
Abstract
Novel perioperative strategies are needed to reduce recurrence rates in patients undergoing nephrectomy for high-risk, non-metastatic clear cell renal cell carcinoma (ccRCC). We conducted a prospective, phase I trial of neoadjuvant nivolumab prior to nephrectomy in 15 evaluable patients with non-metastatic ccRCC. We leveraged tissue from that cohort to elucidate the effects of PD-1 inhibition on immune cell populations in ccRCC and correlate the evolving immune milieu with anti-PD-1 response. We found that nivolumab durably induces a pro-inflammatory state within the primary tumor, and baseline immune infiltration within the primary tumor correlates with nivolumab responsiveness. Nivolumab increases CTLA-4 expression in the primary tumor, and subsequent nephrectomy increases circulating concentrations of sPD-L1, sPD-L3 (sB7-H3), and s4-1BB. These findings form the basis to consider neoadjuvant immune checkpoint inhibition (ICI) for high-risk ccRCC while the tumor remains in situ and provide the rationale for perioperative strategies of novel ICI combinations.
Similar content being viewed by others

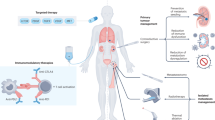
Introduction
The advent of immune checkpoint inhibitors (ICI) revolutionized the treatment paradigm across multiple immunogenic malignancies1, including clear cell renal cell carcinoma (ccRCC)2,3,4,5,6. There has been particular focus on inhibiting the PD-1/PD-L1 (programmed cell death protein 1 and its ligand) axis. Efforts have been underway to characterize mechanisms of ICI response and resistance in ccRCC, with attention to the molecular and immune milieu of the tumor and its microenvironment (TME)7,8,9,10,11,12,13,14. These studies are limited in their ability to disentangle the biological changes associated with inhibition of the PD-1/PD-L1 axis, however, and are subject to limitations from the use of archival biopsy cores, variability in organ sites of sample acquisition, heterogeneity in prior therapeutic exposures, and/or stromal contamination from the destination organ15. Given the increasing biological complexity of combination drug therapies in treating ccRCC, in which additive or potentially synergistic effects may influence their combined efficacy8,16, it is essential to first gain insight into the biology underlying response and resistance to anti-PD-1 monotherapy in order to better understand the effects of these drugs in combination.
Nephrectomy tissue following systemic exposure to therapy offers an ideal platform to prospectively overcome these limitations by encapsulating the effects of therapy on both the entire tumor and the normal adjacent renal parenchyma in situ. One-third of patients with locally advanced RCC are expected to recur following nephrectomy with curative intent17. That PD-1 inhibition may enhance the anti-tumor T cell response induced by the primary tumor while the tumor is still in situ forms the basis to consider neoadjuvant ICI for high risk RCC. As such, our group recently completed a phase I trial demonstrating the safety and feasibility of neoadjuvant nivolumab followed by nephrectomy in 17 patients with non-metastatic high-risk ccRCC18,19. As the first published prospective trial of its kind, we noted that among 15 evaluable patients, all had stable disease on nivolumab with one (6.7%) demonstrating features of an immune-related pathologic response. In earlier institutional series, nephrectomy following ICI remarkably demonstrated an unprecedented complete pathologic response (ypT0) in the primary tumor in 9–18% of cases20,21,22,23.
In our phase I trial, we generated a biobank of clinicopathologically annotated tissue and blood through prospective collection of treatment-naïve renal tumor biopsies, nephrectomy tissue post-nivolumab, and blood samples at baseline (pre-nivolumab), pre-operatively (post-nivolumab), and post-operatively18. Leveraging this unique resource, we herein sought to (1) elucidate the effects of PD-1 inhibition on primary tumor-infiltrating and circulating immune cell populations in ccRCC and (2) correlate TME and circulating immune cell compositions with response to anti-PD-1 therapy.
Results
Nivolumab promotes an inflammatory state within the primary tumor
We first sought to elucidate changes in primary tumor-infiltrating lymphocytes (TIL) in situ induced by neoadjuvant nivolumab. We thus performed fluorescence-activated cell sorting (FACS) analysis on both naive ccRCC tissue and nivolumab-exposed nephrectomy specimens. Using the transcription factors Tbet and eomesodermin (eomes; a paralogue of Tbet) we show that the TME of nivolumab (nivo) treated patients trend towards more effector (Tbet+Eomes+; untreated mean 17.79%, nivo treated mean 27.50%) and have fewer early effector (Tbet+Eomes-; untreated mean 20.86%, nivo treated mean 5.88%) CD8+ T cells. We did not see a difference in the exhausted (Tbet-Eomes+; untreated mean 41.66%, nivo treated mean 46.56%) CD8+ T cells after nivolumab treatment (Fig. 1A,B). An identical analysis was performed on intratumoral CD4+ T cells, but the effects were not found to be as pronounced as in the CD8+ T cell compartment (Supplemental Fig. 1A,B).

Nivolumab enhances effector transcription factor phenotype in CD8 + T cells. (A) Representative fluorescent activated cell sorting (FACS) plots of Tbet and eomes in CD8+ T cells isolated from treatment naïve or nivolumab treated ccRCC tissue specimens obtained from patients undergoing nephrectomy. (B) CD8+ T cells from nivolumab treated ccRCC specimens show decreased Tbet+Eomes- status, while trending towards Tbet+Eomes+ status. (C) Representative FACS plots of Tbet and eomes in CD8+ T cells isolated from peripheral blood mononuclear cell (PBMC) specimens obtained in the treatment naïve or nivolumab-treated setting. (D) CD8+ T cells from the PBMC compartment of nivolumab treated ccRCC patients show decreased Tbet+Eomes– status, while trending towards Tbet+Eomes+ status. (*p < 0.05; ns not significant).
We investigated peripheral blood mononuclear cell (PBMC) CD8+ T cells to determine if the nivolumab-driven effect was specific to the TME or extended to the circulatory compartment as well given the intravenous route of administration. We observed similarities, with nivolumab-treated circulating CD8+ T cells trending towards enhanced effector (untreated mean 21.60%, nivo treated mean 37.55%) and decreased early effector (untreated mean 23.33%, nivo treated mean 8.61%) status. In alignment with the tumor-infiltrating CD8+ T cell data, we saw no difference between the “exhausted” (untreated mean 16.00%, nivo treated mean 19.32%) CD8+ T cells after nivolumab treatment (Fig. 1C,D). An identical analysis was performed on circulating CD4+ T cells, but the effects were not found to be as pronounced as in the CD8+ T cell counterpart (Supplemental Fig. 1C,D).
While not statistically significant, we did note an influx of both CD8+ T cells and CD4+ effector T cells into the TME but did not note a change in the CD4+ T regulatory (Treg) infiltration after nivolumab treatment (Fig. 2A). Consistent with this finding, we calculated the CD8+ T cell-to-Treg ratios for naïve and nivolumab-treated TME and show that nivolumab treatment resulted in an increased ratio (naïve 3.7, nivo 14.2) (Fig. 2B). Given this stark increase in CD8+ T cell-to-Treg ratio, we further explored how nivolumab treatment affected the intratumoral T cell compartment composition. We identified that in naïve TME, both CD8+ T cells and CD4+ effector T cells were present in approximately equal proportions, with Tregs comprising the smallest component; in nivolumab-treated TME, CD8+ T cells were overrepresented relative to both CD4+ effector T cells and Tregs (Fig. 2C,D). The proportion of CD8+ T cells as a part of the T cell compartment was larger in nivolumab-treated vs naïve TME, and importantly, the Treg component comprised a smaller proportion of the T cell compartment in nivolumab-treated vs naïve (Fig. 2E) indicating a reduction in immunosuppressive TIL populations24. It is possible that the change in the CD8+ T cell compartment may be due to either a local expansion of existing intratumoral CD8+ T cells or new infiltration of circulating CD8+ T cells.

