
Abstract
A ternary nanocomposite of plasticized starch (PS), reduced graphene oxide (rGO), and molybdenum disulfide (MoS2) was prepared via a solution casting process, with MoS2 concentrations ranging from 0.25 to 1.00 wt%. The structural, surface morphological, optical, and electrochemical properties of the nanocomposites were studied. FTIR analysis reveals the formation of new chemical bonds between PS, rGO, and MoS2, indicating strong interactions among them. The XRD analysis showed a reduction in the crystallinity of the nanocomposite from 40 to 21% due to the incorporation of nanofiller. FESEM micrograph showed an increment of the surface roughness due to the incorporation of rGO-MoS2 layers. UV–vis spectroscopy demonstrated a reduction of optical bandgap from 4.71 to 2.90 eV, resulting from enhanced charge transfer between the layers and defect states due to the addition of nanofillers. The incorporation of MoS2 increase the specific capacitance of the PS from 2.78 to 124.98 F g−1 at a current density of 0.10 mA g−1. The EIS analysis revealed that the nanofiller significantly reduces the charge transfer resistance from 4574 to 0 Ω, facilitating the ion transportation between the layers. The PS/rGO/MoS2 nanocomposite also exhibited excellent stability, retaining about 85% of its capacitance up to 10,000 charging-discharging cycles. These biocompatible polymer-based nanocomposites with improved electrochemical performance synthesized from an easy and economical route may offer a promising direction to fabricate a nature-friendly electrode material for energy storage applications.
Similar content being viewed by others


Introduction
The world's energy consumption is soaring due to a growing population, economic expansion, and technological advancements across industries, transport, and urban development. However, this surge has led to increased use of finite fossil fuels, emitting hazardous gasses and contributing to global warming and environmental issues1. As a cleaner alternative, renewable energy sources have gained attraction2. Yet, their intermittent nature makes them unpredictable. Energy storage solutions have emerged as vital tools for stabilizing supply and demand by capturing excess electricity at the peak production time and storing it for later use, ensuring a stable and secure electricity supply. Over the past few decades, a multitude of energy storage devices, such as lithium batteries, supercapacitors, and alkali-metal ion batteries, have been developed to effectively store and utilize these energies as required. To conserve natural integrity, the constituent materials of energy storage devices must be biocompatible and biodegradable. Various materials, including 2D materials3, nanotubes4, metal oxides5, transition metals6, polymers7, and biopolymers8, have been effectively employed for storing energy due to their diverse microstructural properties. Apart from nature conservation, however, many of these materials are expensive and require complex manufacturing processes. While starch is not only eco-friendly, it also has ample resources and is easily extractable from various plant sources, including potato, corn, cassava, and even some microorganisms9. The source materials for the starch can be obtained at a very cheap rate of less than $1/kg worldwide. The source materials' easy availability and simple extraction process, make starch a cost-effective choice for the biopolymer matrix.
However, starch films generally exhibit poor charge conductivity, low solubility in various electrolytes, low mechanical properties, and high-water affinity, which limit their commercial use in electronics10. To address these shortcomings, various approaches have been attempted, including chemical modification, blending with cellulose or starch crystals, agar, maltodextrin, xanthan, arabinoxylan, chitosan, and the addition of plasticizers (such as glycerol, sorbitol, and lignin)11,12,13. Some research has focused on the development of composites that incorporate fillers, such as starch/ZnO14, PAni-starch15, starch-rGO16,17, and others18, to enhance the specific properties. The incorporation of rGO into starch was found to improve the capacitive performance of starch, but the addition of more rGO reduces the electrochemical performance due to the agglomeration of rGO layers17. This hinders the possibility of obtaining improved electrochemical performances together with higher specific capacitances from the rGO-incorporated PS matrix. Therefore, one possible route to obtain improve the electrochemical performance of PS/rGO nanocomposite could be the incorporation of a second nanofiller into it by keeping the concentration of the rGO nanofiller low. To the best of our knowledge, no such attempts were made to enhance the electrochemical performance of PS/rGO nanocomposite by incorporating a second nanofiller. The unique 2D structure of transition metal dichalcogenide (TMDs) is a suitable candidate as the second nanofiller for its complementary layer structure and potential synergies. More so, TMDs exhibit tunable band gaps, valley polarization, and strong light-matter interactions and can enhance catalytic activity, charge transport properties, and more19,20,21.
Among the different choices of TMDs nanofillers, nanostructured MoS2 has attracted significant attention among the filler materials of nanocomposites due to its excellent properties, including a narrow band gap (1.75 eV), visible light absorption, large surface area, and unique stacked morphology with strong covalent bonds22,23. Incorporation of MoS2 with rGO may facilitate charge transport through their inter- and intra-layer interactions, leading to faster ion diffusion between the layers and electrolytes24. Consequently, the hybrid layer structure can produce high specific capacitance, high energy density, and improved charge-storage capacity25. Using MoS2 alone may provoke the shuttle effect due to the dissolution of polysulfides in the energy storage performance26. Combining rGO and MoS2 with immobilized support through PS can resolve the dissolution of polysulfides27,28. Therefore, there is a high demand of developing polymer-based ternary nanocomposites by incorporating both MoS2 and graphene nanofillers. To achieve this, a synthesis method that uses mild reaction conditions is time-efficient and requires low energy consumption, which is highly desirable.
In this work, a simple and economical approach was used to enhance the electrochemical performance of the biodegradable PS matrix. A novel ternary PS/rGO/MoS2 nanocomposite has been synthesized via a facile solution casting technique. The solution casting method provides a convenient and cost-effective approach for producing large-scale polymer-based nanocomposites suitable for industrial applications29. The distinct characteristics of the nanocomposite films were probed using Fourier-transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), contact angle (CA) measurements, and analysis of optical properties. The capacitive attributes were assessed through cyclic voltammetry and galvanostatic charging-discharging, coupled with electrochemical impedance spectroscopy (EIS) analysis utilizing an equivalent AC circuit. Enhanced specific capacitance (increased from 2.78 to 124.98 F g−1 at 0.10 mA g−1), remarkable capacitive retention (maintaining 85% capacity after 10,000 cycles), and reduced contact resistance (0 Ω) were observed in the electrochemical assessment. This environmentally friendly electrode material, sourced from nature, non-hazardous, and biodegradable, exhibits improved energy storage properties. The complete study of this biomaterial with improved electrochemical properties may be used in the field of flexible bio-friendly next-generation electronic devices.
Materials and characterization
All methods were carried out in accordance with relevant guidelines and regulations.
