
Abstract
The orchid family has 200,000 species and 700 genera, and it is found worldwide in the tropics and subtropics. In China, there are 1247 species and subspecies of orchids belonging to the Orchidaceae family. Orchidaceae is one of the most diverse plant families in the world, known for their lush look, remarkable ecological tolerance, and capability for reproduction. It has significant decorative and therapeutic value. In terms of evolution, the orchid family is one of the more complicated groups, but up until now, little has been known about its affinities. This study examined the properties of 19 chloroplast (cp) genomes, of which 11 had previously been published and nine had only recently been revealed. Following that, topics such as analysis of selection pressure, codon usage, amino acid frequencies, repeated sequences, and reverse repeat contraction and expansion are covered. The Orchidaceae share similar cp chromosomal characteristics, and we have conducted a preliminary analysis of their evolutionary connections. The cp genome of this family has a typical tepartite structure and a high degree of consistency across species. Platanthera urceolata with more tandem repeats of the cp genome. Similar cp chromosomal traits can be seen in the orchidaceae. Galearis roborowskyi, Neottianthe cucullata, Neottianthe monophylla, Platanthera urceolata and Ponerorchis compacta are the closest cousins, according to phylogenetic study.
Similar content being viewed by others


Introduction
The Orchidaceae, which contains 700 genera and 20,000–35,000 species, is the biggest angiosperm family in the world. It is widespread throughout all terrestrial environments, particularly in the tropics, with the exception of arctic and exceptionally dry deserts. In China, there are 171 genera, 1247 species, and subgenera of those species.Due to their distinctive morphology and ecological adaptations, orchids have a high scientific worth in addition to their high economic value. The natural populations of many orchids in China have declined rapidly and are now in danger as a result of the expansion of orchids there and the deterioration of the ecological environment in recent years1. There has been a lot of research on orchids in recent years2, particularly on their morphology and therapeutic potential, but little has been done on their genetics3,4.
As of July 30, 2022, At the National Center for Biotechnology Information (National center for biotechnology information, NCBI) uploaded a total of 1 228 complete chloroplast genome data for Orchidaceae species, Among them, 153 were Dendrobium Sw., There are 54 Paphiopedilum Pfitz.. There are 28 Phalaenopsis Bl., Dendrobium officinale, Paphiopedilum emersonii, Phalaenopsis aphrodite, Cymbidium dayanum, Apostasia ramifera, Bletilla formosana. The whole chloroplast genome of some orchid species has been published and studied by scholars5.In order to determine the evolutionary time and genome position of various ndh gene deletion, Lin6 of the chloroplast genome of eight kinds of orchid, found that Vanilla shenzhenica, Vanilla planifolia, Galeola faberi and Drakaea elastica truncation or absence of ndh gene, the phenomenon has nothing to do with the known taxonomic or evolutionary relationship.
The Orchidaceae is a complicated evolutionary family, but up until now, inadequate research on its affinities and lack of information of its DNA have made it difficult to analyze.Due to their short sequences, uniparental inheritance, low nucleotide substitution rates, and straightforward, conserved genome structure, chloroplast genes were found to be better suited to the study of genetic relationships in plants, or phylogenetic relationships, after being examined from various angles.
Chloroplasts are crucial organelles for photosynthesis in the plant body and are descended from ancient bacteria that were formerly part of early plants and symbionts like cyanobacteria, which enable plants to contribute positively to Earth’s ecosystem7. The chloroplast is a very significant and active organelle that performs many different metabolic processes, including photosynthesis. The chloroplast genome evolves more slowly8; unlike the frequent changes and recombination in the mitochondrial genome, the single-parent genetic characteristics of the chloroplast genome can effectively reduce the interference of genetic recombination9, so the chloroplast genome becomes an appropriate and effective tool for plant classification and phylogenetic research.
A large single copy (LSC) region and a small single copy (SSC) region are separated by two inverted repeat regions in the chloroplast genome's circular DNA structure10,11.Chloroplast genomes have been used in phylogenetic and kinship studies between plants recently, which may clearly depict and display their evolutionary ties12. In this study, the nine species with the highest species richness—Neottianthe monophylla, Herminium monorchis, Galearis roborowskyi, Ponerorchis chusua, Platanthera urceolata, Malaxis monophyllos, Ponerorchis compacta, Neottia puberula, Neottianthe cucullata—had their cp genomes sequenced, assembled, and Additionally, we carried out a thorough evolutionary analysis on the cp genomes of 19 species belonging to the Orchidaceae subfamily.