Nivolumab increases intratumoral representation of CD8+ T cells. (A) Total CD8+ T cell and CD4 effector cell amounts trend upwards with nivolumab treatment. (B) The ratio of CD8+ T cells to regulatory T cells (Tregs) increases with nivolumab treatment (C) Graphical representation of the T cell compartment from individual naïve or nivolumab treated tumor specimens. (D) Naïve ccRCC specimens show approximately equal ratio of CD8+ and CD4 effector T cells intratumorally, while nivolumab treated ccRCC specimens show enhanced CD8+ T cell representation. (E) Nivolumab treated ccRCC shows higher CD8+ T cell representation and decreased Treg representation relative to naïve ccRCC specimens. (*p < 0.05, **p < 0.01, ns not significant).
Our observations are in alignment with those seen previously in advanced ccRCC, which suggest that ICI remodels the TME through tissue-resident CD8+ T cell expansion14, thereby modifying the interplay between cancer and immune cell populations13. We also corroborate prior preclinical findings from animal models of other malignancies demonstrating an increase in tumor-infiltrating CD8+ T cells following PD-1 blockade25,26. Using murine models of spontaneously metastatic breast cancers, Liu et al. found that neoadjuvant anti-PD-1 therapy resulted in a significant increase in tumor-specific CD8+ T cells, and they noted that tumor-specific CD8+ T cells from neoadjuvantly treated models with regulatory T cell depletion displayed an effector/memory phenotype26. The CD8+ T cell expansion with neoadjuvant therapy was not observed to the same magnitude with adjuvant therapy, supporting the notion that PD-1 inhibition may enhance and rely on the antitumor T cell response by the primary tumor while it is still in situ.
Despite the promising improvement in intratumoral CD8+ T cell effector status, total number, and representation within the T cell compartment, nivolumab-treated CD8+ T cells remained refractory to ex vivo restimulation with αCD3/αCD28 activation beads as evidenced by unchanged production of IFNγ, IL-2, TNFα, and granzyme B (Supplementary Fig. 2A,B). Hence, although the number of T cells increased after receipt of three doses of nivolumab, it remains speculative whether antigent-specific T cell functional activation has been induced. Nevertheless, these cells still produce granzyme B, indicating that they are not completely non-functional and may still be rescuable with additional interventions. This insensitivity to restimulation was reflected by intratumoral CD4+ effector T cells as well (Supplementary Fig. 2C,D). It should be noted that this restimulation approach was utilized to mimic physiologically relevant signaling conditions by engaging receptors that provide Signal 1 and Signal 2 to T cells. Another common approach is to use phorbol 12-myristate 13-acetate (PMA) and ionomycin to provide potent but non-physiologically relevant signaling, which may have been able to restimulate these cells to produce effector cytokines.
As our cumulative FACS analysis indicates that nivolumab drives a partial restoration of an effector T cell status in the TME, we sought to further characterize the TME by performing RNA sequencing (RNAseq) on patient-matched treatment-naïve tumor biopsies and nivolumab-treated nephrectomy specimen. Within our cohort, we found that the most significantly enriched pathway in the nephrectomy specimen compared to the pre-treatment biopsies was MYC targets, variant 1 (false discovery rate (FDR) ≤ 0.05), while no pathways were significantly downregulated (Fig. 3; full gene list in Supplementary Table 1). MYC expression has been seen previously to regulate the immune TME in other malignancies, including hematologic malignancies, liver cancer, non-small cell lung cancer, and neuroblastoma, among others, through modulation of both immune effector cells and immune regulatory cytokines27,28,29,30,31. In particular, MYC has been shown to induce transcription of CD47 and PD-L1, and thus MYC activation may prime tumors to improved response to ICI. MYC pathway activation has also been shown to be essential in the pathogenesis of ccRCC32 and may present an opportunity for targeted therapeutic synergy. Additionally, while other pathways did not reach an FDR ≤ 0.05, we did see enhancement of TNF signaling via NFkB, programmed cell death and general inflammatory signaling pathways after nivolumab treatment when compared to baseline biopsy samples. These pathways are indicative of a broader immunostimulatory effect on the TME after ICI.

Volcano plots and bar charts showing pathways (A) upregulated and (B) downregulated in primary tumor following nivolumab treatment. Significance was achieved for upregulation of MYC targets, variant 1 (FDR ≤ 0.05).
Nivolumab remains durably bound to TIL in the primary tumor
ICI are unique compared to other therapeutic approaches in their utilization of stable and highly specific antibodies. One of the significant benefits to this approach is its durability in vivo well after infusion; however, few studies have provided direct evidence of how long these antibodies can remain effectively bound to their target in the TME of treated patients. Our data show that nivolumab remained bound to intratumoral CD8+, CD4+ effector T cells and Tregs > 7 days after the final infusion, as evidenced by reduced detection of PD-1 on TILs at the time of nephrectomy compared to naïve ccRCC specimens (Fig. 4). This is due to PD-1 epitope blockade, as nivolumab was still bound to PD-1 receptors at the time and inhibited anti-PD-1 FACS antibody binding. This effect was detected on circulating PBMCs as well, but due to their reduced basal expression of PD-1, it was less pronounced and did not reach statistical significance (Supplemental Fig. 3). In the phase II ADAPTeR study, in which 15 patients with metastatic ccRCC were treated with nivolumab, with tissue samples obtained from various sites, Au et al. noted that patients who respond to nivolumab not only exhibit expansion of nivolumab-bound CD8+ T cells, but also importantly demonstrate maintenance of tumor-specific T cell expansion12. Thus, the ongoing antigen-driven stimulation of pre-existing T cells over time is likely a key element in nivolumab’s action and response.

Nivolumab is durably bound to intratumoral T cells 7 days post final infusion. (A) Representative FACS flow plots showing PD-1 expression on intratumoral T cells from either naïve ccRCC or nivolumab treated samples. (B) Tumor infiltrating T cells from naïve ccRCC specimens display high PD-1 signal, while tumor infiltrating T cells from nivolumab treated ccRCC specimens display reduced PD-1 signal, indicating that nivolumab-driven epitope blockade prevented successful anti-PD1 FACS staining. Gating was set using isotype control antibodies for Tbet and Eomes, respectively. (*p < 0.05; **p < 0.01; ***p < 0.001, ns not significant).
This observation is further supported by preclinical studies of breast cancer by Liu et al., who showed that administration of neoadjuvant nivolumab resulted in persistent expansion of CD8+ T cells even 170 days following infusion, which was not seen to the same magnitude following adjuvant administration of nivolumab26. Furthermore, the maintenance of tumor-specific CD8+ T cell expansion was predictive of better survival outcomes compared to those with lower levels of tumor-specific CD8+ T cells, suggesting that the durable increase in tumor-specific T cells systemically in response to nivolumab therapy may rely on the presence of the primary tumor during treatment.
Immune infiltration at baseline predicts response to nivolumab
We next sought to evaluate features in pre-treatment biopsy samples from primary tumors that predict response to PD-1 inhibition. As all 15 evaluable patients in our cohort exhibited stable disease per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and immune-related response criteria (irRC)18, we used a lower radiographic threshold to stratify our cohort into responders (decrease in longest diameter by 5% or more, n = 3) and non-responders. One of those responders also exhibited features of an immune-related pathologic response within the nephrectomy specimen, which was characterized by a regression bed exhibiting histologic features of wound healing, including neovascularization and fibrosis; immune infiltration and cholesterol clefts secondary to tumor cell clearance; and plasma cells and foamy macrophages18.
By whole transcriptomic expression, we found 23 genes to be significantly upregulated (p < 0.01, log2FC > 1) and 114 genes significantly downregulated (p < 0.01, log2FC < – 1) in responders compared to non-responders (Supplementary Fig. 4A,B; full gene list in Supplementary Table 2). Expression for the selected ccRCC driver genes BAP1, PBRM1, and SETD2 did not differ by response in our cohort (Supplementary Fig. 4C). The association of BAP1 and PBRM1 mutations with ICI response in ccRCC have been previously reported11,33,34, though these observations have been largely inconsistent5,9,12,35,36.
We transformed the RNAseq data using the virtual inference of protein activity by enriched regulon analysis (VIPER) algorithm to infer protein activity, which has been shown to reduce noise inherent to RNAseq data and amplify the biological signal from key master regulatory proteins controlling cellular phenotypes37,38. VIPER activity yielded upregulation of 49 proteins and downregulation of 50 proteins in responders compared to non-responders (Fig. 5A,B; full gene list in Supplementary Table 3), with no differences again seen in known ccRCC driver genes (Fig. 5C).

