
Abstract
Abscisic acid (ABA) has been shown to mitigate the deleterious effects of abiotic stresses and to regulate plant growth and development. Salinity is one of the important abiotic stresses affecting plant cell metabolism and physiology, which causes serious damages to crops. In this study, we investigated the protective role of exogenous ABA on leaves in response to salinity stress using rice seedlings (two leaf-one heart) subjected to three treatments: ZCK (control), ZS (50 mM NaCl), and ZSA (5 mg L–1 ABA + 50 mM NaCl). We carried out transcriptomic and metabolomic analyses to identify the molecular mechanisms by which ABA protects plants against salt stress. Results showed that 1159 differentially expressed genes (DEGs) (916 up-regulated, 243 down-regulated) and 63 differentially accumulated metabolites (DAMs) (42 up-regulated, 21 down-regulated) were identified between the ZS and ZSA treatments, respectively. In addition, ABA pretreatment regulated the expression pattern of genes responsible for oxidation redox, starch and sucrose metabolism, and phenylpropanoid biosynthesis. The combined transcriptomic and metabolomic analysis revealed that 16 DEGs and 2 DAMs were involved in Flavonoid biosynthesis and 8 DEGs and 2 DAMs were involved alpha-Linolenic acid metabolism which are responsible for salinity stress tolerance through induced by exogenous ABA. Overall, ABA could enhance rice leaves growth and development mainly by regulating flavonoid biosynthesis and linoleic acid metabolism pathway.
Similar content being viewed by others

Introduction
Rice (Oryza sativa L.) is the most important staple food crops for more than half of the world population. As a salt-sensitive plant, the growth and production of rice is decreasing due to salinity. In fact, salt stress is the most important stress factor among abiotic stresses that not only leads to an imbalance in the Earth’s climatic conditions, but also has destructive effects on plant growth and physiological processes. Salinity in the soil adversely affects root growth and reduces the ability of plants to absorb water and other nutrients from the soil, thus causing delayed plants growth even ultimately lead to plants death. Actually, salt stress leads to membrane damage and stomatal closure, which results in reduced carbon dioxide uptake, hydrolase activity, and increased lipid peroxidation levels, which may stimulate a large number of formation of ROS. Admittedly, antioxidant defense system (mainly includes SOD, POD, CAT, APX) could be effectively eliminated ROS and maintain appropriate equilibrium within the cells 9, which are contributed to improve salt tolerance.
As important small molecules in regulating metabolism, hormones play a crucial role in the response to abiotic stresses1. Abscisic acid (ABA) is a vital endogenous plant hormone which is usually involved in stimulating various aspects of plant growth and development, such as seed dormancy and germination, root structure, leaf senescence, and vegetative growth2. ABA reduces stress and is essential in salt-stressed conditions due to its significant impact on the phenotypic, metabolomic, and transcriptomic responses of plants as well as high efficiency of genes responsible for flavonoid metabolism3. We previous study have demonstrated that exogenous ABA to rice was beneficial to protect membrane lipid peroxidation, the modulation of antioxidant defense systems and endogenous hormonal balance with imposition to salt stress4. In addition to a series of physiological events, many studies also focus on its molecular mechanism at transcriptomic and metabolomics level. Transcriptome analysis showed that ABA control the primary root growth in response to salt stress via inducing the expression of EXPANSIN genes, further investigations indicated that ABA exerts these effects largely through ABA signaling5. Previous study showed that exogenous ABA highly up-regulated the genes involved in the lipid and fatty acid metabolisms and cytoplasmic transport in shoots of rice under salt stress, suggesting that enhanced lipid metabolism might also contribute to enhanced salt tolerance of rice6. Actually, a plenty of genes that confer salt tolerance were reported in recent decades. However, the levels of salt tolerance differ greatly depending on growth conditions, and mechanisms underlying the complicated nature of stress tolerance are far from being fully understood. Additionally, an intrinsic association between genes and metabolites may also exist following salt stress injury.
The inbred rice variety "Huanghuazhan" is an elite indica rice cultivar, which has been grown over 4.5 million ha in southern China, it is a semi-dwarf, super high yield, good eating quality and highly adapt to vast areas of nine provinces in China, and twenty cultivars have been derived from it7. The present study investigated that exogenous ABA on leaves (foliar spraying) in response to salt stress using rice variety ‘Huanghuazhan’ (two leaf-one heart). We carried out transcriptomic and metabolomic analyses to identify the molecular mechanisms by which ABA protects plants against salt stress. By using omics technology, we identified specific genes, secondary metabolites that are targeted for flavonoids and alpha-Linolenic acid. In this regards, we have obtained valuable information for understanding the salt tolerance mechanisms of rice and for the effective control of salinity stress.
Results
Effects of ABA on morphological parameters of rice seedlings under salt stress
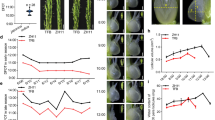
Salt stress significantly decreased rice plant height, stem diameter, leaf length and leaf area when compared with the control. The foliar ABA application significantly increased its stem diameter, leaf length and leaf area (Table 1). Figure 1 shows the phenotype of rice seedlings at the 10th d after salt stress.

