
Abstract
Chagas disease is a leading cause of non-ischemic cardiomyopathy in endemic regions of Central and South America. In Belize, Triatoma dimidiata sensu lato has been identified as the predominate taxon but vectorial transmission of Chagas disease is considered to be rare in the country. We recently identified an acute case of vector-borne Chagas disease in the northern region of Belize. Here we present a subsequent investigation of triatomines collected around the case-patient’s home. We identified yet undescribed species, closely related to Triatoma huehuetenanguensis vector by molecular systematics methods occurring in the peridomestic environment. The identification of a T. cruzi-positive, novel species of Triatoma in Belize indicates an increased risk of transmission to humans in the region and warrants expanded surveillance and further investigation.
Similar content being viewed by others

Introduction
Chagas disease, caused by the parasite Trypanosoma cruzi, is a leading cause of non-ischemic cardiomyopathy in endemic regions of Central and South America1. The parasite is predominately transmitted by vectors belonging to the insect subfamily Triatominae (Hemiptera: Reduviidae), commonly referred to as kissing bugs or conenose bugs. The Triatominae subfamily is comprised of over 150 different described species within 18 genera2,3. While all species within the Triatominae subfamily are thought to be potential vectors of the parasite, the genus Triatoma is one of the most epidemiologically relevant and diverse2.
In most of Central America, the currently predominate vector taxon of human health importance is Triatoma dimidiata sensu lato (Latreille, 1811)4. This is a large geographically diverse species complex, with closely related yet distinct species naturally occurring from southern Mexico to northern Peru. The diversity in morphology and ecology throughout the complex’s distribution has led to disagreement over the taxonomy of the group. Most notably, Lent and Wygodzinsky (1979) studied 160 T. dimidiata s.l. specimens, concluding that the observed clinal variation was not suggestive of more than one species5. More recently, studies have observed significant genetic diversity in the T. dimidiata s.l. complex, but the extent and importance of this diversity is still debated4,6,7,8.
In the past four years, molecular and morphological taxonomy made it possible to describe two new species, closely related to T. dimidiata s.l.9,10. Specifically, in Belize Triatoma mopan was identified as a cave-dwelling species in 201810. In Guatemala, Triatoma huehuetenanguensis was identified from a collection of Triatoma in the department of Huehuetenango9. In Yucatan, Mexico, at least two taxa of the T. dimidiata complex, including what is now referred to as T. huehuetenanguensis, are present in sympatry and frequently hybridize11. Additionally, the recent assignment of a neotype for T. dimidiata, defining morphologic and molecular characteristics of T. dimidiata sensu stricto, was a fundamental start for the understanding of the diversity under the name8.
In Belize, T. dimidiata s.l. has been identified as the predominant taxon, including T. huehuetenanguensis (formerly published as clade III) and hybrids within the complex12,13,14,15,16. Based on studies conducted in the country in 2009, domestic infestation was confirmed with prevalent human blood meals in collected vectors14. Nevertheless, in 2012, PAHO declared the country free of domestic vector-borne T. cruzi transmission, based on lack of detection of human cases during a country-wide seroprevalence study that included >1300 children17. In 2018 we began screening patients (adult and pediatric) presenting with acute febrile illness to polyclinics and hospitals across the country for vector-borne diseases, including Chagas disease. Through this effort, we identified an acute case of Chagas disease in a 7-year-old child residing in Sarteneja, Belize18. After identifying this case-patient, we conducted vector collection around the case-patient’s home. Here, we describe the molecular identity of the vector species collected and their T. cruzi infection status.
Results
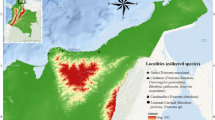
The case-patient’s home was located on the periphery of Sarteneja town and was immediately adjacent to a wooded area (Fig. 1). The structure was comprised of cinderblock and tin siding. Additionally, the dwelling was surrounded by wood piles, debris, and collective animal housing. The construction style of the home, adjacent collective animal housing, and proximity to sylvatic environments were recognizable as ideal peridomestic infestation sites for Triatominae species (Fig. 2).

Map of triatomine collection location. Triatomine collections were conducted at the location of the black dot on the map. The map was created using ArcGIS pro V3.0.