Heatmap of all significantly (A) upregulated and (B) downregulated genes in pre-treatment biopsy samples by response to nivolumab following transformation by VIPER. (C) Gene expression for driver genes BAP1, PBRM1, and SETD2 did not differ by response to nivolumab. (D) Nivolumab responders exhibited enrichment in T-effector and myeloid inflammatory expression and depletion of Angiogenesis expression per IMmotion150 gene signatures. (E) Gene set enrichment analysis revealed enrichment in the CD8+ inflamed subtype signature and depletion of the VEGF immune desert subtype signature in nivolumab responders.
We then asked whether nivolumab responders and non-responders exhibit differential expression of inflammatory and angiogenic gene signatures that have been postulated to hold both prognostic and predictive value7,9. In applying the IMmotion150 gene signatures9 to the pre-treatment biopsy samples, we found that responders were significantly enriched with the T-effector (Teff) and myeloid inflammatory signatures, while they displayed significantly lower expression of the angiogenesis signature (Fig. 5D, Supplementary Fig. 4D). Strikingly, the Teff signature was very highly enriched in the patient who exhibited an immune-related pathologic response. We then applied the four distinct immune-based ccRCC TME cell signatures described by Clark et al. to assess the differential degree of immune infiltration between responders and non-responders7. Concordant with our observations using the IMmotion150 signatures, we found that responders to nivolumab were significantly enriched with the CD8+ inflamed subtype signature in their primary tumors, whereas the VEGF immune desert subtype signature was significantly depleted (Fig. 5E, Supplementary Fig. 4E). Notably, CD8+ inflamed tumors are characterized by high CD8+ T cell infiltration, increased expression of PD-1, PD-L1, PD-L2, and CTLA-4, and higher frequency of chromosome 14 loss and BAP1 mutations7, which have been associated with worse prognosis in ccRCC, but hold implications for response to ICI therapy39,40.
Although Braun et al. did not observe immune infiltrated tumors to differ in their response to or survival following PD-1 blockade compared to immune desert and immune excluded tumors11, our findings are largely in alignment with findings from the ADAPTeR study12. In particular, Au et al. noted that while the exome did not correlate with nivolumab response in their cohort, responders exhibited a higher number of T cell receptor (TCR) clones pre-treatment, suggesting the reliance of anti-PD-1 response on pre-existing immunity, with maintenance of T cell immune activation and upregulation of TCR signaling compared to non-responders. They also applied the IMmotion150 signatures to their cohort and reported an enrichment of the Teffhigh but not the Teffhigh/Myeloidlow among responders, similar to our findings herein within the primary tumor.
These findings are also consistent with those seen in pre-clinical models of breast cancer treated with neoadjuvant anti-PD-1 therapy, in which survival of mice treated with neoadjuvant ICI was greatest among those with the highest level of tumor-specific CD8 + T cells, suggesting that the antitumor efficacy of neoadjuvant ICI may be heavily reliant on the ability to both increase and maintain elevated levels of CD8 + T cells within the primary tumor26.
We noted that myeloid inflammation was also enriched among responders, which was also seen in the ADAPTeR study12. As myeloid inflammation associated with high expression of IL-6, prostaglandins, and the CXCL8 family of chemokines has been implicated in accumulation of myeloid-derived suppressor cells (MDSCs) in tumors and suppression of antitumor immunity41,42,43,44,45,46, we initially hypothesized that higher levels of these circulating cytokines would correlate with decreased clinical response. This would be concordant with the speculation by Krishna et al. that high infiltration of tumor-associated macrophages (TAMs) may limit the efficacy of anti-tumor T cell responses in ICI resistance14. However, we found that responsiveness to nivolumab did not correlate with the concentrations of the majority of circulating factors tested, including IL-6, IL-8/CXCL8, sPD-L3 (sB7-H3), and sPD-L1, either at baseline or following therapy (Supplementary Fig. 5A,B). The exception to this was soluble 4-1BB (s4-1BB), which was found to be more highly expressed in the responders at baseline (Fig. 6F). In earlier work, our group interrogated the immunosuppressive role of infiltrating myeloid-derived cells and PBMCs in treatment-naïve non-metastatic ccRCC and found that TAM populations are not homogeneously immunosuppressive and express distinct transcriptional profiles compared to circulating monocytes that do not strictly fit into the traditional M1-M2 TAM paradigm47,48. As such, we then postulated that pro-inflammatory phenotypes mediated by TAMs may facilitate nivolumab responsiveness, which was supported recently by Bi et al., who noted that TAMs from ICI-treated biopsy specimens shift toward pro-inflammatory states, while also upregulating immunosuppressive markers13.

Neoadjuvant nivolumab followed by nephrectomy primes perioperative synergy for rational ICI combination strategies. (A) Representative FACS plots showing checkpoint receptor expression on intratumoral CD8+ T cells from either naïve ccRCC or nivolumab treated specimen. (B) Nivolumab treatment leads to increased CTLA-4 expression on intratumoral CD8+ T cells and stable expression of LAG-3 and TIM-3. (C) Representative FACS plots showing checkpoint receptor expression on intratumoral Tregs from either naïve ccRCC or nivolumab treated specimen. (D) Nivolumab treatment leads to increased CTLA-4 expression on intratumoral Tregs, stable expression of LAG-3 and a trending increase in TIM-3 expression. (E) Circulating levels of sPD-L1, sPD-L3 (sB7-H3), and s4-1BB were significantly increased 6-months following nephrectomy after treatment with neoadjuvant nivolumab compared to baseline. (F) s4-1BB was more highly expressed in responders (R) than non-responders (NR) at baseline. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns not significant).
Less equivocally, Braun et al. showed that in advancing stages of ccRCC, immune dysfunction appears to be progressive, with enrichment in terminally exhausted CD8 + T cells and a shift in TAMs towards an anti-inflammatory M2-like state with a decrease in inflammatory cytokine production49. Using VIPER in a non-metastatic, treatment-naïve surgical cohort of ccRCC patients, our group also recently uncovered a novel TAM subpopulation undetectable by gene expression analysis alone that is characterized by upregulation of TREM2/APOE/C1Q37. This TAM subpopulation appears to carry prognostic significance, as it was associated with a higher risk of recurrence. Taken together, these findings surrounding the heterogeneous roles for TAMs in both treatment-naïve and ICI-treated cohorts reflect the complex interplay among myeloid inflammation in the TME, ccRCC aggressiveness, and therapeutic responsiveness.
Nivolumab primes perioperative synergy for rational ICI combination strategies
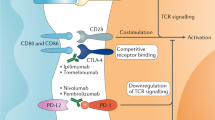
While nivolumab monotherapy gained approval in the second-line setting for metastatic ccRCC based on the CheckMate 025 trial2, currently approved frontline treatments rely on combination strategies of either dual ICI or ICI with anti-angiogenic therapy based on the higher response rates seen with these approaches. The CheckMate 214 trial led to the first approved ICI regimen in the frontline setting for metastatic ccRCC by combining nivolumab with the CTLA-4 inhibitor ipilimumab4. The rationale for this combination derives from complementary mechanisms of CTLA-4 and PD-1 in regulating adaptive immunity, as PD-1 contributes to T cell exhaustion in peripheral tissues, while CTLA-4 inhibits earlier points in T cell activation50,51.
Thus, we investigated the effect of nivolumab on the expression of other immune checkpoints in the primary tumor, as this information could better inform perioperative combination therapeutic strategies to improve response in the primary tumor. Although we did not see an increase in LAG-3 or TIM-3 expression with nivolumab, we found a striking increase in CTLA-4 expression on tumor-infiltrating CD8+ T cells (Fig. 6A–D). This highlights not just an additive effect of combining ipilimumab with nivolumab, but rather an important synergy between PD-1 inhibition and CTLA-4 inhibition, which may conceivably benefit response within the primary tumor in a perioperative setting compared to nivolumab monotherapy alone.