Materials and methods
Sample collection, DNA extraction, and sequencing
Nine species of healthy, fresh leaves (Neottianthe monophylla, Herminium monorchis, Galearis roborowskyi, Ponerorchis chusua, Platanthera urceolata, Malaxis monophyllos, Ponerorchis compacta, Neottia puberula, Neottianthe cucullata) were gathered in the Tibet Autonomous Region and Qinghai Province, then quickly frozen and stored in liquid nitrogen. Through the use of a modified cetyltrimethylammonium bromide (CTAB) procedure13, whole genome DNA was isolated from leaves.The Illumina NovaSeq 6000 platform was used for the sequencing of all DNA sequences. Over 5000 times the coverage of each complete cp genome was provided by the clean reads.Experimental research and field studies on plants including the collection of plant material are comply with relevant guidelines and regulation.The selected materials were identified by professor Yang Zeng of Qinghai Normal University as Ponerorchis chusua and Ponerorchis compacta. Plant collection has been granted permission.
Genome assembly and annotations
Using NOVOPlastyv4.214, we assembled the chloroplast genome, and the more comprehensive results served as the final genome for screening. In addition to manual checks, the newly assembled chloroplast genomes were annotated with the help of PGA (Plastid Genome Annotator)15, the cp genome of Dipterocarpus turbinatus (Genbank: NC_046842.1) served as a reference, and the tRNA genes were verified with the help of ARAGORNv1.2.3816 and tRNAscan-SEv2.0.717. Using OGDRAW, a completely annotated circular plastid map was created (OrganellarGenomeDRAW)18.
Using the online MIcroSAtellite (MISA) 2.1 tool, simple repeat sequences (SSRs) were found in the chloroplast genomes of 19 plant species. There were eight single nucleotides, five dinucleotides, four trinucleotides, and three each of tetranucleotides, pentanucleotides, and hexanucleotides utilized as repeat unit parameters.
Comparative analyses
Taking the Galearis roborowskyi genome as a reference, the homogeneity of the entire chloroplast genomes of 19 species were visualized to examine chloroplast genomic differences, using the shuffle-LAGAN program of mVISTA v2.019. Using IRSCOPE, the borders of the IR, SSC, and LSC areas were also compared.DnaSPv6.12.03, which was used to explore nucleotide diversity (Pi), with the window length set to 600 bp and the step size set to 200 bp, was utilized to extract and analyze the coding and non-coding sections.
The codon adaptation index (CAI), codon bias index (CBI), frequency of optimum codons (Fop), effective number of codons (ENc), GC content of synonymous third codon positions (GC3s), and relative synonymous codon usage values were used to evaluate codon preferences (RSCU)20.
Selective pressure analysis
The selection pressure in orchids was calculated using the ratio of non-synonymous substitutions (Ka) to synonymous substitutions (Ks) (Ka/Ks).Initially, the four chloroplast genomes of orchids were compared, and 79 protein-coding genes were extracted. The non-synonymous substitution rate (Ka) and synonymous substitution rate (Ks) for each gene were calculated, as well as the ratio of the two (Ka/Ks), with positive selection being indicated by a Ka/Ks ratio greater than 1, neutral selection being indicated by a Ka/Ks ratio of 1, and purifying selection by a Ka/Ks ratio less than 1.
Phylogenetic inference
For phylogenetic reconstruction, we collected 10 publically accessible chloroplast genome sequences from the Genbank website (two of which were from external populations). Prior to trimming the lines of comparison for each gene, all single-copy genes from the 19 taxa were extracted. For phylogenetic investigations, they were also joined in a permutational manner.Finally, a phylogenetic tree (ML) was built using TreeBeSTv1.9.221. The support of branches was evaluated by 100 rapid bootstrap replications.
Results
Comparison among chloroplast genomic features in Orchidaceae
Galearis roborowskyi was discovered to have the least chloroplast genome size (149,067 bp), whereas Herminiumm onorchis had the greatest (156,412 bp) (Fig. 1) (support document). The LSC, SSC, and IR lengths for these 11 species are displayed in Table 1. We discovered between 124 and 140 distinct genes, including between 83 and 94 protein-coding genes, 8 rRNA genes, and 38 tRNA genes (Tables 1, 2).