Photographs of case-patient home. Photographs of case-patient home and surroundings were taken during routine triatomine collection. Photograph (A) shows the collective animal housing approximately five meters from the home. Photograph (B) shows the construction materials used for the home.
No Triatoma were collected as a result of active collection efforts by the Baylor College of Medicine or Belize Ministry of Health and Wellness teams. However, the family of the case-patient identified five Triatoma specimens in and around their house between June 2020 and October 2022 and submitted them for analysis. Initial morphologic examination of the submitted Triatoma specimens using the Lent and Wygodzinsky key identified all five as T. dimidiata, though differences in size and coloration were noted5 (Fig. 3).

Photographs of collected triatomine samples. Photographs of each specimen collected were taken using a stereoscope with camera adapter. KBBZ001 and KBBZ003-005 are most closely related to T. huehuetenanguensis and KBBZ002 is most closely related to T. dimidiata s.l.
The phylogenetic reconstructions for each marker showed sample KBBZ002 to be closely related to T. dimidiata s.l., while the other four were closely related to T. huehuetenanguensis (Fig. 4A, B). While samples KBBZ001 and KBBZ003-005 were found to be closely related to T. huehuetenanguensis, comparison of the K2P genetic distances between the sequences showed a divergence consistent with interspecific relationship (CytB: mean = 2.57 Std = 0.15; ITS-2: mean = 0.29 Std = 0.15; Table 1), as previously noted8. Comparison between KBBZ001 and KBB003-005 sequences were consistent with intraspecific relationships (CytB: mean = 0.91 Std = 0.24; ITS-2: mean = 0.24 Std = 0.15; Table 1)8.


Maximum-likelihood phylogeny of (A) CytB and (B) ITS-2 sequences. Numbers on clades represent bootstrap support higher than 50. Samples from triatomine collected in this study are in red.
Mitogenomes were assembled for all collected specimens (Supplemental Fig. 1). All assemblies had the canonical 13 protein coding genes, 22 tRNAs, and 2 rRNAs. Only the KBBZ002 mitogenome was predicted to be circular; however, based on the depth of coverage of the sequence data, all five assemblies were likely incomplete. The read coverage data indicated a substantial increase in read depth within the control region, with unresolved repetitive elements.
Parasite testing data were available for three of the five Triatoma samples. Two Triatoma specimen closely related to T. huehuetenanguensis (KBBZ004-005) did not have the abdomen processed for parasite testing in order to preserve the sample for future morphologic characterization. Our testing identified only one Triatoma specimen, belonging to the undescribed group closely related to T. huehuetenanguensis (KBBZ003), to be infected with T. cruzi.
The genotyping of parasite DNA from KBBZ003 resulted in the detection of T. cruzi TcIV discrete typing unit (DTU). After mapping using all the DTUs as reference, a total of 89,746 sequences of good quality mapped only to the TcIV DTU. The phylogenetic analysis indicated four unique TcIV min-exon sequence haplotypes, which were most closely related to the TcIV T. cruzi sequences identified from T. dimidiata vectors previously sequenced from southern Belize13 as well as the acute Chagas disease pediatric case-patient18 represented in this study. Yet they were distinct from the T. cruzi TcIV reference, CanIII, from Brazil (Fig. 5). Reference strains used in the phylogenetic analysis are detailed in Supplemental Table 1.

Phylogenetic analysis of TcIV Trypanosoma cruzi mini-exon sequences. Mini-exon sequences obtained from the triatomine KBBZ003 (in red) were compared with sequences from reference T. cruzi strains (asterisk) and TcIV strains from other triatomine insect vectors, mammals, and the pediatric case of acute Chagas disease (plus sign) using maximum likelihood. Bootstrap support is indicated for the main nodes of the tree for clarity. Percentage values indicated within sequence names refer to the relative proportion of each haplotype identified within the triatomine.
Discussion
In this paper, we show the importance of accurately identifying Triatoma vectors through molecular techniques, particularly those collected in domestic and peridomestic environments with known vector-human transmission of the parasite T. cruzi. From the five Triatoma specimens collected (three tested), one was found positive for T. cruzi, with the parasite representing a molecular match with the case-patient. Through our investigation, we found this Triatoma vector belongs to a taxon that has not yet been formally described; suggesting that simply stating that T. dimidiata s.l. or T. huehuetenanguensis is the vector is insufficient for the region. Clear observable morphological differences to T. huehuetenanguensis, coupled with the high dissimilarity between the sequences provides evidence that this is a new species, which our group is now characterizing for formal description.