Next, we asked whether removal of the primary tumor antigen following nivolumab “priming” could influence circulating cytokine levels to rationally direct a combination neoadjuvant/adjuvant strategy. Remarkably, compared to baseline, we found that levels of circulating sPD-L1, sPD-L3 (sB7-H3), and s4-1BB were significantly increased 6 months following nephrectomy (Fig. 6E), while these levels were not significantly impacted following nivolumab treatment alone prior to nephrectomy. These observations suggest that targeting these upregulated cytokines post-nephrectomy following neoadjuvant administration of nivolumab may improve systemic response and theoretically reduce recurrence in these high-risk populations.
Currently, only two adjuvant therapies are approved for high-risk, non-metastatic ccRCC including sunitinib and, more recently, the PD-1 inhibitor pembrolizumab based on improvements in disease-free survival; however, adjuvant sunitinib failed to show an overall survival benefit52, while the overall survival data for adjuvant pembrolizumab currently remains immature53. The phase III, randomized, controlled PROSPER RCC (ClinicalTrials.gov identifier NCT03055013) trial notably entails continuation of neoadjuvant nivolumab in the post-nephrectomy setting for an additional 9 months following surgery54. While speculative, our findings herein support the shift to a different ICI agent adjuvantly for a more precisely informed approach, such as one targeting PD-L1 (e.g. avelumab, atezolizumab, durvalumab), B7-H3 (e.g. enoblituzumab, omburtamab), or the 4-1BB/4-1BBL complex (e.g. utomilumab, urelumab). Among these candidate targets, avelumab is the only approved agent in metastatic ccRCC (in combination with axitinib)5, while inhibitors of B7-H3 and agonists of 4-1BB/4-1BBL remain largely unexplored in RCC. Bi et al. also explored different subpopulations of CD8 + T cells in RCC and postulated a predictive role of 4-1BB expression on CD8 + T cells in the differential response to anti-PD-1 therapy13.
Discussion
We report on the correlative results from a phase I trial investigating neoadjuvant nivolumab for high-risk, non-metastatic ccRCC18. In particular, we elucidated the isolated effects of PD-1 inhibition on primary TIL and circulating immune cell populations in ccRCC, and we correlated the evolving immune milieu with anti-PD-1 response. Compared to prior studies, we uniquely leveraged nephrectomy tissue exposed to systemic PD-1 inhibition while the tumor and kidney remained in situ in the non-metastatic setting. We compared direct changes in the immune microenvironment as a result of nivolumab treatment to matched treatment-naïve tumor tissue along with changes in PBMCs and circulating cytokines at multiple timepoints throughout treatment.
We found that PD-1 inhibition durably induces a pro-inflammatory state within the primary tumor in situ, as evidenced by a sustained increase in the effector T cell phenotype and decreased representation of immunosuppressive regulatory T cell subsets both in TILs and in PBMCs following exposure to nivolumab. Furthermore, baseline immune infiltration within the primary tumor, as evidenced by enriched Teff and myeloid inflammatory signatures along with angiogenic depletion in pretreatment biopsy specimen, appears to correlate with response to nivolumab. These observations are largely concordant with other recent studies linking functional CD8 + T cells and pro-inflammatory TAMs with ICI response in ccRCC13,14,49,55. The influence of myeloid inflammation in modulating tumor aggressiveness and therapeutic response remains more complex, however, as we continue to gain insight into the heterogeneous roles that TAMs play in ccRCC13,37,47,48,49.
In light of these observations, at first it may seem paradoxical that ccRCC tumors with a high infiltration by CD8 + T cells are classically associated with worse prognosis yet respond better to ICI, while those that are more angiogenic and less inclined to respond to ICI have tended to exhibit more favorable outcomes. It may be true that the inherent biological aggressiveness and natural history of these tumors are dictated by the balance of inflammation and angiogenesis8. However, prognostic outcomes have been largely dictated by response to targeted therapies in the pre-ICI era, many of which target angiogenic pathways. Likewise, validated prognostic risk models for metastatic RCC that were derived in the pre-ICI era may hold little relevance in predicting survival in patients treated with ICI. As such, we are now witnessing unprecedented improvements in survival outcomes for patients with inherently inflamed tumors, including those exhibiting sarcomatoid or rhabdoid differentiation, and updated prognostic models applicable to the ICI era are needed56.
We did not encounter any pathologic complete responses in the primary tumor after anti-PD-1 monotherapy. It is conceivable that three doses of nivolumab may be insufficient to induce adequate antitumor immunity. Nevertheless, we observed that nivolumab does appear to “prime” the immune microenvironment for rational synergistic combination approaches to enhance response. For example, CTLA-4 expression increased in tumors after nivolumab, supporting the rationale for combining anti-CTLA-4 agents with anti-PD-1 agents in the neoadjuvant setting—the first dual ICI combination approved for the frontline treatment of metastatic ccRCC. Findings from retrospective series that have shown ypT0 following combination ICI, including ipilimumab with nivolumab, add further support to this notion20,21. Following nephrectomy, there appears to be an increase in other soluble immune checkpoints in circulation, including sPD-L1, sPD-L3 (sB7-H3), and s4-1BB, supporting a role for synergistic combination strategies of neoadjuvant and adjuvant ICI. Future studies of novel adjuvant ICI following neoadjuvant PD-1 inhibition will be needed to test this speculation.
Within our cohort, these circulating cytokines increased only after nephrectomy, but not following nivolumab treatment alone, highlighting a potential systemic response from removing the primary tumor antigen. This may hold relevance in the metastatic setting, for which the optimal role and timing of cytoreductive nephrectomy continue to evolve in the contemporary ICI era57. A recent study demonstrated that upfront cytoreduction may yield systemic benefits in patients with metastatic RCC58, while other series have also shown oncologic benefits of combining cytoreductive nephrectomy with ICI23,59. Ongoing and future trials combining cytoreductive nephrectomy with ICI will shed further light on the oncologic role of surgery in these settings.
Our results are limited by the small sample size with no patients exhibiting a pathologic complete response or radiographic response by RECIST v1.1 criteria. Notably, the lack of ypT0 was also seen in the only other published prospective trial of neoadjuvant nivolumab for high-risk, non-metastatic ccRCC60. Likewise the phase III PROSPER RCC trial, which was powered to assess recurrence-free survival with perioperative (neoadjuvant and adjuvant) nivolumab for patients with high-risk, non-metastatic RCC compared to surgery alone, did not achieve its primary outcome. Taken together, these results support the role of combination, rather than monotherapy, approaches of perioperative ICI and/or a longer duration of preoperative therapy. Curiously, in patients with resectable stage III or IV melanoma, the administration of 3 cycles pembrolizumab neoadjuvantly followed by 15 cycles pembrolizumab adjuvantly yielded a significantly longer event-free survival compared to 18 cycles adjuvant pembrolizumab alone61. While differential biology between RCC and melanoma, differences in activity between nivolumab and pembrolizumab, and differences in therapeutic duration may explain these discrepant observations, the addition of neoadjuvant PD-1 inhibition resulted in improved outcomes for patients with melanoma, supporting continued investigation of neoadjuvant ICI in RCC.
Furthermore, tumor biopsies obtained prior to nivolumab administration are subject to under-sampling and limited tissue quantity and quality; instead, multi-regional sampling would better capture the intra-tumoral heterogeneity of geographically distinct tumor regions. Although our phase I, single-arm trial is not powered for oncologic outcomes and lacks a comparator group, our study is strengthened by its prospective nature of enrollment, standardized approach to treatment, and acquisition of tissue and blood at pre-defined timepoints with matched pre- and post-nivolumab treated tissue available for comparison. Despite the limited responses to nivolumab seen in the primary tumor, our study provides important hypothesis-generating insights into the biology underlying response and resistance to anti-PD-1 monotherapy, which is crucial to better understand the synergistic effects of combination drug therapies in treating ccRCC. Future correlative studies from the PROSPER RCC trial will be helpful to overcome some of these limitations.