Effects of ABA on morphology of rice seedlings after 10 d of salt stress. ZCK treatment of water, ZA treatment with ABA lonely, ZS treatment with NaCl, ZSA treatment with ABA + NaCl in Huanghuazhan. The current study mainly investigate ZCK, ZS and ZSA.
Salt stress significantly decreased root length, root surface area, root volume and root average diameter in rice when compared with the control. Foliar spraying of ABA greatly increased these root morphology indicators (Table 2).
Effects of ABA on rice seedling biomass under salt stress
Compared with CK, salt stress significantly reduced the shoot dry weight and root dry weight of rice. ABA + NaCl significantly increased its shoot dry weight and root dry weight compared with the salt treatment alone (Fig. 2).

Effects of ABA on shoot dry weight (A) and root dry weight (B) of rice seedlings under salt stress. Different letters indicate significant difference at the five percent significant level according to Duncan’s multiple range tests.
DEGs and DAMs induced by ABA pretreatment in response to salt stress
To investigate the effects of ABA treatment on gene transcription under salt stress, mRNA sequencing was performed. Nine samples were sequenced through BGISEQ. An average of 95.08% clean reads were generated in each sample, corresponding to a total of 6.46G Gb clean bases. Total 83.18% to 84.37% of clean reads were mapped to reference genome (Table S2). We identified 165 DEGs (24 up and 141 down) by comparing NaCl and CK treatments, while 1159 DEGs (916 up and 243 down) were identified by comparing the ABA + NaCl and NaCl treatments (Fig. 3). These results suggested that the genes expressed to exogenous ABA would be considered important candidates gene for further analysis.

Venn diagram (A) and the volcano maps (B,C) of DEGs under salt stress and ABA treatment. The X axis represents the log2 transformed difference-fold value, and the Y axis represents the-log10 transformed significance value. Red represents up-regulated DEG, blue represents down-regulated DEG, and gray represents non-DEG.
GO analysis of the DEGs revealed that control, NaCl, and NaCl + ABA treatments enriched GO terms biological processes, cellular components and molecular function (Fig. 4). In the category of biological process, the DEGs of the control and salinity treatment, salinity and NaCl + ABA treatment group mainly distributed in cellular, metabolic process and response to stimulus while the DEGs in all the treatments had the highest proportion in cell and cell part in the category of cellular components. Additionally, DEGs mainly distributed in binding and catalytic activity in the category of molecular function (Fig. 4).

GO classification of differentially expressed genes between the control and NaCl group (A), NaCl and NaCl + ABA treatment group (B) after 72 h of foliar sprayed ABA.
GO annotations were used to classify the possible functions of the rice genes. Enriched analysis were performed to obtain Q-value (≤ 0.05 significant) through the phy.per function in the R software. The GO enrichment analysis of 165 and 1159 DEGs in ZCK vs. ZS and ZS vs. ZSA, respectively, classified GO into three categories such as biological process (BP), cellular component (CC), and molecular function (MF). GO analysis of the DEGs indicated significant enriched GO terms in BP, CC and MF (Table 3). GO terms in the MF and BP were significantly enriched in ZCK vs. ZS and ZS vs. ZSA. Similarly, compared to ZCK vs. ZS, the number of enriched GO terms in the MF and BP was found higher in ZS vs. ZSA. These results indicated that the application of ABA plays an important role in regulating MF and BP gene expression under salinity stress. Additionally, combined ZCK vs. ZS and ZS vs. ZSA, the number of GO terms in the BP category were found higher among the three categories. It is obvious that the biological process related to defense response, catabolic process and carbohydrate metabolism were most influenced by salinity stress.
Furthermore, we performed a KEGG pathway analysis for DEGs. The 165 DEGs (ZCK vs. ZS) with KEGG annotation were assigned to 18 pathways and 1159 DEGs (ZS vs. ZSA) were assigned to 21 KEGG pathways. The annotation results were classified by pathway type and it was observed that the pathways of DEGs were basically related to metabolism (Fig. 5). Comparative analysis of ZCK vs. ZS and ZS vs. ZSA, showed that DEGs were significantly differ in environmental adaptation, carbon metabolism processes and biosynthesis of other secondary metabolites. We explored key genes differentially expressed in rice leaves after salt stress and ABA treatment from these metabolic pathways.