Identification and characterization of novel species is critical for understanding the risk for Chagas disease transmission in the region. Genetic variance in distinct species can signal differences in vector ecology and their ability to transmit disease to humans. Even within the T. dimidiata species complex, there have been varying vector ecology, transmission dynamics, and host preferences identified. This newly identified species represents a further divergence from the species complex and represents an undefined risk of disease transmission and ecologic preferences. Such noted diversity may suggest a benefit to further study of triatomine vector species in Central America.
Historically in Belize, the described T. dimidiata s.l. and T. huehuetenanguensis (formerly published as clade III T. dimidiata s.l.) were thought to be predominately sylvatic in nature, but able to seasonally infest houses, and hybrids have also been identified12,13,15. While our findings from this small sampling are not robust enough to draw significant conclusions about vector ecology of this potential new species, we made some observations of note. This vector was found in or around a home, was found positive for the parasite, and was also associated with a confirmed case of acute transmission of disease.
The analysis of parasite sequences from this Triatoma specimen indicates the presence of the DTU TcIV, which was also detected in the case-patient in whose home the vectors were collected18, is closely related to the TcIV reported in North America and distinct from the TcIV from South America. Furthermore, multiple (four) DTU sequence haplotypes of the parasite were found, indicating a high diversity of the parasite circulating in this area and the novel Triatoma vector’s potential involvement in transmission.
This study has some noteworthy limitations. Our small sample size of collected triatomine from one geographic area limits our ability to understand the range, prevalence of pathogen, and their vector ecology. Further study is needed to define the risk of human transmission of Chagas disease from this novel species and its distribution in Belize.
Conclusion
This study represents the first documented identification of a new species closely related to T. huehuetenanguensis. Despite previous research in the region that described a lack of domestic infection and vector-human transmission in this area, our analysis found this novel species in a peridomestic setting which was in close proximity to the home of a case-patient diagnosed with acute Chagas disease. The identification of a T. cruzi positive, novel species of Triatoma in Belize indicates a potential increased risk of transmission to humans in the region and warrants expanded surveillance and further investigation. Local public health officials should consider suggesting preventative actions until risk can be defined.
Methods
Sample collection
The Belize Ministry of Health and Wellness vector control division and a team from Baylor College of Medicine conducted an environmental investigation for Triatoma around the case-patient’s home at multiple time points between 2020 and 2022. Active collections were conducted around the case-patient’s home three times, twice by the Baylor College of Medicine team (October 2021 and June 2022) and once by the Belize Ministry of Health and Wellness team (May 2020). During collection visits the teams educated the family about how to safely collect the vectors if observed. The triatomine samples collected were shipped to the laboratory at Baylor College of Medicine for processing. This study was reviewed and approved by the Institutional Review Board at Baylor College of Medicine (H-44070) and the Belize Ministry and Health and Wellness.
Sample processing
All samples were initially identified morphologically under a stereoscope using the Lent and Wygodzinsky 1979 keys5. Three of the kissing bugs (KBBZ001-003) had the bottom third of the abdomen removed and two (KBBZ004-005) had a leg removed using a sterile scalpel for DNA extraction. For abdominal samples, the removed portion of the abdomen was incubated overnight at 56ºC in Buffer AL and Proteinase K from the Qiagen Blood and Tissue Kit (Qiagen, Germany). For leg samples, the removed portion was frozen in liquid nitrogen and ground using a pestle. Extraction was then completed using Qiagen Blood and Tissue Kit (Qiagen, Germany) per protocol with a final elution in 90 µl. Extracted DNA from abdominal samples was used for T. cruzi testing, including DTU identification. Extracted DNA from both abdominal and leg samples was used for molecular confirmation of species.