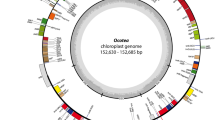
A genetic map of Herminium monorchis chloroplast genome. Genes inside the circle have their transcription going clockwise, whereas those outside the circle have it going the other way. Different colors that code for genes designate various functional groupings. The amount of guanine-cytosine (GC) is represented by the grey-black portion of the inner circle, while the amount of adenine–thymine (AT) is shown by the light grey portion. The inner circle displays the reverse repeat (IRa, IRb) regions as well as the small single-copy region (SSC) and large single-copy region (LSC).
The cp genome of Galearis roborowskyi was then further compared using the mVISTA method, revealing a similar pattern of sequences throughout the chloroplast genome, which comprises 17 orchid species and two outgroups (Aloe maculata and Aloe vera) (Fig. 2, support document).The findings demonstrate that, in contrast to the two outgroups, the chloroplast genomes of 17 species of orchid have comparable architecture and gene sequences. All studied species had more conserved coding regions than non-coding regions. The LSC region also diverged from the SSC region more so than it did from the IR region. The alignment revealed that genes, particularly the psbA matK and rps16 genes, were less conserved in the genomes of Neottianthe monophylla.


Using the shufe-LAGAN program, the chloroplast genomes of 23 distinct species were examined. The horizontal axis displays the location in the chloroplast genome, and the same proportions are displayed in the vertical direction at a scale of 50 to 100%.The gene being labeled and the direction of transcription are represented by each arrow. Exons, tRNA, conserved non-coding sequences, and mRNA are designated by different colors as genomic areas (support document).
Guanine-cytosine (GC) content in orchids ranged from 29.8% (Platanthera contigua) to 48.1%. (Orchis militaris). GC content in the LSC, SSC and IR regions was 33.8–35.4%, 28.6–30.7% and 41.3–43.2%, respectively, Compared to LSC and SSC, the GC content in IR was substantially higher (Table 1; Fig. 3).

Changes in chloroplast GC content of all 17 species.
Divergence hotspots
DnaSPv6.12.03 was used to analyze the whole cp genomes of 17 orchid species and two Peripheral species. At a window length of 600 bp, the nucleotide diversity (Pi) between sequences was determined and analyzed. There were 623 mutant sites in the aligned gene sequences, with Pi values ranging from 0.00051 to 0.42575 and a mean value of 0.06999. With Pi values over 0.16, four extremely variable locations were found. TrnS-GCU-trnG-UCC, trnT-GU-psbD, trnI-GAU-rrn16, and rpl2 are some of these regions. The LSC area contains two of these gene fragments, while the IR region contains the other two.Rpl2 is among them, whereas the rest are found in the non-coding region. Compared to the non-coding area, the genes that code for proteins are more conserved. The region of trnI-GAU-rrn16 included the difference with the highest value, which was 0.42575. (Fig. 4).

Nucleotide polymorphism of the chloroplast genomes of Orchidaceae species.
Contraction and expansion of inverted repeats
The size of the cp genome can alter due to the contraction and growth of the IR area, which also has an impact on the rate at which cp genes evolve22,23. A comparison of the IR boundaries of 28 species of orchids revealed that the IR boundary locations with the most pronounced alterations were IRb/SSC, SSC/IRa, and IRa/LSC (Fig. 5).The LSC/IRa and LSC/IRb edges of the Orchidaceae chloroplast genome are substantially conserved, with virtually the same genes flanking them. The rps3 gene is found on IRb at the junction of LSC and IRb. The rpl22 gene spans the LSC/IRb area in most species, with Neottia suzukii and Neottia ovata having the highest expansion, with the exception of Aloe vera、 Aloe maculata and Hancockia uniflora.


Comparison of the borders of the all regions among 28 chloroplast genomes of Orchidaceae.
Codon usage and amino acid frequency
The degree of relative synonymous codon usage, sometimes referred to as codon usage preference, measures how frequently a specific codon is utilized in the codon that codes the corresponding amino acid. When RSCU > 1, the codon is used more frequently. This codon has no preference in the case of RSCU = 1; if it is used very infrequently, RSCU1 (support document).
To assess codon usage in the Orchidaceae, we analyzed codon usage deviations for genes in the cp genomes of nine orchid species (Tables 3 and 4). Codon use deviations were derived using relative synonymous codon usage (RSCU). Six codons that encode the amino acids arginine (Arg), leucine (Leu), and serine (Ser) were shown to have the highest preference in this investigation. The arginine was determined to have the highest (2.00 to 2.20) and lowest (0.40 to 0.46 RSCU levels). Other RSCU-related metrics, such as the codon adaptation index (CAI), the codon utilisation index (CBI), the optimal codon frequency (Fop), the effective codon number (ENc), and the synonymous codon 3 position (GC3s), were more moderate.