In summary, we leverage a phase I trial to elucidate the effects of PD-1 inhibition on immune cell populations in ccRCC and correlate the evolving immune milieu with anti-PD-1 response. Using a cohort of patients who received neoadjuvant nivolumab prior to nephrectomy, we found that nivolumab induces a durable inflammatory state within the primary tumor, and patients with more highly inflamed tumors at baseline exhibit better responses to nivolumab. We note that nivolumab modulates immune changes within the primary tumor, and subsequent nephrectomy alters circulating inflammatory cytokine concentrations. These findings form the basis to consider neoadjuvant ICI for high-risk ccRCC while the tumor is still in situ and provide the rationale for perioperative (neoadjuvant and adjuvant) strategies of novel ICI combinations.
Methods
Patients
We utilized tissue from all evaluable patients enrolled in our recent prospective, open-label, single-arm phase I trial designed to assess the primary endpoint of safety and tolerability of neoadjuvant nivolumab in patients with non-metastatic high-risk RCC (ClinicalTrials.gov identifier NCT02575222, first registration 13/10/2015)18. All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by our institutional review board under protocol IRB00068726 (J15179). Informed consent was obtained from all subjects for participation in this study. Complete methodological details of the trial protocol, including complete inclusion/exclusion criteria and clinicopathologic characteristics of the cohort, are available in the original publication18. Briefly, patients with biopsy-confirmed, non-metastatic, high-risk ccRCC (T2a-T4NanyM0 or TanyN1M0) planned to undergo radical or partial nephrectomy were enrolled between February 2016 and June 2018. A run-in phase of 5 patients was followed by continuous safety monitoring with stopping rules until 15 patients were enrolled with evaluable data for the primary and secondary endpoints of the trial. Out of 23 patients assessed for eligibility, 17 enrolled in the study, of whom 15 patients constituted the final evaluable cohort. Patients received neoadjuvant nivolumab (3 mg/kg) on day 1 of each of three consecutive 14-day cycles of therapy for a total of three doses. Dosing calculations were based on the body weight assessed during the previous cycle, with screening body weight used for dosing of cycle 1. Within 7 days of completion of cycle 3, patients underwent a partial or radical nephrectomy at the discretion of the treating surgeon. All 15 patients exhibited stable disease per the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and immune-related response criteria (irRC). Notably, one patient exhibited a 15.7% decrease in the long-axis tumor diameter with corresponding features of an immune-related pathologic response within the nephrectomy specimen.
Sample acquisition
Six image-guided percutaneous biopsy cores of primary tumors were acquired prior to starting nivolumab for each patient. One core was sent for standard-of-care histological analysis for clinical diagnosis, while the other 5 cores were retained for research, including 2 cores that were placed in RNAlater for downstream RNA extraction and 3 cores that were paraffin-embedded.
Following treatment with nivolumab, 3 non-necrotic pieces of tumor from the nephrectomy specimen, each measuring 1 × 1 × 0.5 cm, were harvested for research use, including one piece that was paraffin-embedded, one freshly processed for lymphocyte isolation, and one placed in RNAlater for downstream RNA extraction.
Venous blood for biomarker analysis was collected using standard phlebotomy techniques. Up to 200 mL of whole blood was obtained at the following timepoints: (1) baseline (prior to initiation of nivolumab therapy), (2) pre-operatively on the day of surgery (following completion of neoadjuvant nivolumab therapy per protocol), (3) 1 month post-operatively, (4) 3 months post-operatively, (5) 6 months post-operatively, and (6) 12 months post-operatively. Of the 200 mL collected, 10 mL was collected in a serum-separating tube, and up to 190 mL was collected in heparinized syringes or tubes. Blood samples were processed within 2 h after acquisition to optimize results.
Whole blood sample preparation
Whole blood was collected in BD Vacutainer Glass Blood Collection Tubes with Sodium Heparin (BD Biosciences, Cat # 366480) or in 60 mL syringes (BD Biosciences, Cat #309654) with Heparin Sodium (1000 [USP’U]/mL) to prevent clotting. The whole blood was then transferred to a 50 mL conical tube (BD Biosciences, Cat # 352097) and diluted with Hanks Balanced Salt Solution 1X without calcium, Magnesium and phenol red (Corning, Cat # 21-022-cv), and underplayed with 10 mL of Ficoll-Paque PLUS density gradient media (GE Healthcare Life Sciences, Cat # 17144003) using glass pasture pipettes (Thermo Fisher Scientific, Cat # 13-678-6B). Samples then underwent centrifugation at 2000 RPM (845 RCF) for 20 min and allowed to gently come to a stop without the application of the braking system as to maintain the density gradient layer. Peripheral blood mononuclear cells (PBMCs) were then isolated using sterile fine tip transfer pipettes (Thermo Fisher Scientific, Cat # 232-1S) and washed using 30 mL of phosphate buffered saline (PBS) (Quality Biological, Cat # 114-058-101). PBMCs were then centrifuged at 1300 RPM (357 RCF) for 10 min and allowed to come to a stop with the application of the break. These cells were then resuspended in 10 mL of complete RPMI (RPMI 1640, 10% FBS, 1% MEM non-essential amino acids, 1% sodium pyruvate, 1% antibiotic) (Corning, Cat # 10-040-CV; Gemini Bio-Products, Cat # 100-106; Thermo Fisher Scientific, Cat # 11140076; Sigma-Aldrich, Cat # S8636; Quality Biological, Cat # 120-095-721) and counted on a hemocytometer.
Tumor tissue preparation for fluorescence-activated cell sorting (FACS)
Non-necrotic tumor tissue which was transported from the grossing suit to the laboratory in 1X PBS at room temperature (RT). The sample was removed from the 1X PBS and mechanically separated into 2-4 mm cubes before being placed into a c-tube (Miltenyi Biotec, Cat # 130-093-237) containing 4.7 mL of RPMI 1640 (Corning, Cat # 10-040-CV) and three digestive enzymes (enzyme A, enzyme D, enzyme I). The tissue sample then underwent enzymatic dissociation in accordance with the Human Tissue Dissociation Kit (Miltenyi Biotec, Cat # 130-095-929), using the gentleMACS Octo Dissociator (Miltenyi Biotec, Cat # 130-096-427) to provide appropriate mechanical dissociation and application of heating cycles for enzymatic dissociation. The resulting single cell suspension was then filtered using a 100-micron filter and washed using 1 × PBS before being counted on a hemocytometer.
Positive selection
The single cell suspensions of both PBMC and whole tumor were enriched for T-cells through use of Dynabeads CD4 Positive Isolation Kit (Life Technologies, Cat # 11331D) and Dynabeads CD8 Positive Isolation Kit (Life Technologies, Cat # 11333D). These kits were used in parallel to isolate t-cells as opposed to a CD3 + T-cell positive isolation kit to prevent accidental activation of these cells through engagement of the CD3 receptor. Cells were prepared in accordance with the kit protocol provided.
Ex vivo re-stimulation and FACS cytokine staining
A fraction of enriched PBMCs and enriched tumor-infiltrating lymphocytes (TIL) were plated in a 96 well U-bottom plate and stimulated with Dynabeads Human T-Activator CD3/CD28 for T cell Expansion and Activation (Thermo Fisher Scientific, Cat #11132D) at a ratio of 1 bead:1 cell for 12 h in cMedia in the presence of protein transport inhibitor cocktail 500X (Thermo Fisher Scientific, Cat #00-4980-03). The samples were washed with 1X PBS, and underwent staining with Fixable Viability Dye (eFluor 780 Thermo Fisher Scientific, Cat # 65-0865-14), followed by fluorophore conjugated antibodies specific for human CD4 (BV570 Biolegend, Cat 300534), CD8 (BV650 Biolegend, Cat # 301041), PD-1 (PE-Cy7 Biolegend, Cat # 329918) and CD3 (AF700 BD Biosciences, Cat # 557943). The samples then underwent fixation and permeabilization (eBioscience, Cat # 88-8824-00), after which they stained by fluorophore conjugated antibodies specific for human FoxP3 (Pac Blue BD Biosciences, Cat # 560460), IFNγ (FITC BD Biosciences, Cat # 554700), IL-2 (PE Thermo Fisher Scientific, Cat # 562462) and TNFα (APC Biolegend, Cat # 502913). Manufacturer specified isotype control antibodies were used to determine positive signal and gating strategies. Data were analyzed using FlowJo Software (Tree Star, Mac version 9.9.4).