Materials
Natural flake graphite (98%, Loba Chemie, India), potassium permanganate (KMnO4) (99%, Merck, Germany), sodium nitrate (NaNO3) (99%, Qualikems, India), concentrated sulfuric acid (H2SO4) (98%, Merck, Germany), hydrogen peroxide (H2O2) (30%, Merck, India), glycerol (Propane-1, 2, 3-triol) (Merck, India), sodium molybdate dehydrates (Na2MoO2·2H2O) (98%, Merck, Germany), thiourea (CH4N2O) (98%, Research-Lab, India), dimethyl sulfoxide (DMSO, C2H6OS) (99.5%, Merck Germany).
Sample preparation
Extraction of starch
The fresh potato purchased from the local market was cleaned, peeled, and grated. The grated potato was mixed with DI water in a beaker to remove debris. Stirring separated the starch particles, which settled as sediment. The crumpled potato was removed before the liquid changed color. After 15 min, the white milky starch settled in the beaker. The starch was collected, washed, dried at 60 °C, and ground to obtain a fine powder. Please see the Supplementary section for the details.
Synthesis of reduced graphene oxide
Graphene oxide (GO) was prepared before reducing oxygen from it to obtain reduced graphene oxide (rGO). GO was prepared by the modified Hummer method, which involves the oxidation and exfoliation of graphite30. Then rGO was synthesized by using the hydrothermal method. In this method 5 mg of GO powder was added to 15 mL of DI water to form a 0.3 mg/mL solution, which was sonicated for 30 min. The dispersion was transferred to a Teflon-lined stainless-steel autoclave, heated at 150 °C for 6 h, and cooled naturally. The product was washed, dried at 40 °C overnight, and ground to obtain homogeneous rGO powder.
The XRD spectrum of rGO is presented in Supplementary Fig. S2a. The well-resolved broad diffraction peaks of the (0 0 2) and (0 0 1) planes were observed at 2θ of 24.8°, and 43.12°, indicating the formation of rGO. The broad peak at (0 0 2) corresponds to a few layers of rGO sheets in the particles. The peak at 2θ of 43.12° in rGO is related to the intercalation of oxygen groups or water molecules, causing an increase of interlayer spacing.
For the rGO spectrum, the intense peak located at 1542 cm−1, 1689 cm−1 can be attributed to the stretching bands C=C, C=O, respectively. The very weak peak located at 2321 cm−1 corresponds to the C–O stretching. In addition, the broad peak at 3433 cm−1 indicates the presence of –O–H bond31.
Synthesis of molybdenum disulfide
Molybdenum disulfide (MoS2) was also synthesized by hydrothermal method. First, Sodium molybdate dihydrate (Na2MoO2·2H2O) and thiourea (CH4N2O) were dissolved in 120 mL distilled water. The solution was stirred vigorously until the solution became transparent, stirring increases the reaction rate. The transparent solution was transferred to an airtight Teflon jar for high-pressure treatment in an autoclave at 200 °C for 24 h, yielding a blackish precipitate. After centrifugation with DI water and ethanol, the precipitate was dried at 60 °C and ground to obtain fine MoS2 powder.
The XRD spectrum of MoS2 is presented in Supplementary Fig. S3a. In the XRD spectrum, the diffraction peaks at 2θ values of 13.99, 33.14, 39.46, 40.8 and 58.77 are observed that correspond to the (0 0 2), (1 0 0), (1 0 3), (1 0 5) and (1 1 0) reflections of the hexagonal structure of MoS2 respectively32.
The FTIR spectrum of MoS2 is presented in Supplementary Fig. S3b. The peak at 620 cm−1 can be attributed to the Mo–S stretching vibration. The multiple peaks at 700–1150 cm−1 can be assigned to the sulfate groups, while the sharp peaks at 3440 and 1610 cm−1 can be ascribed to the O–H stretching and water bending, respectively33.
Preparation of nanocomposite
To prepare the PS/rGO/MoS2 nanocomposite, at first, an aqueous solution of rGO/MoS2 was made, followed by sonication with DMSO as a 1:3 ratio of nanofiller and DMSO to have a uniformly dispersed solution. Three different concentrations of nanofillers were used, where rGO concentration remained fixed (1%), and MoS2 concentration varied from 0.25 to 1%. To prepare the film, sonicated rGO/MoS2, 5 g starch powder, and DI water were mixed. Then, this blend was stirred. Glycerol was added into the fine mixed solution and stirred at 90 °C. When the solution was dense enough to form a film, it was poured into a petri dish for drying at 50 °C. As biofilm is easily attracted by fungus and humidity, the dried biofilm was preserved in a warm environment. Finally, five different films had been prepared with varying concentrations of MoS2. Throughout this paper, nanocomposites A, B, C, D, and E denote the Plasticized Starch (PS), PS/rGO (1%), PS/rGO/MoS2 (0.25%), PS/rGO/MoS2 (0.5%) and PS/rGO/MoS2 (1%) nanocomposite respectively.
Characterizations
After their synthesis, various characterization methods were used to analyze the physical properties of the PS/rGO/MoS2 nanocomposites. Fourier Transform Infrared Spectroscopy (FTIR) spectra of the as-prepared nanocomposite were studied in the range of 400–4000 cm−1 spectra to investigate the presence of the functional group. The FTIR analysis was conducted using a Shimadzu IRSpirit spectrophotometer in attenuated total reflection (ATR) mode. X-ray diffraction (XRD) analysis has been done to check the crystalline phase by using 3040XPert PRO, Philip system with monochromatic CuKα (λ = 1.54 Å) radiation at a scanning rate of 2°/min from 5° to 90°. The microstructure and surface morphology were analyzed by the Field Emission Scanning Electronic Microscope using JSM-7600F, JEOL FESEM machine at an accelerating voltage of 5 kV. The optical properties were studied using a SHIMADZU UV 2600 spectrometer, where BaSO4 was used for baseline correction in the wavelength range of 200–900 nm at ambient conditions. The contact angle was assessed with a contact angle meter (Apex, India) through the sessile drop technique under room temperature and controlled humidity conditions. A 10 µL water droplet was employed for the measurements on the film surface. Three separate measurements of the contact angle were taken on distinct locations of the surface for each film, and the resulting average value was adopted. The electrochemical characteristics were assessed using a CS310 Electrochemical Working Station (corrtest, China) in a three-electrode configuration. This setup comprised a glassy carbon electrode employed as the working electrode, a counter electrode consisting of a platinum foil plate (1 cm × 1 cm), and a reference electrode of Ag/AgCl. Additionally, a 0.1 M KCl solution was used as the ionic aqueous electrolyte.
To prepare the homogenous slur of electrode material, first, the surface area of the working electrode was wiped with ethanol. This exposed area is where the material will be deposited and dried at 60 °C for around 1 h. In the meantime, a tiny portion of the film (a few mm2) was cut, then measured, and took only the desired mass. After that, the selected portion of the film was heated and stirred with DI water until the film melted and became sticky. This slurry composite was then deposited on the electrode with a micropipette. This deposited electrode was dried at room temperature for a few hours to gluttonize on the surface.