Repeat analyses
Platanthera urceolata’s plastid genome had 255 SSRs in total, more than any of the other eight orchid families shown in Fig. 6. The bulk of SSRs in the cp genome (81.9%) were single nucleotides, with poly T and poly A predominating (Figs. 6 and 7). The findings of the analysis of the SSR frequency in the genomes of nine Orchidaceae species are depicted in Fig. 4. All species’ genomes had the dinucleotides AT/AT and AG/CT, however only Herminium monorchis’ dinucleotide AC/GT was absent from the genomes of the other eight species. Furthermore, only Neottianthe monophylla, Platanthera urceolata, and Ponerorchis chusua were found to have five trinucleotides (AAT/ATT, AAG/CTT, ACT/AGT, AGG/CCT and ATC/ATG), nine tetra-repeats(AAAG/CTTT, AAAT/ATTT, AAGT/ACTT, AATC/ATTG, AATG/ATTC, AATT/AATT, ACAG/CTGT, ACAT/ATGT and AGAT/ATCT), eight pentanucleotides(AAAAT/ATTTT, AAAGT/ACTTT, AACAT/ATGTT, AATAG/ATTCT, AATAT/ATATT, ACATC/ATGTG, ACGAT/ATCGT and ACTAT/AGTAT) and six hexanucleotides(AAATAT/ATATTT, AACTAT/AGTTAT, AAGAGG/CCTCTT, AAGCTG/AGCTTC, AATATT/AATATT, AATTGC/AATTGC) (Fig. 7).

Frequency of diferent microsatellite motifs in diferent repeat types of nine Orchidaceae plastome genomes.

Number of different SSR types in the nine Orchidaceae chloroplast genomes.
Selective pressure analysis
DnaSP software was used to determine the chloroplast codon Ka/Ks in order to compare the Ka/Ks of 21 distinct species pairs and further examine the selection pressure on chloroplast genes in orchids during evolution (Fig. 8). For each of the 21 species pairs, Ka/Ks ratios were computed. For the pairs Herminium monorchis-Platanthera urceolata and Galearis roborowskyi-Neottianthe monophylla, higher Ka/Ks ratios were found. The photosynthesis-related genes atpF, ndhD, ndhE, and ndhH, the expression-related genes rpl22, rpoC1, rpoC2, accD, and ycf1 of other functional genes, and the genes related to expression-related genes rps18 and rpoC2 all showed Ka/Ks > 1, indicating that these genes were under positive selection during evolution (Table 5).

Ka/Ks values of different functional genes.
Phylogenetic relationships among Orchidaceae
Using Aloe maculata and Aloe vera as outgroups, the evolutionary relationships of the cp genes in 28 orchid species were investigated. Using the ML、NJ and MP technique, a developmental tree of 50 single-copy genes was created (Fig. 9). The cp genomes of 28 species of orchids were used in this study to infer phylogenetic relationships, and the ML, NJ and MP analysis was used to compare those relationships to outgroups like Aloe macrophylla and Aloe vera. The tree was created with 26 nodes. All phylogenetic trees have the same topology (the three trees are presented together), and most of the nodes have 100% bootstrap support, indicating high analysis confidence. The 28 orchid species studied are mainly divided into several large clades, of which Neottianthe, Platanthera, Hetaeria, and Neottia all clearly clustered into one clade, indicating that their congeners are more closely related.

Phylogenetic tree of 28 species based on chloroplast genomes,The topology is indicated with ML/NJ/MP bootstrap support values at each node.
Discussion
Kim et al. compared the chloroplast genome size of 30 orchids and their gene loss, and found that most of the genes associated in heterotrophic orchids had been lost (ndh), while most of the housekeeping genes retained24. This result was also verified in Kim et al.25.Chloroplast genomes have strong conservation in plant evolution. First, it is a typical four-segment structure, and then it is highly conserved26,27 in gene content and gene order, which is why chloroplast genomes are widely used for phylogenetic research.