FACS staining for checkpoint molecules
A fraction of enriched PBMCs and enriched TIL were plated in a 96 well U-bottom plate. The samples were washed with 1X PBS and underwent staining with Fixable Viability Dye (eFluor 780 Thermo Fisher Scientific, Cat # 65-0865-14), followed by fluorophore conjugated antibodies specific for human CD45RO (BV570 Biolegend, Cat # 304226), CD8 (BV650 Biolegend, Cat # 301041), LAG-3 (FITC BMS), TIM-3 (PE Biolegend, Cat # 345005), CD4 (PE-Texas Red BD Biosciences, Cat # 562316), CD45RA (PerCP-Cy5.5 Biolegend, Cat # 304122), PD-1 (PE-Cy7 BD Biosciences, Cat # 561272) and CD3 (AF700 BD Biosciences, Cat # 557943). The samples then underwent fixation and permeabilization (eBioscience, Cat # 88-8824-00), after which they stained by fluorophore conjugated antibodies specific for human FoxP3 (Pac Blue BD Biosciences, Cat # 560460) and CTLA-4 (APC BD Biosciences, Cat # 560938). Manufacturer specified isotype control antibodies were used to determine positive signal and gating strategies. Data were analyzed using FlowJo Software (Tree Star, Mac version 9.9.4).
FACS staining for transcription factors
A fraction of enriched PBMCs and enriched TIL were plated in a 96 well U-bottom plate. The samples were washed with 1X PBS, and underwent staining with fixable viability dye (eFluor 780 Thermo Fisher Scientific, Cat # 65-0865-14), followed by fluorophore conjugated antibodies specific for human CD45RO (BV570 Biolegend, Cat # 304226), CD8 (BV650 Biolegend, Cat # 301041), CD4 (Pacific Blue Thermo Fisher Scientific, Cat # MHCD0428), CD45RA (PerCP-Cy5.5 Biolegend, Cat # 304122), PD-1 (PE-Cy7 Biolegend, Cat # 329918) and CD3 (AF700 BD Biosciences, Cat # 557943). The samples then underwent fixation and permeabilization (eBioscience, Cat # 88-8824-00), after which they stained by fluorophore conjugated antibodies specific for human, Ki-67 (FITC BD Bioscience, Cat # 561165), T-Bet (PE Thermo Fisher Scientific, Cat # 12-5825-82) and Eomesodermin (APC Thermo Fisher Scientific, Cat # 50-4877-42). Manufacturer specified isotype control antibodies were used to determine positive signal and gating strategies. Data were analyzed using FlowJo Software (Tree Star, Mac version 9.9.4).
Enzyme-linked immunosorbent assay (ELISA) for circulating cytokines
Custom Luminex ELISA was performed as per manufacturer’s instructions (R&D Systems, a Bio-Techne brand) on serum samples obtained at various timepoints as previously described62. The analytes included in these studies were soluble PD-L3 (B7-H3), soluble PD-L1, soluble Galectin-3, soluble 4-1BB, interferon-γ, interleukin-2 (IL-2), Il-1b, IL-5, IL-7, IL-13, IL-17, IL-36b, IL-33, IL-6, IL-8, IL-15, IL-23, and TNF-a.
RNA extraction from tumor tissue
Formalin-fixed, paraffin-embedded (FFPE) slides were freshly cut and stored at – 20 °C until nucleic acid extraction was performed. Samples were processed in accordance with the AllPrep DNA/RNA FFPE kit (Qiagen, Cat# 80234). Briefly, samples undergo a quick lysis, followed by a cooling step and centrifugation to release and collect RNA. Additional steps are taken to reverse crosslinking and facilitate binding to RNA purification columns. An on-column DNase step was performed on the RNA purification column, to remove any contaminating DNA. After wash steps, RNA was eluted from the columns. Nucleic acid yield and quality were first assessed using a NanoDrop ND-1000 spectrophotometer and later by quantifying the abundance of ribosomal RNA fractions with Experion (Bio-Rad) and/or Agilent 2100 Bioanalyzer.
RNA sequencing and analysis
RNAseq libraries were prepared using the TruSeq RNA Exome kit (Illumina). The libraries were multiplexed three per lane and sequenced on the NovaSeq6000 platform to obtain, on average 150 bp per sample. CASAVA 1.8.4 (Illumina) was used to convert BCL files to FASTQ files using default parameters. trimgalore v0.6.3 was used to trim the reads. rsem-1.3.0 was used for running the alignments as well as generating gene and transcript expression levels. The “rsem-calculate-expression” module was used, and the RNAseq data was aligned to human reference genome hg38 and normalized to transcripts per million (TPM).
Clinical annotations for each sample included identification of patients as responders or non-responders to therapy as well as identification of pre-treatment biopsy versus post-treatment nephrectomy specimens. Notably, since all patients on the phase I trial exhibited stable disease by radiographic criteria, responders were defined by a 5% or greater shrinkage in the longest diameter of the primary tumor radiographically from baseline following therapy, while all others were designated non-responders. Differential gene expression between paired biopsy and nephrectomy specimens collected from the same patient was assessed by paired Wilcox test, with p-values corrected for multiple hypothesis testing with the Benjamini–Hochberg method. Data tables were generated of raw and corrected p-value, log2-fold-change, and mean expression for each gene in nephrectomy and biopsy specimens. Genes were considered significantly differentially expressed at corrected p-value less than 0.05. A volcano plot was further generated showing log2-fold-change against -log10(corrected p-value). Differential expression was similarly assessed comparing pre-treatment biopsy specimens in therapy responder versus non-responder patients, with statistical significance computed by unpaired Wilcox test, with Benjamini–Hochberg p-value correction. In addition to the differential expression data table and volcano plot, heatmaps were generated showing the genes most strongly upregulated in responders (p-value < 0.01, log2-fold-change > 1) and the genes most strongly downregulated in responders (p-value < 0.01, log2-fold-change < -1). Furthermore, heatmaps were also generated showing expression of genes with prognostic significance in ccRCC (BAP1, PBRM1, SETD2), genes involved in angiogenesis (VEGFA, KDR, ESM1, PECAM1, ANGPTL4, CD34), genes associated with effector T-cell response (CD8A, EOMES, PRF1, IFNG, CD274), and genes associated with myeloid inflammation (IL-6, CXCL1, CXCL2, CXCL3, CXCL8, PTGS2)9.
Protein activity inference and differential activity
Protein activity was inferred from the entire dataset of RNAseq specimen by the virtual inference of protein activity by enriched regulon analysis (VIPER) algorithm using a previously published gene regulatory network inferred on kidney cancer data from The Cancer Genome Atlas (TCGA) to infer activity of upstream signaling and transcriptional regulatory proteins from the expression of their downstream targets (algorithm for the reconstruction of accurate cellular networks, ARACNe)63,64. This has been demonstrated to significantly reduce noise inherent to RNA-sequencing data and amplify the biological signal from key master regulatory proteins controlling cellular phenotypes37,38. Gene expression input to VIPER consisted of TPM gene expression matrices scaled to gene by gene z-scores. Differential protein activity scores were then computed by paired Wilcox test for biopsy versus nephrectomy specimens, and by unpaired Wilcox test for pre-treatment responder versus non-responder patients, with multiple hypothesis test correction as described above in the differential gene expression analysis. Violin plots and heatmaps were similarly generated for inferred protein activity.
Evaluation of ccRCC subtype signature enrichment
Enrichment of gene sets associated with specific ccRCC proteogenomic subtypes as described by Clark et al.7 was assessed by gene set enrichment analysis (GSEA), with genes ranked by log-fold-change in gene expression or VIPER-inferred protein activity comparing responders vs. non-responders. Normalized enrichment scores and p-values for each gene set were determined by 1000 stochastic permutations of the gene labels. GSEA plots were generated for each gene set for both differential expression and differential protein activity, corresponding to the following ccRCC subtypes: CD8- inflamed, CD8 + inflamed, Metabolic Immune Desert, and VEGF Immune Desert.