Results and discussion
Chemical bonding properties
FTIR study was performed to assess the chemical changes in PS due to the addition of rGO and MoS2 nanofiller. Figure 1 illustrates the FTIR spectra of all the nanocomposite ranges from 500 to 4000 cm−1. For curve A (PS), the peak at 837 cm−1 1026 cm−1 corresponds to the ring vibration and asymmetric stretching C–O–C, respectively, and are a signature of the organic starch polymer34. The broad peaks at 1026 cm−1, and 3279 cm−1 correspond to the O–H stretching and bending. The absorption bands observed in the 1382 cm−1 and 1647 cm−1 revealed the presence of carboxylic and epoxy groups, specifically C–O bending and C=O stretching. The peak at 2929 cm−1 is attributed to the C–H stretching. For the PS/rGO, the C–O–C peak at 837 cm−1 diminished. This is attributed to the fact that in the vicinity of the starch granule, parts of its carbon ring bonds were removed, and oxygen groups were substituted by hydroxyl groups13,35. In the FTIR spectra, three new peaks at 1522 cm−1, 2017 cm−1, and 3682 cm−1 have been found due to the addition of rGO, which corresponds to C–H bending, C=C bending, and C–H stretching, respectively.

FTIR spectra of PS/RGO/MoS2 nanocomposite with the variation of MoS2 filler content. Inset represents the zoom in image for the selected portion of the FTIR spectrum.
For curve E (PS/rGO/MoS2), the peak intensity at 1522 cm−1, and 2017 cm−1 gradually increased with the increase of MoS2 concentration. This suggests that improved interaction between the nanofiller and the PS matrix occurred due to the addition of nanofillers. Due to the addition, the intensity of the O–H bond gets reduced and the peak was found to be shifted towards a higher wavenumber. The intensity reduction and this shifting of peak come from the relative contribution of overlapped bands and the change of surface chemistry with the change of internal orientation due to the addition of nanofillers.
The presence of different polar groups (–COOH, C=O) and the presence of water (–OH functional group) suggests the strong intercalation between the electrolyte and the nanocomposite, which is one reason for the improved charge storing performance.
Structural properties
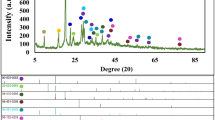
The microstructure of the nanocomposites is investigated by analyzing their XRD spectra, which are presented in Fig. 2. The characterization peak at 2θ values of 14.86°, 17.09°, 19.88°, 22.04°, 24.01°, 25.89°, 31.19°, 34.12°, and 37.89° confirms the B-type crystalline structure of starch36. In the XRD spectra of PS/rGO, the tiny peaks of starch disappeared, which indicates the bonding between PS and rGO occurs. With the incorporation of MoS2, the intensity of the peaks gets reduced further. This suggests a reduction in the crystallinity of the nanocomposite due to the addition of nanofiller. To evaluate the effect of nanofiller on the crystallinity of the polymer matrix, the degree of crystallinity of the samples was measured using the Hermans–Weidinger approach37:
where Ac = Crystalline peak area, Aa = Total area.

X-ray diffraction (XRD) spectra of PS, PS/rGO and PS/rGO/MoS2 nanocomposite.
The degree of crystallinity obtained for the different nanocomposites is presented in Table 1, which clearly shows a reduction in crystallinity due to the introduction of rGO and MoS2 nanofiller. The incorporation of rGO reduces the crystallinity of PS from 40 to 29%. These reductions are due to the gelatinating of PS/rGO nanocomposites and the swelling of rGO into the starch chain through the interaction of hydrogen bonds. This swelling reordered the molecular arrangement of the polymer and together with the demobilization capability of hydrogen bond, reduced the crystallinity38. The sheet-like structure of rGO hinders the molecular movement of PS during crystallization, which is also responsible for the decrease in crystallinity.
Adding MoS2 further reduces the crystallinity of the nanocomposites (from 29 to 21%). When crystalline materials are added to the amorphous chain molecule, the small crystalline regions of ordered structure within an amorphous matrix are destroyed. The hexagonal MoS2 can introduce sulfur vacancy, and the stacked layer creates dislocations in the starch matrix. These defects and dislocation can act as sites of stress concentration and hinder the growth of crystalline regions. As a result, the overall crystallinity of the starch decreases.
The d-spacing, crystallite size, and the micro-strain of the nanocomposites were also estimated from the XRD data and are tabulated in Table 1. The d-spacing can be estimated by Braggs law39:
Crystallite size and Lattice strain are also studied using the Debye–Scherrer approach40. Crystallite size refers to the size of the individual crystalline domains or grains within a polycrystalline material. The crystallite size was also studied following the relation:
where \(\beta\) is the full-width half maximum (FWHM).
Crystallite size provides information about the presence of imperfections, defects, or boundaries between different grain domains41. The crystallite size for PS is 12 nm, which increases up to 23 nm due to the incorporation of MoS2 in the polymer matrix. Larger crystallites can result in fewer grain boundaries, reducing the impedance to the flow of electrons, and thus lowering electrical resistance, which helps improve the charge carrier transportation42.
The lattice strain of the nanocomposites was calculated by the following relation:
Lattice strain is the broadening of the XRD peak of crystal structure due to imperfection and distortion.
Surface morphology analysis
The FESEM study was performed to understand the morphology of the PS/rGO/MoS2 nanocomposites, which is presented in Fig. 3. The FESEM image of PS (in Fig. 3a) shows that it consists of different shape of granules, which confirm the B-type of the starch43. Figure 3b represented the FESEM micrograph of rGO consisting of a thin sheets-like structure. The rGO sheets are randomly aggregated containing wrinkled textures with distinct folding edges. Figure 3c shows the FESEM images of MoS2 microspheres composed of numerous thin petals that aligned themselves to a common center to form the shape of a flower. The petals of the nanoflower are several nm thick, and the diameter of flowers varies between 2 and 5 μm44.

FESEM images of (a) PS (b) rGO (c) MoS2 nanoflower (d) PS/rGO, and (e) PS/rGO/MoS2 nanocomposite.