Nine Orchidaceae species’ chloroplast genome lengths were examined in this study. Nine Orchidaceae have full chloroplast genomes, with an average length of 153,330 bp and sizes ranging from 149,067 bp (Galearis roborowskyi) to 156,412 bp (Herminiumm onorchis).The tetrad structure of the chloroplast genome in land plants makes it highly conserved under normal conditions. The majority of the 74 protein-coding genes in the angiosperm chloroplast genome are present, but there are also instances of gene capture, gene rearrangement, and gene loss in various families and species28,29. Comparative analysis makes it simple to locate mutation hotspots because of the plant cp genome's highly conserved structure. In population genetic or phylogenetic investigations, these mutational hotspots surrounding by conserved sequences are frequently utilized as DNA barcodes28,47. We discovered that sequence variation in Orchidaceae primarily occurs in the non-coding regions via a combined analysis of mVISTA sequence variation and DnaSP-inferred nucleotide variation.Three unique areas (psbA, matK, and rps16) were identified in the investigation of the sequence variation in the cp genome (Fig. 2). TrnS-GCU-trnG-UCC, trnT-GGU-psbD, trnI-GAU-rrn16, and rpl2 were all shown to be highly variable according to our sliding window analysis. To determine which of these high variation genes or gene spacers could be utilized as accurate and trustworthy DNA barcodes in the genus Orchidaceae, more research is required.
We hypothesize that the nine Orchidaceae species' diverse chloroplast genome lengths may result from the expansion and contraction of the boundary between the SC region and the IR sections29. The results further demonstrated the existence of IR areas, as well as their extension and contraction, by comparing and evaluating the IR/SC boundary sections of the nine species of chloroplast genome.The findings demonstrate that all nine Orchidaceae species exhibit the characteristic chloroplast tetrad structure, which is structured in the form of two SC areas and two IR regions at regular intervals.
Mutation pressure30 and selection pressure31 are the main influencing factors leading to codon preference, but also affected by other factors, such as gene expression level32, gene length33 and tRNA abundance34, etc.Our findings demonstrated that codon usage bias was preserved across species in the Orchidaceae35, and that codon usage alterations play a significant role in the evolution of the cp genome. Additionally, the majority of codons preferred to end in A/U with RSCU1, indicating that the cp genome’s adaptive evolution may have contributed to some degenerate codon usage bias36. Additionally, all ENc values are greater than 54.85, and the values for CAI, CBI, and Fop are significantly lower than one, showing that all eleven species have very low codon use biases.Liu Jiangfeng selected 47 protein coding sequences from the garlic chloroplast genome of sickle wing, analyzed codon usage patterns, and found that codon preference was affected by selection and mutation, as well as some other influencing factors.
Previous studies showed that polymorphism at the SSR locus is useful in studying population genetics37,38,39.In this study, the minimum repeats of one, two, three, four, five, and six nucleotides were set to 8, 4, 4, 3, 3 and 3 using the MISA software. A total of 255 SSRs, including 226 SSRs made up of the A/T, AT/TA, AAT/ATT, AAAT/ATTT, and AATT/AATT, were found in Platanthera urceolata. This confirms that the SSRs in the chloroplast genome are primarily made up of short tandem repeats of the A and T, which is similar to the findings published for other plant chloroplast genomes. Previous research has shown that A/T repeat types predominate among all repeat units in many plant chloroplast genomes, and this phenomenon has also contributed to the extremely high AT content in chloroplast genomes40.
Numerous studies have demonstrated that understanding the adaptive genetic evolution of the chloroplast genome is crucial to understanding changes in gene structure and function41. It is common practice to utilize the ratio of non-synonymous substitution rates (Ka) to synonymous substitution rates (Ks) as a measure of selection pressure between various species at the sequence level42. In most genes in an organism, synonymous nucleotide substitutions occur more frequently than non-synonymous changes; as a result, Ka/Ks values are typically lower than one43. This study’s findings indicated that 10 genes had undergone positive selection. Rpl22, RpoC1, RpoC2, and Rps18 were connected to gene expression among these genes.Similarly to the findings of most plant research, the photosynthesis-related genes atpF, ndhD, ndhE, and ndhH as well as additional functional genes accD and ycf1 all showed Ka/Ks greater than 1 in most species, indicating the presence of positive selection pressure on these genes.