Statistical analysis
All quantitative and statistical analyses were performed using the R computational environment and packages described above. One-way ANOVA was used to compare cytokine levels between patients and timepoints. A Gaussian distribution was assumed, and a Sidak correction for multiple comparisons was performed. For samples that were missing cytokine data (i.e. sample from a given timepoint was below the limit of detection), a mixed-effects analysis was performed; a Gaussian distribution was assumed, and a Tukey correction for multiple comparisons was performed. As all patients exhibited stable disease by radiographic criteria, associations with clinical response were analyzed according to radiographic change in the longest diameter, with responders designated as those exhibiting a decrease in diameter by 5% or more (n = 3), and non-responders as all others. Non-responders who exhibited an increase in the longest diameter by 5% or more radiographically were additionally designated as poor responders (n = 2). Statistical comparisons of gene signature enrichment scores were performed by non-parametric Wilcox test with Benjamini–Hochberg multiple testing correction where appropriate. In all cases, statistical significance was defined as an adjusted p-value less than 0.05.
Data availability
Sequencing data for consenting patients has been deposited in GEO under accession ID GSE245372 and has been made publicly available.
References
Ribas, A. & Wolchok, J. D. Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355. https://doi.org/10.1126/science.aar4060 (2018).
Motzer, R. J. et al. Nivolumab versus Everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1803–1813. https://doi.org/10.1056/NEJMoa1510665 (2015).
Motzer, R. J. et al. NCCN guidelines insights: Kidney cancer, version 2.2020. J. Natl. Compr. Cancer Netw. 17, 1278–1285. https://doi.org/10.6004/jnccn.2019.0054 (2019).
Motzer, R. J. et al. Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 378, 1277–1290. https://doi.org/10.1056/NEJMoa1712126 (2018).
Motzer, R. J. et al. Avelumab plus Axitinib versus Sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1103–1115. https://doi.org/10.1056/NEJMoa1816047 (2019).
Rini, B. I. et al. Pembrolizumab plus Axitinib versus Sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1116–1127. https://doi.org/10.1056/NEJMoa1816714 (2019).
Clark, D. J. et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 179, 964-983e931. https://doi.org/10.1016/j.cell.2019.10.007 (2019).
Singla, N. Progress toward precision medicine in frontline treatment of metastatic renal cell carcinoma. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2019.4716 (2019).
McDermott, D. F. et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 24, 749–757. https://doi.org/10.1038/s41591-018-0053-3 (2018).
Singla, N. Re: Brian I. Rini, Thomas Powles, Michael B. Atkins, et al. Atezolizumab plus Bevacizumab Versus Sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019;393:2404-15: Putting IMmotion into motion: Personalizing frontline treatment for metastatic renal cell carcinoma. Eur. Urol. 77, e168–e169. https://doi.org/10.1016/j.eururo.2019.09.015 (2020).
Braun, D. A. et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 26, 909–918. https://doi.org/10.1038/s41591-020-0839-y (2020).
Au, L. et al. Determinants of anti-PD-1 response and resistance in clear cell renal cell carcinoma. Cancer Cell 39, 1497-1518e1411. https://doi.org/10.1016/j.ccell.2021.10.001 (2021).
Bi, K. et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649-661e645. https://doi.org/10.1016/j.ccell.2021.02.015 (2021).
Krishna, C. et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 39, 662-677e666. https://doi.org/10.1016/j.ccell.2021.03.007 (2021).
Singla, N. et al. Pancreatic tropism of metastatic renal cell carcinoma. JCI Insight https://doi.org/10.1172/jci.insight.134564 (2020).
Palmer, A. C. & Sorger, P. K. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 171, 1678-1691e1613. https://doi.org/10.1016/j.cell.2017.11.009 (2017).
Chin, A. I., Lam, J. S., Figlin, R. A. & Belldegrun, A. S. Surveillance strategies for renal cell carcinoma patients following nephrectomy. Rev. Urol. 8, 1–7 (2006).
Gorin, M. A. et al. Neoadjuvant nivolumab in patients with high-risk nonmetastatic renal cell carcinoma. Eur. Urol. Oncol. https://doi.org/10.1016/j.euo.2021.04.002 (2021).
Singla, N. & Margulis, V. Locally advanced kidney cancer: A new space for immunotherapy?. Eur. Urol. Oncol. https://doi.org/10.1016/j.euo.2021.05.003 (2021).
Singla, N. et al. Pathologic response and surgical outcomes in patients undergoing nephrectomy following receipt of immune checkpoint inhibitors for renal cell carcinoma. Urol. Oncol. 37, 924–931. https://doi.org/10.1016/j.urolonc.2019.08.012 (2019).
Pignot, G. et al. Nephrectomy after complete response to immune checkpoint inhibitors for metastatic renal cell carcinoma: A new surgical challenge?. Eur. Urol. 77, 761–763. https://doi.org/10.1016/j.eururo.2019.12.018 (2020).
Singla, N. & Margulis, V. Re: Geraldine Pignot, Antoine Thiery-Vuillemin, Jochen Walz, et al. Nephrectomy after complete response to immune checkpoint inhibitors for metastatic renal cell carcinoma: A new surgical challenge? Eur Urol. In press. https://doi.org/10.1016/j.eururo.2019.12.018: The next surgical frontier in kidney cancer: Nephrectomy after immune checkpoint inhibition. Eur. Urol. 78, e79–e80. https://doi.org/10.1016/j.eururo.2020.04.025 (2020).
Singla, N. et al. Improved survival after cytoreductive nephrectomy for metastatic renal cell carcinoma in the contemporary immunotherapy era: An analysis of the National Cancer Database. Urol. Oncol. 38(604), e609-604e617. https://doi.org/10.1016/j.urolonc.2020.02.029 (2020).
Teng, M. W., Ritchie, D. S., Neeson, P. & Smyth, M. J. Biology and clinical observations of regulatory T cells in cancer immunology. Curr. Top. Microbiol. Immunol. 344, 61–95. https://doi.org/10.1007/82_2010_50 (2011).
Woo, S. R. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927. https://doi.org/10.1158/0008-5472.CAN-11-1620 (2012).
Liu, J. et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 6, 1382–1399. https://doi.org/10.1158/2159-8290.CD-16-0577 (2016).
Kortlever, R. M. et al. Myc cooperates with ras by programming inflammation and immune suppression. Cell 171, 1301-1315e1314. https://doi.org/10.1016/j.cell.2017.11.013 (2017).
Casey, S. C., Baylot, V. & Felsher, D. W. The MYC oncogene is a global regulator of the immune response. Blood 131, 2007–2015. https://doi.org/10.1182/blood-2017-11-742577 (2018).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. https://doi.org/10.1126/science.aac9935 (2016).
Kim, E. Y., Kim, A., Kim, S. K. & Chang, Y. S. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 110, 63–67. https://doi.org/10.1016/j.lungcan.2017.06.006 (2017).
Melaiu, O. et al. PD-L1 is a therapeutic target of the bromodomain inhibitor JQ1 and combined with HLA class I, a promising prognostic biomarker in neuroblastoma. Clin. Cancer Res. 23, 4462–4472. https://doi.org/10.1158/1078-0432.CCR-16-2601 (2017).
Tang, S. W. et al. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 273, 35–43. https://doi.org/10.1016/j.canlet.2008.07.038 (2009).
Braun, D. A. et al. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. https://doi.org/10.1001/jamaoncol.2019.3158 (2019).
Miao, D. et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. https://doi.org/10.1126/science.aan5951 (2018).
Motzer, R. J. et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell 38, 803-817e804. https://doi.org/10.1016/j.ccell.2020.10.011 (2020).
Abou Alaiwi, S. et al. Mammalian SWI/SNF complex genomic alterations and immune checkpoint blockade in solid tumors. Cancer Immunol. Res. 8, 1075–1084. https://doi.org/10.1158/2326-6066.CIR-19-0866 (2020).
Obradovic, A. et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 184, 2988-3005e2916. https://doi.org/10.1016/j.cell.2021.04.038 (2021).
Obradovic, A. et al. PISCES: A pipeline for the systematic, protein activity-based analysis of single cell RNA sequencing data. bioRxiv https://doi.org/10.1101/2021.05.20.445002 (2022).
Chen, L. et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 8, 1156–1175. https://doi.org/10.1158/2159-8290.CD-17-1033 (2018).