Molecular investigation of triatomine samples
Molecular confirmation of species was conducted using Illumina sequencing and subsequent bioinformatics analyses. Specifically, extracted DNA was sent to SEQCENTER (Pittsburgh, PA, USA) for whole genome sequencing. The DNA library was prepared using the Illumina DNA library prep kit (2 × 151 bp reads) and sequenced on an Illumina NextSeq 2000. A minimum of 1Gbp was sequenced for each sample. Adapter sequences as well as sequence with phred scores lower than 20 (−q 20) were filtered using fastp (v0.20.1). Mitogenome sequences were assembled using the filtered Illumina data and the MitoZ pipeline (v3.4) using the ‘all’, ‘--assembler megahit’, ‘--clade Arthropoda’ –genetic_code 5’ options19. The annotations generated by this method were manually corrected in Geneious Prime 2022.1.1 to limit gene feature overlaps.
The ITS-2 sequences were determined by performing a separate megahit (v1.2.9) assembly using default parameters for each samples' read data. The contigs of these assemblies were mapped against ITS-2 sequences either from T. huehuetenanguensis (MG947605.1) or T. dimidiata (MT556666.1 and AM286710.1) using minimap2 (v2.22-r1101)28. The contigs that mapped to these ITS-2 sequences were then blasted against GenBank's non-redundant nucleotide database and the ITS-2 sequence was extracted from the contig based on these alignments. The ITS-2, and CytB sequences were blasted against the GenBank's nt database for identity confirmation. Subsequently, these sequences were aligned to all corresponding available ITS-2 and CytB sequences, listed on GenBank as T. dimidiata, T. huehuetenanguensis and T. mopan.
Pairwise Kimura 2-parameter genetic distances were calculated in R, using the package ape20,21. Maximum likelihood phylogenies were reconstructed for each alignment, in order to visualize the evolutionary position of our samples. For this, Geneious version Prime 2021.2.2., and the RaxMl implementation, with GTR model and 1000 bootstrap pseudoreplicates. A summarized distance matrix can be found in Table 1, while the whole matrix and phylogenies are displayed in Fig. 4A, B.
Trypanosoma cruzi infection detection
Samples were tested for the presence of T. cruzi using the TcZ1/TcZ2 primer set that targets the pathogens repetitive genomic satellite DNA (satDNA), using PCR conditions described previously22,23. The sample found positive by initial screening had the DTU identified. Specifically, parasite genotyping was performed using two PCR primer sets targeting the mini-exon gene sequence, including Souto primers24, which give PCR products of different sizes according to the DTU, as well as newly designed primers that amplify a larger fragment of 500 bp of this marker25,26. Two reference strains were used as positive controls, WB1 (TcI) and Esmeraldo (TcII), and no template DNA (molecular grade water) was used as a negative control. PCRs were performed using previously reported PCR conditions24,25. PCR products were separated on a 2% agarose gel stained with ethidium bromide and purified using a PureLink Quick PCR Purification Kit (Invitrogen, Waltham, MA, US). Amplicons were processed for Nanopore library preparation using the Rapid Barcoding Sequencing (SQK-RBK004) protocol and sequenced using the MinION sequencing platform (Oxford Nanopore Technologies, Oxford, UK) on a R9.4.1 flow cell, and basecalled using MinKNOW with high accuracy setting. Raw reads had an average phred score of 16.2, and only consensus haplotypes of mini-exon sequences representing > 1% of amplicon reads were retained.
Trypanosoma cruzi Phylogenetic analysis
Parasite genotyping read quality was assessed using the FastQC tool v0.11.9 with default parameters27. Sequence reads were then separately mapped to T. cruzi mini-exon reference sequences representing 7 DTUs using minimap2 with default settings28. Manual inspection of aligned reads was done with the Integrative Genomics Viewer (IGV) browser to ensure complete coverage mapping with DTU reference sequences. FreeBayes SNP/variant calling tool was used to distinguish sequence variants from sequence artifacts, as implemented in Geneious Prime 2022.1.1, and only sequences representing ≥ 1% of the total sample were included in further analyses. Muscle alignments were performed with all haplotypes and reference strains from all DTUs to precisely identify DTUs, and separate analyses were performed for the TcIV DTU to improve comparisons of closely related sequences. Phylogenetic trees based on maximum likelihood were constructed using the Phylogeny.fr platform29.