Numerous scientists have conducted in-depth phylogenetic analyses of chloroplast genome sequences in recent years. This is critical to our comprehension of how angiosperms evolved from other organisms44,45. Complete chloroplast genomes have been utilized by several researchers to examine the evolutionary relationships and relatedness of plants46,47. For this study, 17 published orchid chloroplast sequences were chosen in order to better understand the evolutionary links among orchids. The phylogenetic relationships of orchids were modeled using Aloe vera and Aloe maculata as outgroups.The findings suggest that the species were split into two groups. Galearis roborowskyi, Neottianthe cucullata, Neottianthe monophylla, Platanthera urceolata and Ponerorchis compacta all belonged to the same group, and the clustering map made it evident how closely related the Orchidaceae species are to one another. This study gives some theoretical support for the detailed investigation of the phylogeny of orchids and demonstrates the effectiveness of the chloroplast genome in separating out the phylogenetic links of orchid species. In this paper, nine complete orchid chloroplast genomes are revealed.
Conclusions
An examination of the cp genome sequences of 17 species of orchids revealed that their cp genome organization, gene sequencing, codon use, and repetitive sequence traits are remarkably comparable.In general, these structures are conserved, although the constriction and infrared regions are observed for the expansion of this region associated with plastid sequence variation. Analysis of positive selection of genes in the chloroplast genome of this family suggests that atpF, ndhD, ndhE, and ndhH may play a role in the growth of most Orchaceae species to strongly light environments.These genomic data provide new insights into the interspecific relationships of the Orchidaceae species. The phylogenetic analysis of the chloroplast genome’s single-copy genes revealed that 19 species may be separated into two groups, which offers some theoretical support for a thorough investigation of the phylogeny of the orchidaceae. This study sets the foundation for further exploration of the taxonomic, phylogenetic and evolutionary history of Orchidaceae.
Data availability
Further questions can be sent to the respective authors, whose original contributions are mentioned in the article/supplementary material. The chloroplast genome sequence mentioning species has been uploaded to NCBI with accession numbers shown below:
Species | Accession number | |
|---|---|---|
1 | Galearis roborowskyi | PRJNA997927 |
2 | Ponerorchis compacta | PRJNA998340 |
3 | Neottia puberula | PRJNA998753 |
4 | Neottianthe cucullata | PRJNA999017 |
5 | Ponerorchis chusua | PRJNA999044 |
6 | Herminium monorchis | PRJNA999615 |
7 | Platanthera urceolata | PRJNA999050 |
8 | Neottianthe monophylla | PRJNA999948 |
9 | Malaxis monophyllos | PRJNA999172 |
The sequence of other closely related and outer related species used in the analysis were downloaded from NCBI with the following accession numbers:
Species | Accession number | |
|---|---|---|
1 | Aloe maculata | NC_035505.1 |
2 | Aloe vera | NC_035506.1 |
3 | Bletilla sinensis | NC_060362.1 |
4 | Changnienia amoena | NC_045402.1 |
5 | Galearis cyclochila | NC_046818.1 |
6 | Hancockia uniflora | OK012601.1 |
7 | Hemipilia yajiangensis | NC_067080.1 |
8 | Hetaeria anomala | MW589524.1 |
9 | Hetaeria oblongifolia | MW589525.1 |
10 | Hetaeria youngsayei | MW589526.1 |
11 | Neottia japonica | NC_041446.1 |
12 | Neottia ovata | NC_030712.1 |
13 | Neottia suzukii | NC_041447.1 |
14 | Pecteilis hawkesiana | NC_082102.1 |
15 | Platanthera chlorantha | NC_044626.1 |
16 | Platanthera japonica | NC_037440.2 |
17 | Platanthera ussuriensis | MN686021.1 |
18 | Ponerorchis gracilis | NC_046810.1 |
19 | Thrixspermum centipeda | NC_054174.1 |
References
Van den Berg, C. et al. An overview of the phylogenetic relationships within Epidendroideae inferred from multiple DNA regions and recircumscription of Epidendreae and Arethuseae (Orchidaceae). AJB 92, 13–24. https://doi.org/10.3732/ajb.92.4.613 (2005).
Mendonca, M. P. & Lins, L. V. Revisao das Listas das Especies da Flora eda Fauna Ameaçadas de Extincao do Estado de Minas Gerais. Fund. Biodivers. BeloHoriz. 12, 33–45 (2007).
Cameron, K. M. et al. A phylogenetic analysis of the Orchidaceae: Evidence from rbcL nucleotide. AJB 86, 8–24. https://doi.org/10.2307/2656938 (1999).
Van den Berg, C. et al. A phylogenetic study of Laeliinae (Orchidaceae) based on combined nuclear and plastid DNA sequences. AOB 104, 17–30. https://doi.org/10.1093/aob/mcp101 (2009).