The FESEM image of the cross-section of the PS/rGO is presented in Fig. 3d. The granules structure of PS disappears after the addition of rGO and is reformed into a thin sheet-like structure which is randomly aggregated. This emphasized the uniform dispersion of rGO in the PS matrix. The deformation comes from the contribution of rGO nanosheets as distinct edges, wrinkled, and folded surfaces, and the SP2 hybridization is present within. The folded structure of PS/rGO is also the result of the loss of the oxygen functional group, which comes from the exfoliation process of GO. In Fig. 3e, for the PS/rGO/MoS2, a more folded layer network is observed, which suggests a more compact nature of the composite followed by self-ordering of the high-aspect ratio of MoS2 layers. This suggests an increase in the specific surface area of the nanocomposite due to the incorporation of MoS2 nanoflower into the polymer matrix. The enhanced surface area may provide more active surface sites for electrochemical reactions to occur.
Optical properties
Figure 4a illustrates how the optical absorption of the nanocomposites is influenced by rGO and MoS2. The presence of rGO and MoS2 in the nanocomposite led to an enhancement in optical absorbance; notably, as the concentration of MoS2 increased, the UV light absorption of the nanocomposites also exhibited an increment. The improved UV shielding performance can be ascribed to two contributing factors. Firstly, the even distribution of MoS2 nanoflower enables efficient UV light absorption, leading to its conversion into heat. Secondly, the UV light may suffer scattering between the PS/rGO/MoS2 interfaces. Through the utilization of absorbance (A) data, the absorption coefficient (α) is computed using the Beer-Lambert equation:
where d is the thickness of the nanocomposites. Figure 4b illustrates how the absorbance changes with wavelength for the PS/rGO/MoS2 nanocomposite. The band gap of the nanocomposite can be ascertained from the absorption coefficient following Tauc’s equation45:
where h is Planck’s constant, β is a specimen structure-dependent constant, \(E_{g}\) is the band gap energy, and n is an empirical index. To ascertain the band gap, (αhν)1/n was plotted as a function of hν. The optimal fit for the current optical data corresponds to n = 1/2, as depicted in Fig. 4b. The indirect band gap of the polymer nanocomposites is estimated from the extrapolation of linear portions to the energy axis. The calculated values for the band gaps of the PS/rGO/MoS2 nanocomposites at different concentrations of MoS2 nanofillers are presented in Table 2. From the Table, a downtrend of bandgap from 4.71 to 2.8 eV is observed with the increasing concentration of MoS2. The incorporation of MoS2 may give rise to additional energy levels in between the conduction and valence bands, consequently leading to a decrease in the nanocomposite's bandgap.

(a) Tauc plot for band gap calculation (b) band gap variation with the varying concentration of MoS2 of PS/rGO/MoS2 nanocomposite.
Urbach energy of the nanocomposites was studied to further explore the effect of MoS2 and rGO nanofiller on the optical bandgap of the starch. During optical transition, the charge carrier may encounter disorders that comes from defect centers and thermal vibration. These disorders create the density of states tailing in between the forbidden energy gap. The defect states tailing is associated with energy which is called Urbach energy, EU. EU is related to the absorption coefficient by46.
where \(\alpha_{0}\) is a constant, hν is the photon energy, \(E_{g}\) is the bandgap. EU can be determined from the log(α(ν)) versus hν graph. The inverse of the slopes of the straight-line portion of the graphs gives the EU. The value of EU obtained for different samples is provided in Table 2 and plotted in Fig. 5. Figure 5 shows that the incorporation of rGO/MoS2 nanofillers causes a gradual increase of EU value, from 0.23 to 0.47 eV. This increase in EU indicates an increase in defect states into the composite nanomaterials due to the incorporation of nanofillers.

(a) Plot of ln(α) versus energy to evaluate the Urbach energy of PS/rGO/MoS2 nanocomposites (b) variation of Urbach energy as a function MoS2 concentration.
The rise in EU resulting due to the integration of rGO can be ascribed to the formation of ionic complexes and the disruption and distortions within the host PS polymer. These effects come from the interactions between diverse surface oxygenated functional groups on rGO and PS. This increment also suggests the existence of localized states that may act as trap centers for charge carriers and facilitate the optical transition47. The trend of increment of EU also continues with the loading of MoS2 concentrations. This can be attributed to the sulfur vacancy pairs of MoS2, which may introduce additional shallow trap states into the host PS matrix. Furthermore, the Mo and S act as interstitial impurities which occupy the interstitial spaces between the PS matrix. The defects originated by the nanofillers introduce additional energy levels between the forbidden gaps, reducing the optical bandgap.
Surface wettability
The contact angle (CA) is used to measure the surface wettability of a material is a critical factor in determining the accessibility of aqueous electrolyte into the electrode. The CA of different samples is presented in Fig. 6. According to Fig. 6, PS shows a 0 °CA, suggesting high surface wettability and surface energy of PS. When it falls onto the PS film, the water gets absorbed immediately due to the roughness. The hydrophilic polar radicals of the constituent molecules of PS immediately absorbed the water that came into the contact. The incorporation of rGO increased the CA of the nanocomposite from 0° to 48.32°. The rGO nanofiller is enriched with non-polar C–C and C=C bonds. These non-polar bonds reduce the affinity of the material for water molecules, leading to the increase of CA48. Then, due to the incorporation of MoS2 nanofiller,, the value of CA was found to reduce gradually from 48.32° to 22.80°. The observed decrease in CA can be attributed to the presence of a negative charge on the surface of MoS2, which comes from their sulfur vacancies and other defects49. This negative charge attracts and interacts with the positively charged hydrogen atoms in water molecules, facilitating the adsorption of water and enhancing hydrophilicity. Also, MoS2 offers a high surface roughness and is composed of stacked layers that are held together by weak van der Waals forces at the nanoscale20. The layers and roughness accommodate water molecules between them, and the water molecules adhere to the surface. So, together with this dispersive interaction of the non-polar functional group, which is driven by the Van der Waals force, increases the surface wettability, leading to hydrophilic behavior.

Contact angle (CA) of as-prepared PS/rGO/MoS2 nanocomposites, where MoS2 concentration were varied.
Electrochemical properties
The cyclic voltammetry (CV) measurements were done at different scan rates (from 5 to 200 mV s−1) to identify the charge storage and charge transfer behaviors of nanocomposite. The CV curve describes the Nernst equation which relates the potential of an electrochemical cell (E) to the standard potential (E°) and the equation is50:
Figure 7a–e shows the CV curves for PS, PS/rGO, and PS/rGO/MoS2 nanocomposites. All of the curves show the sharp increment of current density in the forward scan rate and drop sharply in the reverse scan. This behavior suggests the composites are electrochemically stable as electrode material. The CV curves were found to be changed from a leaf-like shape to a rectangular shape with the addition of rGO and then MoS2 into PS. Additionally, a hump of redox peak is observed at 0.138 V with the 0.25% concentration of MoS2 (Fig. 7c). This presence of this redox peak suggests the existence of pseudo capacitance51. This redox peak was absent in PS and PS/rGO. The rise in the redox peak comes from the active site provided by MoS2 for redox reaction via vacancy and defect states. Then for the 0.50% concentration of MoS2, the hump is slightly shifted and becomes wide, indicating an increase in the redox reaction rate. But, after the addition of 1% MoS2, the curve becomes more rectangular, which is caused by EDLC behavior. This distortion confirms the applicability of a pseudo capacitor and EDLC52.