Data availability
The authors declare that all data supporting the findings of the study are available in this article and its Supplementary files, or from the corresponding authors. Sequencing data generated from the triatomine samples are associated with the BioProject PRJNA874442 and BioSamples SAMN30498312-SAMN30498316. The Illumina sequencing reads generated from the Triatoma in the Sequence Read Archive (SRA accession #SRR21389601-SRR21389605). The mitogenome assemblies were deposited in GenBank OP345447-OP345451.
References
Perez-Molina, J. A. & Molina, I. Chagas disease. Lancet 391(10115), 82–94 (2018).
Justi, S. A. & Galvao, C. The evolutionary origin of diversity in Chagas disease vectors. Trends Parasitol. 33(1), 42–52 (2017).
Alevi, K. C. C., de Oliveira, J., da Silva, R. D. & Galvao, C. Trends in taxonomy of Chagas disease vectors (Hemiptera, Reduviidae, Triatominae): From Linnaean to integrative taxonomy. Pathogens 10(12), 122 (2021).
Dorn, P. L., Monroy, C. & Curtis, A. Triatoma dimidiata (Latreille, 1811): A review of its diversity across its geographic range and the relationship among populations. Infect. Genet. Evol. 7(2), 343–352 (2007).
Lent, H. & Wygodzinsky, P. Revision of the Triatominae (Hemiptera, Reduviidae), and their significance as vectors of Chagas’ disease. Bull. AMNH. 163(3), 83 (1979).
Bargues, M. D. et al. Phylogeography and genetic variation of Triatoma dimidiata, the main Chagas disease vector in Central America, and its position within the genus Triatoma. PLoS Negl. Trop. Dis. 2(5), e233 (2008).
Gomez-Palacio, A., Arboleda, S., Dumonteil, E. & Townsend, P. A. Ecological niche and geographic distribution of the Chagas disease vector, Triatoma dimidiata (Reduviidae: Triatominae): Evidence for niche differentiation among cryptic species. Infect. Genet. Evol. 36, 15–22 (2015).
Justi, S. A. & Dale, C. Designation of the neotype of Triatoma dimidiata (Latreille, 1811) (Hemiptera, Reduviidae, Triatominae), with full integrated redescription including mitogenome and nuclear ITS-2 sequences. Zookeys 1076, 9–24 (2021).
Lima-Cordon, R. A. et al. Description of Triatoma huehuetenanguensis sp. n., a potential Chagas disease vector (Hemiptera, Reduviidae, Triatominae). Zookeys 820, 51–70 (2019).
Dorn, P. L. et al. Description of Triatoma mopan sp. n. from a cave in Belize (Hemiptera, Reduviidae, Triatominae). Zookeys 775, 69–95 (2018).
Herrera-Aguilar, M. et al. Identification of a large hybrid zone between sympatric sibling species of Triatoma dimidiata in the Yucatan peninsula, Mexico, and its epidemiological importance. Infect. Genet. Evol. 9(6), 1345–1351 (2009).
Petana, W. B. American trypanosomiasis in British Honduras. Ann. Trop. Med. Parasit. 65(2), 169–178 (1971).
Polonio, R., Lopez-Dominguez, J., Herrera, C. & Dumonteil, E. Molecular ecology of Triatoma dimidiata in southern Belize reveals risk for human infection and the local differentiation of Trypanosoma cruzi parasites. Int. J. Infect. Dis. 108, 320–329 (2021).
Polonio, R., Ramirez-Sierra, M. J. & Dumonteil, E. Dynamics and distribution of house infestation by Triatoma dimidiata in central and southern Belize. Vector Borne Zoonotic Dis. 9(1), 19–24 (2009).
Caranci, A. T. et al. Distribution of Triatoma dimidiata sensu lato (Reduviidae: Triatominae) and risk factors associated with household invasion in Northern Belize, Central America. J. Med. Entomol. 59(2), 764–771 (2022).
Dorn, P. L. et al. Two distinct Triatoma dimidiata (Latreille, 1811) taxa are found in sympatry in Guatemala and Mexico. PLoS Negl. Trop. Dis. 3(3), e393 (2009).