Haoyu Liu, Likuan Liu, Zhenglei Wang, et al. Progress in chloroplast genome of orchid aceae . Wild plant resources in China, 42(07): 73–79 (2023). (刘浩宇,刘力宽,王郑雷等.兰科植物叶绿体基因组研究进展 .中国野生植物资源, 2023, 42(07): 73–79.)
Lin, C. S. et al. The location and translocation of ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 5, 9040 (2015).
Brunkard, J. O., Runkel, A. M. & Zambryski, P. C. Chloroplasts extend stromules independently and in response to internal redox signals. PNAS 112, 10044–10049. https://doi.org/10.1073/pnas.1511570112 (2015).
Birky, C. W. Jr. The inheritance of genes in mitochondria and chloroplasts: Laws, mechanisms, and models. Annu. Rev. Genet. 35(1), 125–148 (2001).
Wolfe, K. H., Li, W. H. & Sharp, P. M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84(24), 9054–9058 (1987).
Raubeson, L. A. & Jansen, R. K. “Chloroplast genomes of plants”, in plant diversity and evolution: Genotypic and phenotypic variation in higher plants. Taxon 10, 45–68. https://doi.org/10.1079/9780851999043.0000 (2011).
Shetty, S. M. et al. Complete chloroplast genome sequence of corroborates structural heterogeneity of inverted repeats in wild progenitors of cultivated bananas and plantains. Plant Genome-Us https://doi.org/10.3835/plantgenome2015.09.0089 (2016).
Cai, J. et al. Complete plastid genome sequencing of four Tilia species (Malvaceae): A comparative analysis and phylogenetic implications. PLoS One https://doi.org/10.1371/journal.pone.0142705 (2015).
Porebski, S., Bailey, L. G. & Baum, B. R. Modifcation of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 15(1), 8–15. https://doi.org/10.1007/BF02772108 (1997).
Dierckxsens, N., Mardulyn, P. & Smits, G. NOVOPlasty: Denovo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4), e18–e18. https://doi.org/10.1093/nar/gkw955 (2016).
Qu, X.-J., Moore, M. J., Li, D.-Z. & Yi, T.-S. PGA: A software package for rapid, accurate, and fexible batch annotation of plastomes. Plant Methods 15(1), 50. https://doi.org/10.1186/s13007-019-0435-7 (2019).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic acids Res. 32(1), 11–16. https://doi.org/10.1093/NAR/GKH152 (2004).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1), W54-7. https://doi.org/10.1093/nar/gkw413 (2016).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47(W1), W59-64. https://doi.org/10.1093/nar/gkz238 (2019).
Brudno, M. et al. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 19(suppl_1), i54-62. https://doi.org/10.1093/BIOINFORMATICS/BTG1005 (2003).
Lin, D. et al. Codon usage variation of Zika virus: The potential roles of NS2B and NS4A in its global pandemic. Virus Res. 247, 71–83. https://doi.org/10.1016/j.virusres.2018.01.014 (2018).
Saitou, N. & Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4(4), 406–425. https://doi.org/10.1093/OXFORDJOURNALS.MOLBEV.A04045 (1987).
Zhang, H., Li, C., Miao, H. & Xiong, S. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PloS One 8(11), e80508. https://doi.org/10.1371/journal.pone.0080508 (2013).
Choi, K. S. et al. Two Korean endemic Clematis chloroplast genomes: Inversion, reposition, expansion of the inverted repeat region, phylogenetic analysis, and nucleotide substitution rates. Plants 10(2), 397. https://doi.org/10.3390/plants10020397 (2021).
Young-Kee, K. et al. Extensive losses of photosynthesis genes in the plastome of a Mycoheterotrophic Orchid, Cyrtosia septentrionalis (Vanilloideae: Orchidaceae). Genome Biol. Evol. 11(2), 565–571 (2019).
Kim, Y. K. et al. Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences. Front. Plant Sci. https://doi.org/10.3389/fpls.2020.00022 (2020).
Thomas, B., Maria, K. & Saša, S. Plastid genome evolution across the genus Cuscuta (Convolvulaceae): Two clades within subgenus Grammica exhibit extensive gene loss. J. Exp. Bot. 64(4), 977–989 (2013).
Logacheva, M. D. et al. The plastid genome of mycoheterotrophic monocot Petrosavia stellaris exhibits both gene losses and multiple rearrangements. Genome Biol. Evol. 6(1), 238–246 (2014).