Cyclic voltammetry curve obtained using three electrode system in 0.1 M aqueous solution of KCl within the potential window of 0.2–0.4 V of (a) PS (b) PS/rGO (1%) (c) PS/rGO/MoS2 (0.25%) (d) PS/rGO/MoS2 (0.5%) and (e) PS/rGO/MoS2 (1%) at different scan rate (f) CV data at 5 mV s−1.
Figure 7f shows the CV curves for all the nanocomposites at a fixed scan rate of 5 mV s−1. The quasi-rectangle CV curves of the PS/rGO/MoS2 nanocomposite are greater than that of pure PS and PS/rGO, which indicates that the MoS2 boosts capacitive performance compared to pure PS. The intercalating complex reaction through the layer structure enhanced the spacing of MoS2, which decreased the energy barrier of ion diffusion, increased the ion intercalation, and boosted the capability of charge storage53.
Figure 8 demonstrates the galvanostatic charging-discharging (GCD) curves at various current densities in the range of 0.1–3 mA g−1. The discharge time is increasing with the decrease of current density for all five nanocomposites. This is because, at lower current density, a smaller number of ions can come close to a certain portion of surface area and can diffuse easily within electrolytes54. On the other hand, at high current density, ion traffic occurs at the same surface; thus, the charging-discharging rate accelerates, and a short discharge time is observed. In addition, with the lower current density, the electrolyte can take place at the empty sites of the electrode; hence the smooth ion-electrolyte intercalation also takes part in the increment of discharge time and eventually higher capacitive behavior55. It was observed from Fig. 8f that the discharge time extends as the MoS2 content increases. Additionally, all the GCD curves deviate from the symmetric triangular shape, suggesting the presence of a pseudocapacitive charge storage mechanism.

Galvanostatic charging discharging curve of (a) PS (b) PS/rGO (1%) (c) PS/rGO/MoS2 (0.25%) (d) PS/rGO/MoS2 (0.5%) and (e) PS/rGO/MoS2 (1%) at different current density (f) GCD curve at a current density of 0.1 mA g−1.
From Table 3 and Fig. 8, a comparison of energy storage performance has been estimated. Specific capacitance was calculated by the following equation:
where \(C_{s}\) = Specific capacitance, I = Current, Δt = Discharge time, m = Deposited mass, ΔV = Potential window.
Energy density (E) and power density (P) were also calculated by calculation of specific capacitance using the following relation:
These values for each nanocomposite have been presented in Table 3. The specific capacitance of PS was estimated to be 2.78 F g−1. The granule microstructure highly resists ion transportation. The various range of granules, together with the grain boundary of PS, hinders the charge transportation process56. In the plasticizing process, the disruption of the amylose and amylopectin chain occurs due to the high temperature. This retrogradation may have an impact on the specific capacitance of starch. Then, with the addition of rGO, the specific capacitance increases from 2.78 to 35.67 F g−1. The layer structure and its wide surface facilitate charge transportation. This accessible area between the electrolyte and active material improves the capacitive performance. Additionally, the layered structure of rGO reduces the diffusion length of electrolyte ions, further boosting capacitance57.
The incorporation of MoS2 nanofiller was found to improve the specific capacitance of the nanocomposite. Also, the specific capacitance was found to increase with the concentration of MoS2 loading into the PS matrix. The incorporation of 1% MoS2 nanoflower was found to provide specific capacitance as high as 124 F g−1, which is 62 times higher than that of the PS. The specific capacitance obtained for the ternary PS/rGO/MoS2 nanocomposite is also larger than that of the binary PS/rGO nanocomposites estimated by Mollick et al.17. Such an improvement of the specific capacitance of the PS matrix due to the incorporation of MoS2 can be attributed to a number of factors: Firstly, the combined higher specific surface area of rGO and MoS2 together expands the functional area of the interface between the nanocomposite and electrolyte58. Also, the porous structure of MoS2 nanoflower can enhance the specific capacitance. Micropores and mesopores can enlarge the electroactive region and accelerate ion diffusion59. Furthermore, an increase in MoS2 concentration creates more active sites, which give the free pathway of ions, hence, enhancing the electrode reaction kinetics60. Additionally, MoS2 concentration also offers improved surface wettability which enhances the electrical conductivity.
Additional insights were gathered from Electrical Impedance Spectroscopy (EIS) to further analysis of the ion migration and charge transfer process. The impedance (Z) is related to alternating voltage as such:
where ω = angular frequency = 2πf, f = frequency.
The alternating potential and the corresponding current create sinusoidal output and can be expressed as:
Here, φ = Phase difference between the current and voltage.
Then the impedance become:
Using Euler theorem this expression become:
where \(Z^{\prime } \left( \omega \right)\) = Real impedance, \(Z^{\prime \prime } \left( \omega \right)\) = Imaginary impedance.
Therefore, Fig. 9a is representing the EIS Nyquist plot where real impedance (Z′) is plotted against corresponding imaginary impedance (Z″). The dotted line represents the experimental data, whereas the simulation data is presented as a solid line. The equivalent circuit for each nanocomposite was simulated from the plot and presented in the inset of Fig. 9. Zview software was used for the simulation and circuit fitting. Corresponding circuit parameters of the equivalent circuit are presented in Table 4.

(a) The Nyquist plot with the best fitted simulation curve (b) circuit of PS, PS/rGO, (c) circuit of PS/rGO/MoS2 (0.25 and 0.5%), (d) circuit of PS/rGO/MoS2 (1%) nanocomposites performed at 0.01 Hz to 106 Hz.