PAHO. Certification of the Interruption of Vectoral Transmission of Trypanosoma cruzi in Belize 2012. https://www3.paho.org/hq/index.php?option=com_content&view=article&id=8788:2013-certification-interruption-vectoral-transmission-trypanosoma-cruzi-belize&Itemid=40353&lang=en (2012).
Murray, K. O. et al. Diagnosis of acute Chagas disease in a Belizean child with evidence of a multiclonal Trypanosoma cruzi infection. Am. J. Trop. Med. Hyg. 107(5), 992–995 (2022).
Meng, G., Li, Y., Yang, C. & Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11), e63 (2019).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34(18), 3094–3100 (2018).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16(2), 111–120 (1980).
Paradis, E. & Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35(3), 526–528 (2019).
Moser, D. R., Kirchhoff, L. V. & Donelson, J. E. Detection of Trypanosoma cruzi by DNA amplification using the polymerase chain reaction. J. Clin. Microbiol. 27(7), 1477–1482 (1989).
Gorchakov, R. et al. Trypanosoma cruzi infection prevalence and bloodmeal analysis in triatomine vectors of Chagas disease from rural peridomestic locations in Texas, 2013–2014. J. Med. Entomol. 53(4), 911–918 (2016).
Souto, R. P., Fernandes, O., Macedo, A. M., Campbell, D. A. & Zingales, B. DNA markers define two major phylogenetic lineages of Trypanosoma cruzi. Mol. Biochem. Parasitol. 83(2), 141–152 (1996).
Majeau, A., Herrera, C. & Dumonteil, E. An improved approach to Trypanosoma cruzi molecular genotyping by next-generation sequencing of the mini-exon gene. Methods Mol. Biol. 1955, 47–60 (2019).
Dumonteil, E. et al. Interactions among Triatoma sanguisuga blood feeding sources, gut microbiota and Trypanosoma cruzi diversity in southern Louisiana. Mol. Ecol. 29(19), 3747–3761 (2020).
Babraham Bioinformatics. FastQC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Dereeper, A. et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 (2008).
Pedersen, B. S. & Quinlan, A. R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 34(5), 867–868 (2018).
Acknowledgements
This study was funded by a grant from the Centers for Disease Control and Prevention (U01GH002235). This study was conducted while SAJ held National Research Council Research Associateship at the Walter Reed Army Institute of Research; partially funded through the Armed Forces Health Surveillance Division-Global Emerging Infectious Disease Surveillance (AFHSD-GEIS) Project P0065_22_WR (to Yvonne-Marie Linton). Material contained within this publication has been reviewed by the Walter Reed Army Institute of Research and the opinions or assertions contained in this study are the private views of the authors and are not to be construed as official or as reflecting true views of the Department of the Army, or the Department of Defense. The findings and conclusions of this paper are those of the authors and do not necessarily represent the official position of the US Centers for Disease Control and Prevention (CDC).
Author information
Authors and Affiliations
Contributions
S.M.G.: Conceptualization, methodology, formal analysis, writing-original draft, visualization; A.N.: methodology, visualization, writing-original draft; A.R.K.: methodology, software, data curation, formal analysis, writing-original draft; S.A.J.: methodology, software, data curation, writing-original draft, formal analysis; R.M.: conceptualization, methodology, writing-review & edit; E.Z.-G.: conceptualization, writing-review & edit; C.H.: methodology, formal analysis, data curation, writing-review & edit; J.T.: investigation, formal analysis, data curation; R.M.: investigation, formal analysis, data curation; H.D.: investigation, formal analysis, data curation; A.M.: investigation, project administration; K.B.: Investigation, methodology; S.E.R.: conceptualization; F.M.: conceptualization, methodology; R.C.F.: conceptualization, methodology; B.L.: conceptualization, methodology; E.D.: methodology, formal analysis, data curation, writing-review & edit; Gerhaldine H.M.: conceptualization, methodology, project administration; K.O.M.: Conceptualization, methodology, formal analysis, writing-original draft, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gunter, S.M., Nelson, A., Kneubehl, A.R. et al. Novel species of Triatoma (Hemiptera: Reduviidae) identified in a case of vectorial transmission of Chagas disease in northern Belize. Sci Rep 14, 1412 (2024). https://doi.org/10.1038/s41598-023-50109-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-50109-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