Sade-Feldman, M. et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998-1013e1020. https://doi.org/10.1016/j.cell.2018.10.038 (2018).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867. https://doi.org/10.1038/nature01322 (2002).
Liu, Q. et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 31, 61–71. https://doi.org/10.1016/j.cytogfr.2016.08.002 (2016).
Najjar, Y. G. et al. Myeloid-derived suppressor cell subset accumulation in renal cell carcinoma parenchyma is associated with intratumoral expression of IL1beta, IL8, CXCL5, and Mip-1alpha. Clin. Cancer Res. 23, 2346–2355. https://doi.org/10.1158/1078-0432.CCR-15-1823 (2017).
Prima, V., Kaliberova, L. N., Kaliberov, S., Curiel, D. T. & Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. U.S.A. 114, 1117–1122. https://doi.org/10.1073/pnas.1612920114 (2017).
Sumida, K. et al. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur. J. Immunol. 42, 2060–2072. https://doi.org/10.1002/eji.201142335 (2012).
Yuan, M. et al. Tumor-derived CXCL1 promotes lung cancer growth via recruitment of tumor-associated neutrophils. J. Immunol. Res. 2016, 6530410. https://doi.org/10.1155/2016/6530410 (2016).
Nirschl, T. R. et al. Transcriptional profiling of tumor associated macrophages in human renal cell carcinoma reveals significant heterogeneity and opportunity for immunomodulation. Am. J. Clin. Exp. Urol. 8, 48–58 (2020).
Theodros, D. et al. High-dimensional cytometry (ExCYT) and mass spectrometry of myeloid infiltrate in clinically localized clear cell renal cell carcinoma identifies novel potential myeloid targets for immunotherapy. Mol. Cell. Proteom. https://doi.org/10.1074/mcp.RA120.002049 (2020).
Braun, D. A. et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 39, 632-648e638. https://doi.org/10.1016/j.ccell.2021.02.013 (2021).
Curran, M. A., Montalvo, W., Yagita, H. & Allison, J. P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U.S.A. 107, 4275–4280. https://doi.org/10.1073/pnas.0915174107 (2010).
Parry, R. V. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553. https://doi.org/10.1128/MCB.25.21.9543-9553.2005 (2005).
Ravaud, A. et al. Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. N. Engl. J. Med. 375, 2246–2254. https://doi.org/10.1056/NEJMoa1611406 (2016).
Choueiri, T. K. et al. Adjuvant pembrolizumab after nephrectomy in renal-cell carcinoma. N. Engl. J. Med. 385, 683–694. https://doi.org/10.1056/NEJMoa2106391 (2021).
Harshman, L. C. et al. Transforming the perioperative treatment paradigm in non-metastatic RCC-A possible path forward. Kidney Cancer 1, 31–40. https://doi.org/10.3233/KCA-170010 (2017).
Koh, M. Y., Sayegh, N. & Agarwal, N. Seeing the forest for the trees-single-cell atlases link CD8(+) T cells and macrophages to disease progression and treatment response in kidney cancer. Cancer Cell 39, 594–596. https://doi.org/10.1016/j.ccell.2021.03.008 (2021).
Singla, N. A new therapeutic era for metastatic renal cell carcinoma: Call for a new prognostic model. JAMA Oncol. 6, 633–634. https://doi.org/10.1001/jamaoncol.2019.6862 (2020).
Singla, N., Ghandour, R. A. & Margulis, V. Is cytoreductive nephrectomy relevant in the immunotherapy era?. Curr. Opin. Urol. 29, 526–530. https://doi.org/10.1097/MOU.0000000000000659 (2019).
Silagy, A. W. et al. Evolving biological associations of upfront cytoreductive nephrectomy in metastatic renal cell carcinoma. Cancer 127, 3946–3956. https://doi.org/10.1002/cncr.33790 (2021).
Pignot, G. et al. Oncological outcomes of delayed nephrectomy after optimal response to immune checkpoint inhibitors for metastatic renal cell carcinoma. Eur. Urol. Oncol. https://doi.org/10.1016/j.euo.2022.07.002 (2022).
Carlo, M. I. et al. Phase II study of neoadjuvant nivolumab in patients with locally advanced clear cell renal cell carcinoma undergoing nephrectomy. Eur. Urol. 81, 570–573. https://doi.org/10.1016/j.eururo.2022.01.043 (2022).
Patel, S. P. et al. Neoadjuvant-adjuvant or adjuvant-only pembrolizumab in advanced melanoma. N. Engl. J. Med. 388, 813–823. https://doi.org/10.1056/NEJMoa2211437 (2023).
Shenderov, E. et al. Nivolumab plus ipilimumab, with or without enzalutamide, in AR-V7-expressing metastatic castration-resistant prostate cancer: A phase-2 nonrandomized clinical trial. Prostate 81, 326–338. https://doi.org/10.1002/pros.24110 (2021).
Alvarez, M. J. et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 48, 838–847. https://doi.org/10.1038/ng.3593 (2016).
Margolin, A. A. et al. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. https://doi.org/10.1186/1471-2105-7-S1-S7 (2006).
Funding
This study was supported by funding from Bristol-Myers Squibb. No funder/sponsor had a role in the collection, management, analysis, or interpretation of the data. The preparation, review, approval, and decision to submit the manuscript for publication was made solely by the study’s authors.
Author information
Authors and Affiliations
Contributions
N.S. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: N.S., C.G.D., H.J.H., B.J.T., M.E.A. Acquisition of data: N.S., T.R.N., A.Z.O., E.S., K.L., X.L., A.P., M.A.G., S.P.R., H.J.H., P.M.P., T.J.B., J.S.D., T.L.L., J.M.T., C.G.D., M.E.A. Analysis and interpretation of data: N.S., T.R.N., A.Z.O., E.S., J.C.Z., S.P.R., J.S.D., J.M.T., T.L.L., Y.M.A.G., M.A.G., C.G.D., M.E.A. Drafting of the manuscript: N.S., T.R.N. Critical revision of the manuscript for important intellectual content: all authors Statistical analysis: T.R.N., B.J.T., A.Z.O. Obtaining funding: M.E.A., C.G.D. Administrative, technical, or material support: K.L., X.L., A.P., J.C.Z., T.R.N. Supervision: N.S., M.E.A., C.G.D. Other: None.
Corresponding author
Ethics declarations
Competing interests
Financial disclosures: Nirmish Singla certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (e.g. employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Steven P. Rowe: research funding and consulting agreement with Progenics Pharmaceuticals, Inc., Precision Molecular, Inc., PlenaryAI, Inc.; stock options in D&D Pharmatech. Hans J. Hammers: paid consultant/advisor for ARMO Biosciences, Inc., Bayer AG, Bristol-Myers Squibb, Corvus Pharmaceuticals, Exelixis, Inc., Eli Lilly and Company, Merck & Co., Inc., Pfizer, Inc., Novartis International AG, and Surface Oncology, Inc.; research/grant support from Bristol-Myers Squibb and Merck & Co., Inc. Tamara L. Lotan: research funding from DeepBio, AIRA Matrix and Exact Sciences. Janis M. Taube: paid consultant/advisor for Akoya Biosciences, Inc., AstraZeneca, plc., Bristol-Myers Squibb, Compugen, Ltd., and Merck & Co., Inc.; equipment loan, reagent provision, and stock options with Akoya Biosciences, Inc. Yasser M. A. Ged: advisory board consultation fees from Bristol Meyer Squib, Exelixis, BostonGene, and Aveo. Michael A. Gorin: consultant for RSIP Vision, Corbin Clinical Resources (Perineologic), Blue Earth Diagnostics Limited, KOELIS, Inc., Point Biopharma, Merck, Ambu A/S; speakers bureau and consult for Lantheus Holdings, Inc.; patent licenses for Precision Molecular. Charles G. Drake: employment at Janssen Pharmaceuticals, Inc.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singla, N., Nirschl, T.R., Obradovic, A.Z. et al. Immunomodulatory response to neoadjuvant nivolumab in non-metastatic clear cell renal cell carcinoma. Sci Rep 14, 1458 (2024). https://doi.org/10.1038/s41598-024-51889-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51889-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