Figure 9b presents the fitted circuit for PS and PS/rGO. The equivalent circuit consists of a series resistance, Rs is connected to a constant phase element, CPE which is parallelly connected to a double layer resistance, Rdl, and charge transfer resistance, Rct. CPE arises due to the nonideal capacitive behavior of the electrochemical system. The Rdl comes from the grain contribution, and Rct comes from the grain boundary contribution. High Rct is observed for PS compared to PS/rGO which is 4574 Ω and 1958 Ω, respectively. This high resistance of PS comes from the rough and granule structure which creates boundaries that interrupt the free movement of ion transportation55,61. Additionally, PS consists of chained amylose and amylopectin, which reduce the area of ion movement62. On the contrary, the layer structure and the wide surface area of rGO reduce the boundary and facilitate ion transportation63. Then Fig. 9c represents the equivalent circuit for the PS/rGO/MoS2 (0.25%, and 0.5%). The fitted circuit changed with a fourth component, the Warburg coefficient (Wo). The Warburg coefficient (Wo) represents the degree of diffusion resistance within the inhibitor PS film. The gradual decrease of Rct is also observed from 497 to 293 Ω with the loading of 0.25%, and 0.50% of MoS2 concentration respectively. Figure 9d represents the equivalent circuit for PS/rGO/MoS2 (1%). The deletion of the third component is observed, which indicates a zero Rct value. This suggests a gradual decrease of the grain resistance due to the incorporation of MoS2 nanofiller for charge transferring within the electrolyte or inter-intra layer64. This decrement of resistance is attributed to the fact that with the addition of MoS2, the functioning area is gradually increased, thus facilitating the charge transportation and decrease, eventually diminishing the contact resistance65. The improved wettability also facilitates the intercalation with electrolytes51. The reduction of Wo is also found compared to PS/rGO/MoS2 (0.25% and 0.5% films). The lower the value, the less difficult it is for ions to diffuse through the pores in the inhibitor PS films. This suggests that the PS films are less porous or can have pores with smaller equivalent diameters. It also suggests the material is less corrosive66.
Capacitive retention is the measure of the ability of a device to retain stored energy during an open circuit test. Figure 10a depicts the cyclic stability: Coulombic efficiency and capacity retention of PS/rGO/MoS2 nanocomposites as an electrode material. Coulombic efficiency (η) is calculated by the following equation:

(a) The coulombic efficiency and capacitive retention for 10,000 cycles from GCD (b) discharging time for 1st and 10,000th cycle and (c) electrochemical impedance spectra (EIS) curve for the 1st and 10,000th cycle of GCD curve.
Then capacitive retention was calculated:
It is observed from the Fig. 10a that 100% efficiency was achieved after completing 10,000 cycles. However, cyclic retention gets reduced to 86% after 100 cycles, and then after 2000 cycles, it reduces to ~ 80%. However, this 80% was consistent till 10,000 cycles. This first reduction to 86% can be attributed to the fact of the saturation of active site on the PS/rGO/MoS2 film surface during the charge–discharge cycle. Furthermore, the slight decrease to 80% is due to the structural breakdown and delamination of the film surface in the aqueous solution of KCl, and the ion may have been trapped in the layer of rGO/MoS2 due to the repetition of charging and discharging cycles67. It has been observed that a 100% Coulombic efficiency was achieved even after completing 10,000 cycles.
Figure 10b represents the EIS curve of the 1st and 10,000th cycle. After undergoing 10,000 cycles of the electrode material, the radius of the impedance spectrum's semi-circle was found to be diminished compared to that observed during the first cycle. This reduction of radius suggests the reduction of internal resistance. The findings indicate that up to 10,000 cycles of operation, this electrode displays an increased electrical conductivity and demonstrates rapid electron-conducting capabilities at the interface. This reduction of internal resistance can be ascribed to the electrochemical stimulation of the active material, decreased aggregation of the active material throughout cycling, and an enhanced interface68. From Fig. 10c, it is observed that the discharge time drains faster after uninterrupted 10,000 cycles.
From this study, it is observed that the incorporation of MoS2 nanoflower produces defect sites into the nanostructure that offer more active sites. Furthermore, the enhancement of the surface area due to the MoS nanofiller helps facilitate the increased number of active sites and improves the electrochemical performance of the nanocomposites. Thus forming a ternary nanocomposite by incorporating MoS2 nanofillers into the PS/rGO composites will offer enhanced capacitive performance by keeping the concentration of rGO low. The improved electrochemical performance of nanocomposite can therefore be attributed to the synergistic effect of the layered structured MoS2 and rGO nanofillers, which improve the crystallinity, incorporate defect sites, reduce the contact resistance, and provide a large surface area.
Conclusions
In conclusion, biodegradable PS/rGO/MoS2 nanocomposites have been prepared following a facile solution casting method. The structural and surface study demonstrates improved interaction between PS and rGO/MoS2, which leads to the reduction of composite crystallinity and reveals the presence of defects. From the UV–vis study, the band gap reduction was found together with the increment of defects, which was confirmed by the Urbach energy analysis that comes from the contribution of MoS2 loading. Contact angle study shows the improvement of surface wettability due to the incorporation of MoS2. The PS/rGO/MoS2 nanocomposites provide specific capacitance as high as 124 F g−1, about 60 times higher than that of PS. The nanocomposite also provides excellent cyclic stability of about ~ 85% retention of the capacitance up to 10,000 cycles of operation. The notable decrease in bandgap and crystallinity leads to the adsorption of electrons and ions into the biopolymer matrix, resulting in enhanced electrochemical performance. Furthermore, the structural and sulfur drive defect that comes from the stacked MoS2/rGO nanosheets leading to very high surface area, enhanced electrochemical conductivity, and interaction with the –OH functional group provided by the nanofiller improved the electrochemical performance of the nanocomposite. This research has the potential to provide a straightforward and cost-effective method for creating environmentally friendly energy storage devices, which are essential for ensuring a sustainable energy future.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Nerini, F. F., Julia, T. & Long, S. T. Mapping synergies and trade-offs between energy and the sustainable development goals. Nat. Energy 3, 10–15 (2018).
Gielen, D. et al. The role of renewable energy in the global energy transformation. Energy Strategy Rev. 24, 38–50 (2019).
Wang, J. et al. Electrochemical energy storage performance of 2D nanoarchitecture hybrid materials. Nat. Commun. 12, 3563–3579 (2021).
Yin, Q., Ye, M., Jia, H., de Araújo Melo, A. F. A. & Ji, Q. Sequentially optimizing carbon nanotubes framework towards flexible and compact capacitive energy storage. J. Mater. Chem. A 10, 21430–21445 (2022).
Fang, S., Bresser, D. & Passerini, S. Transition metal oxide anodes for electrochemical energy storage in lithium- and sodium-ion batteries. Adv. Energy Mater. 10, 923–935 (2020).
Asif, H. et al. Recent advances, properties, fabrication and opportunities in two-dimensional materials for their potential sustainable applications. Energy Storage Mater. 59, 102780–102797 (2023).
Dalwadi, S., Goel, A. C., Kapetanakis, C., Salas, D. L. C. & Hu, X. The integration of biopolymer-based materials for energy storage applications: A review. Int. J. Mol. Sci. 7, 612–620 (2023).
Meera, K., Arun, K. & Ramesan, M. T. High performance biopolymer blend nanocomposites derived from cashew gum/polyvinyl alcohol/boehmite for flexible electronic devices. J. Appl. Polym. Sci. 140, 1–17 (2023).