Kim, Y. K., Park, C. W. & Kim, K. J. Complete chloroplast DNA sequence from a Korean endemic genus, Megaleranthis saniculifolia, and its evolutionary implications. Mol. Cells 27, 365–381. https://doi.org/10.1007/s10059-009-0047-6 (2009).
Millen, R. S. et al. Many parallel losses of infA from Chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. https://doi.org/10.1105/tpc.13.3.645 (2001).
Héctor, R., Alejandro, Z. & Héctor, M. Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 28(10), 2084–2090 (2000).
Bierne, N. & Eyrewalker, A. The problem of counting sites in the estimation of the synonymous and nonsynonymous substitution rates: Implications for the correlation between the synonymous substitution rate and codon usage bias. Genetics 165(3), 1587–1597 (2003).
Collins, J. J. et al. Noise in eukaryotic gene expression. Nature 422(6932), 633–637 (2003).
Ingvarsson, P. K. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol. Biol. Evol. 24(3), 836 (2007).
Moriyama, E. N. Codon usage bias and tRNA abundance in Drosophila. J. Mol. Evol. 45(5), 514–523 (1997).
Wang, W. & Messing, J. High-throughput sequencing of three lemnoideae (Duckweeds) chloroplast genomes from total DNA. PloS One 6, 659–670. https://doi.org/10.1371/journal.pone.0024670 (2011).
Yan, C., Du, J., Gao, L., Li, Y. & Hou, X. The complete chloroplast genome sequence of watercress (Nasturtium ofcinale RBr): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. https://doi.org/10.1016/j.gene.2019.02.075 (2019).
Grassi, F. et al. Chloroplast SSR markers to assess DNA diversity in wild and cultivated grapevines. Vitis-Geilweilerhof- 41(3), 157–158 (2002).
Provan, J. et al. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trend Ecolevolut. 16(3), 142–147 (2001).
Kuang, D. Y. et al. Complete chloroplast genome sequence of magnolia kwangsiensis (magnoliaceae): Implication for DNA barcoding and population genetics. Genome 54(8), 663–673 (2011).
Nie, X. J. et al. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS One 7, e36869. https://doi.org/10.1371/journal.pone.0036869 (2012).
Nei, M. & Kumar, S. Molecular evolution and phylogenetics. OUnP https://doi.org/10.1007/BF02772108 (2000).
Wang J. Study on the evolution of chloroplast genome of Hippophae, Thesis for M.S, Chinese Academy of Forestry Sciences, Supervisor: Zeng Y.F., pp.3–4. (王婧, 2020, 沙棘属植物叶绿体基因组的进化研究, 硕士学位论文, 中国林业科学院, 导师: 曾艳飞, pp.3–4) (2020).
Makalowski, W. & Boguski, M. S. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2820 orthologous rodent and human sequences. Proc. Natl. Acad. Sci. 95(16), 9407–9412. https://doi.org/10.1073/PNAS.95.16.9407 (1998).
Leebens-Mack, J. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the Felsenstein zone. Mol. Biol. Evol. 22, 1948–1963. https://doi.org/10.1093/MOLBEV/MSI191 (2005).
Moore, M. J. et al. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. PNAS 104, 19363–19368. https://doi.org/10.1073/pnas.0708072104 (2007).
Barrett, C. F. Plastid genomes reveal support for deep phylogenetic relationships and extensive rate variation among palms and other commelinid monocots. New Phytol. 209, 855–870. https://doi.org/10.1111/nph.13617 (2016).
Bock, D. G. et al. Genome skimming reveals the origin of the Jerusalem artichoke tuber crop species: Neither from jerusalem nor an artichoke. New Phytol. 201, 1021–1030. https://doi.org/10.1111/nph.12560 (2014).
Funding
This research was funded by [the Science and Technology International Cooperation Program] grant number [2022-HZ-815]. We are very grateful for their generous funding.
Author information
Authors and Affiliations
Contributions
J.D. and L.L. both contributed equally to this work, and they are both listed as the first authors. The main data search and processing effort was done by L.L. and Z.L., while J.D. worked on the manuscript authoring. The concepts for the manuscript were put forth by W.Z. and Z.W., who also revised the document. J.L. and Y.Z. provided insightful feedback on the draft. The manuscript’s published form was approved by all authors after they had read it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, L., Du, J., Liu, Z. et al. Comparative and phylogenetic analyses of nine complete chloroplast genomes of Orchidaceae. Sci Rep 13, 21403 (2023). https://doi.org/10.1038/s41598-023-48043-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-48043-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
