti-inflammatory and cholinesterase inhibitory activity. Eur. J. Med. Chem. 241, 114616 (2022).
Hamoud, M. M. et al. Design and synthesis of novel 1, 3, 4-oxadiazole and 1, 2, 4-triazole derivatives as cyclooxygenase-2 inhibitors with anti-inflammatory and antioxidant activity in lps-stimulated RAW2647 macrophages. Bioorganic Chem. 124, 105808 (2022).
Mahdavi, M. et al. Synthesis of new benzimidazole-1,2,3-triazole hybrids as tyrosinase inhibitors. Chem. Biodiver. 15(7), e1800120 (2018).
Ranjbar, S. et al. 1, 2, 3-Triazole-linked 5-benzylidene (thio) barbiturates as novel tyrosinase inhibitors and free-radical scavengers. Archiv der Pharmazie 353(10), 2000058 (2020).
Hosseinpoor, H. et al. Anti-melanogenesis and anti-tyrosinase properties of aryl-substituted acetamides of phenoxy methyl triazole conjugated with thiosemicarbazide: Design, synthesis and biological evaluations. Bioorganic Chem. 114, 104979 (2021).
Azizian, H. et al. Docking study, molecular dynamic, synthesis, anti-α-glucosidase assessment, and ADMET prediction of new benzimidazole-Schiff base derivatives. Sci. Rep. 12(1), 14870 (2022).
Yousefnejad, F. et al. Design, synthesis, in vitro, and in silico evaluations of benzo[d]imidazole-amide-1,2,3-triazole-N-arylacetamide hybrids as new antidiabetic agents targeting α-glucosidase. Sci. Rep. 13(1), 12397 (2023).
Lipinski, C. A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discovery Today: Technol. 1(4), 337–341 (2004).
Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58(9), 4066–4072 (2015).
Abulkhair, H. S. et al. In vivo- and in silico-driven identification of novel synthetic quinoxalines as anticonvulsants and AMPA inhibitors. Archiv der Pharmazie 354(5), 2000449 (2021).
El-Adl, K. et al. Design, synthesis, docking, ADMET profile, and anticancer evaluations of novel thiazolidine-2,4-dione derivatives as VEGFR-2 inhibitors. Archiv der Pharmazie 354(7), 2000491 (2021).
Noori, M. et al. Thioquinoline derivatives conjugated to thiosemicarbazide as potent tyrosinase inhibitors with anti-melanogenesis properties. Sci. Rep. 13(1), 2578 (2023).
Hashemi, A. et al. Synthesis and tyrosinase inhibitory activities of novel isopropylquinazolinones. BMC Chem. 17(1), 65 (2023).
Ghasemi, N., Moradi, S., Iraji, A. & Mahdavi, M. Thiazolopyrimidine derivatives as novel class of small molecule tyrosinase inhibitor. BMC Chem. 17(1), 156 (2023).
Najafi, Z. et al. Design, synthesis, and molecular dynamics simulation studies of new chalcone-based 2-Arylidene-1,3-indandiones as Tyrosinase inhibitors. Chem. Select 8(33), e202302192 (2023).
Iraji, A. et al. Design, synthesis, spectroscopic characterization, in vitro tyrosinase inhibition, antioxidant evaluation, in silico and kinetic studies of substituted indole-carbohydrazides. Bioorganic Chem 129, 106140 (2022).
Funding
The authors wish to thank the support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (grant number: IR.SUMS.REC.1402.324).
Author information
Authors and Affiliations
Contributions
A.B., and S.M. synthesized and characterized compounds. A.I. performed biological tests and in silico study and contributed to the manuscript preparation. M.M. supervised all phases of the study. All authors read and revised the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bagheri, A., Moradi, S., Iraji, A. et al. Structure-based development of 3,5-dihydroxybenzoyl-hydrazineylidene as tyrosinase inhibitor; in vitro and in silico study. Sci Rep 14, 1540 (2024). https://doi.org/10.1038/s41598-024-52022-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52022-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