Apriyanto, A., Compart, J. & Fettke, J. A review of starch, a unique biopolymer–structure, metabolism and in planta modifications. Plant Sci. 318, 1012–1023 (2022).
Morales, N. J., Candal, R., Famá, L., Goyanes, S. & Rubiolo, G. H. Improving the physical properties of starch using a new kind of water dispersible nano-hybrid reinforcement. Carbohydr. Polym. 127, 291–299 (2015).
Din, Z., Xiong, H. & Fei, P. Physical and chemical modification of starches: A review. Crit. Rev. Food Sci. Nutr. 7, 1–68 (2015).
Meera, K. & Ramesan, M. T. Tailoring the performance of boehmite nanoparticles reinforced carboxymethyl chitosan/cashew gum blend nanocomposites via green synthesis. Polymer 268, 125706–125726 (2023).
Salem, A. M., Mohamed, A. R. & Yassin, A. Y. The effect of low concentrations of polypyrrole on the structural, thermal, and dielectric characteristics of CMC/PPy blends. J. Mater. Sci. Mater. Electron. 34, 1522–1535 (2023).
Ma, J., Zhu, W., Tian, Y. & Wang, Z. Preparation of zinc oxide-starch nanocomposite and its application on coating. Nanoscale Res. Lett. 11, 1–18 (2016).
Chia, M. R., Ahmad, I. & Phang, S. W. Starch/polyaniline biopolymer film as potential intelligent food packaging with colorimetric ammonia sensor. Polymers 14, 60–70 (2022).
Ma, T., Chang, P. R., Zheng, P. & Ma, X. The composites based on plasticized starch and graphene oxide/reduced graphene oxide. Carbohydr. Polym. 94, 63–70 (2013).
Mollik, S. I., Ahmad, M. H., Alam, R. B., Bari, M. W. & Islam, M. R. Improved thermal, mechanical, and electrochemical performance of bio-degradable starch/reduced graphene oxide nanocomposites. AIP Adv. 11, 095312 (2021).
Yassin, A. Y. et al. Structural, optical and antibacterial activity studies on CMC/PVA blend filled with three different types of green synthesized ZnO nanoparticles. J. Inorg. Organomet. Polym. 33, 1855–1867 (2023).
Piacentini, A., Daus, A., Wang, Z., Lemme, M. C. & Neumaier, D. Potential of transition metal dichalcogenide transistors for flexible electronics applications. Adv. Electron. Mater. 9, 2381–2409 (2023).
Manzeli, S., Ovchinnikov, D., Pasquier, D., Yazyev, O. V. & Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2, 1–15 (2017).
Wilson, J. A. & Yoffe, A. D. The transition metal dichalcogenides discussion and interpretation of the observed optical, electrical and structural properties. Adv. Phys. 18, 193–335 (1969).
Choudhary, N. et al. Two-dimensional lateral heterojunction through bandgap engineering of MoS2 via oxygen plasma. J. Phys. Condens. Matter 28, 36–48 (2016).
Abdel, M. I. A. MoS2-based nanocomposites: Synthesis, structure, and applications in water remediation and energy storage: A review. Environ. Chem. Lett. 101, 1–37 (2021).
Humberto, R. G. Two-dimensional layered materials offering expanded applications in flatland. ACS Appl. Nano Mater. 3, 6134–6139 (2020).
Liu, C., Bai, Y., Zhao, Y., Yao, H. & Pang, H. MoS2/graphene composites: Fabrication and electrochemical energy storage. Energy Storage Mater. 33, 470–502 (2020).
Lei, T., Xinhai, L., Zhixing, W., Huajun, G. & Jiexi, W. Lightweight reduced graphene oxide@MoS2 interlayer as polysulfide barrier for high-performance lithium–sulfur batteries. ACS Appl. Mater. Interfaces 10, 3707–3713 (2018).
Stanisz, M., Klapiszewski, Ł & Jesionowski, T. Recent advances in the fabrication and application of biopolymer-based micro- and nanostructures: A comprehensive review. J. Chem. Eng. 397, 125409 (2020).
Balakrishnan, P., Sreekala, M. S., Kunaver, M., Huskić, M. & Thomas, S. Morphology, transport characteristics and viscoelastic polymer chain confinement in nanocomposites based on thermoplastic potato starch and cellulose nanofibers from pineapple leaf. Carbohydr. Polym. 169, 176–188 (2017).
Nagaraj, N., Chia, J. L. & Takao, M. Polymer based thermoelectric nanocomposite materials and devices: Fabrication and characteristics. Nano Energy 78, 105186–105215 (2020).
Hummers, W. S. & Offeman, R. E. Preparation of graphitic oxide. J. Am. Chem. Soc. 80, 1339–1353 (1958).
Aragaw, B. A. Reduced graphene oxide-intercalated graphene oxide nano-hybrid for enhanced photoelectrochemical water reduction. J. Nanostruct. Chem. 10, 9–18 (2020).
Chen, M. et al. Smart combination of three-dimensional-flower-like MoS2 nanospheres/interconnected carbon nanotubes for application in supercapacitor with enhanced electrochemical performance. J. Alloys Compd. 9, 696–712 (2017).
Maugé, F., Lamotte, J., Nesterenko, N. S., Manoilova, O. & Tsyganenko, A. A. FT-IR study of surface properties of unsupported MoS2. Catal. Today 70, 271–284 (2001).
Peregrino, P. P. Thermal and electrical properties of starch–graphene oxide nanocomposites improved by photochemical treatment. Carbohydr. Polym. 106, 305–311 (2014).
Li, R., Liu, C. & Ma, J. Studies on the properties of graphene oxide-reinforced starch bio composites. Carbohydr. Polym. 84, 631–637 (2011).
Guinesi, L. S. et al. Kinetics of thermal degradation applied to starches from different botanical origins by non-isothermal procedures. Thermochim. Acta 447, 190–196 (2006).
Hermans, P. H. & Weidinger, A. X-ray studies on the crystallinity of cellulose. J. Polym. Sci. 4, 135–144 (1949).
Fachbereich, D. & Verfahrenstechnik, M. On the effect of nanofillers on the environmental stress cracking resistance of glassy polymers. Ind. Eng. Chem. Res. 8, 1–57 (2019).
Elton, L. R. B. & Daphne, F. J. X-ray diffraction and the Bragg law. Am. J. Phys. 34, 1036–1038 (1966).
Debye, P. & Scherrer, P. Interference on inordinate orientated particles in x-ray light. III. Phys. Z. 18, 291–301 (1917).
Yassin, A. Y. Synthesized polymeric nanocomposites with enhanced optical and electrical properties based on gold nanoparticles for optoelectronic applications. J. Mater. Sci. Mater. Electron. 34, 46–64 (2023).
Al-Muntaser, A. A. et al. Boosting the optical, structural, electrical, and dielectric properties of polystyrene using a hybrid GNP/Cu nanofiller: Novel nanocomposites for energy storage applications. J. Mater. Sci. Mater. Electron. 34, 678–692 (2023).
Beauson, J., Schillani, G., van der Schueren, L. & Goutianos, S. The effect of processing conditions and polymer crystallinity on the mechanical properties of unidirectional self-reinforced PLA composites. Compos. Part A Appl. Sci. Manuf. 152, 20–28 (2022).
Ahmad, M. H., Alam, R. B., Ul-hamid, A., Farhad, S. F. U. & Islam, M. R. Hydrothermal synthesis of Co3O4 nanoparticles decorated three dimensional MoS2 nanoflower for exceptionally stable supercapacitor electrode with improved capacitive performance. J. Energy Storage 47, 1–12 (2022).
Tauc, J., Grigorovici, R. & Vancu, A. Optical properties and electronic structure of Ge optical properties and electronic structure of amorphous Germanium. ACS Appl. Energy Mater. 4, 10877–10885 (1996).
Urbach, F. The long-wavelength edge of photographic sensitivity and of the electronic absorption of solids. Phys. Rev. 92, 1324–1324 (1953).
Lizu, A. H., Bari, M. W., Gulshan, F. & Islam, M. R. GO based PVA nanocomposites: Tailoring of optical and structural properties of PVA with low percentage of GO nanofillers. Heliyon 7, 6983–6893 (2021).
Kozbial, A. Study on the surface energy of graphene by contact angle measurements. Langmuir 30, 8598–8606 (2014).
Gao, J. et al. Aging of transition metal dichalcogenide monolayers. ACS Nano 10, 2628–2635 (2016).
Ciribelli, B. N., Colmati, F. & de Souza, E. C. Nernst equation applied to electrochemical systems and centenary of his Nobel Prize in chemistry. Int. J. Innov. Educ. Res. 8, 670–683 (2020).
Liu, Y., Jiang, S. P. & Shao, Z. Intercalation pseudo capacitance in electrochemical energy storage: Recent advances in fundamental understanding and materials development. Mater. Today Adv. 7, 100072 (2020).
Gogotsi, Y. & Penner, R. M. Energy storage in nanomaterials -capacitive, pseudocapacitive, or battery-like?. ACS Nano 12, 2081–2083 (2018).
Wang, S. Three-dimensional MoS2@CNT/rGO network composites for high-performance flexible supercapacitors. Eur. J. Chem. 23, 3438–3446 (2017).
Zeng, R. et al. Swollen ammoniated MoS2 with 1T/2H hybrid phases for high-rate electrochemical energy storage. ACS Sustain. Chem. Eng. 7, 11540–11549 (2019).
Mohd, Z. A., Nazish, P., Dip, K. N. & Ruhul, R. Enhanced activity of highly conformal and layered tin sulfide (SnSx) prepared by atomic layer deposition (ALD) on 3D metal scaffold towards high performance supercapacitor electrode. Sci. Rep. 9, 1–16 (2019).
Bertoft, E. Understanding starch structure: recent progress. Agronomy 7, 3–56 (2017).
Rapisarda, M., Marken, F. & Meo, M. Graphene oxide and starch gel as a hybrid binder for environmentally friendly high-performance supercapacitors. Commun. Chem. 4, 20–31 (2021).
Thangavelu, K., Aubry, C. & Zou, L. Amphiphilic Janus 3D MoS2/rGO nanocomposite for removing oil from wastewater. Ind. Eng. Chem. Res. 60, 1266–1273 (2021).
Wang, X. & Wang, H. Designing carbon anodes for advanced potassium-ion batteries: Materials, modifications, and mechanisms. Adv. Powder Mater. 1, 100057–100079 (2022).
Manchala, S., Tandava, V. S. R. K., Jampaiah, D., Bhargava, S. K. & Shanker, V. Novel and highly efficient strategy for the green synthesis of soluble graphene by aqueous polyphenol extracts of eucalyptus bark and its applications in high-performance supercapacitors. ACS Sustain. Chem. Eng. 7, 11612–11620 (2019).
Xu, J. et al. Nitrogen-doped reduced graphene oxide–polyaniline composite materials: Hydrothermal treatment, characterization and super capacitive properties. J. Energy Storage 43, 1031–1095 (2021).
Alam, R. B., Ahmad, M. H. & Islam, M. R. Bio-inspired gelatin/single-walled carbon nanotube nanocomposite for transient electrochemical energy storage: An approach towards eco-friendly and sustainable energy system. Heliyon 438, 1–9 (2021).
Wang, Q. et al. Facile strategy for mass production of polymer-supported film electrodes for high performance flexible symmetric solid-state supercapacitors. Appl. Surf. Sci. 487, 295–303 (2019).
Shuaifeng, L. et al. Insights into interfacial effect and local lithium-ion transport in polycrystalline cathodes of solid-state batteries. Nat. Commun. 11, 5700–5710 (2020).
Xiao, L. & Hongwei, Z. Two-dimensional MoS2: Properties, preparation, and applications. J. of Materiomics 1, 33–44 (2015).
Hong, T., Sun, Y. H. & Jepson, W. P. Study on corrosion inhibitor in large pipelines under multiphase flow using EIS. Corros. Sci. 44, 1–112 (2002).
Ramakrishnan, K., Nithya, C. & Karvembu, R. Heterostructure of two different 2D materials based on MoS2 nanoflowers@ rGO: An electrode material for sodium-ion capacitors. Nanoscale Adv. 1, 334–341 (2019).
Lin, L. et al. Green fabrication of agglomeration-reductive and electrochemical-active reduced graphene oxide/polymerized proanthocyanins electrode for high-performance supercapacitor. ACS Appl. Energy Mater 1, 369–379 (2023).
Acknowledgements
One of the authors, MRI gratefully acknowledges the financial support from the Committee for Advanced Studies and Research (CASR), Bangladesh University of Engineering and Technology, under the Grant DAERS/R-01/CASR-337th/2021. EM and MRI are grateful to the University Grant Commission, Bangladesh for providing financial support under the Grant Order: 37.01.0000.073.07.045.21.944.
Author information
Authors and Affiliations
Contributions
E.M. took the data, E.M. and M.R.I. analyzed the data, E.M. and M.R.I. wrote the main manuscript text.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahmud, E., Islam, M.R. Improved electrochemical performance of bio-derived plasticized starch/ reduced graphene oxide/ molybdenum disulfide ternary nanocomposite for flexible energy storage applications. Sci Rep 13, 20967 (2023). https://doi.org/10.1038/s41598-023-48326-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48326-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