KEGG classification of differentially expressed genes between the control and NaCl group (A), NaCl and NaCl + ABA treatment group (B) after 72 h of foliar sprayed ABA.
For the KEGG enrichment analysis, pathways with a Q value ≤ 0.05 were regarded as significantly changed in response to salinity stress with or without ABA treatment. The top 20 enriched KEGG pathways are presented in Fig. 6. In ZCK vs. ZS, the pathways among the DEGs including plant-pathogen interaction (Os04626), with 13 genes down-regulated in ZS (Supplementary Table S3), Monobactam biosynthesis (Os00261), with 2 genes down-regulated in ZS, and Lysine biosynthesis (Os00300), with 2 genes down-regulated in ZS (Supplementary Table S4), were the major enriched pathways among the DEGs. In ZS vs. ZSA, including Glutathione metabolism (Os00480), with 19 genes (17 genes up-regulated and 2 genes down-regulated in ZSA) (Supplementary Table S5), phenylpropanoid biosynthesis (Os00940), with 43 genes (39 genes up-regulated and 4 genes down-regulated in ZSA) (Supplementary Table S6), Flavonoid biosynthesis (Os00941), with 16 genes (with 13 genes up-regulated and 3 genes down-regulated in ZSA) (Supplementary Table S7), Photosynthesis-antenna proteins (Os00300), with 4 genes down-regulated in ZSA, alpha-Linolenic acid metabolism (Os00592), with 10 genes (8 genes up-regulated and 2 genes down-regulated in ZSA) (Supplementary Table S8) were the major enriched pathways among the DEGs.

Functional analysis of DEGs based on KEGG pathway annotations. Pathways with a Q-value ≤ 0.05 that were significantly enriched in DEGs between the control and NaCl group (A), NaCl and NaCl + ABA treatment group (B) after 72 h of foliar sprayed ABA, were analyzed with the KEGG database.
Confirmation of gene expression patterns by qRT-PCR
The expression patterns of randomized eight candidate genes that related to phenylpropanoid biosynthesis, starch and sucrose metabolism, alpha-Linolenic acid metabolism, and JA biosynthesis were examined by qRT-PCR to validate the RNA-sequencing results (Fig. 7; Supplementary Table S9). The expression patterns determined by qRT-PCR were compared with those of the RNA-seq assay. The qRT-PCR and RNA-seq results showed a positive correlation coefficient (Pearson coefficient R2 = 0.910), indicating that the transcriptome data were able to reflect transcript abundance in our study.

Linear regression between the levels of qRT-PCR data and transcript expression.
DAM analysis of ABA pretreatment in response to salinity stress
We performed metabolite profiling of rice samples (ZCK, ZS, and ZSA) to assess the overall metabolic effects of pretreatment with ABA on rice seedling leaves under salt stress. Defined the metabolites that differential fold change (FC) ≥ 1.2 or FC ≤ 0.83 and Variable Importance in the Projectior (VIP) ≥ 1 as differential metabolites. 47 differential metabolites were detected in ZCK vs. ZS, with 25 up-regulated and 22 down-regulated, the levels of 1-(3,5-Dihydroxyphenyl)-12-hydroxy-2-tridecanyl acetate, Tetranor-12r-hete and 4-Benzyl-N-(3,5-dimethyl-4-isoxazolyl) tetrahydro-1(2H)-pyrazinecarboxamide, among others, increased at least two fold, whereas D-Sphingosine, 2-Naphthalenesulfonic acid, Rehmannioside C and 3,4,5-Trimethoxycinnamic acid, among others, decreased under salt stress. Between the ZS and ZSA treatments, 63 differential metabolites were detected, with 42 up-regulated and 21 down-regulated; Cyclamic acid, 12-Hydroxydodecanoic acid, 1-(1-Hydroxybutyl)-1,3,4,5,6,7-hexahydro-2-benzofuran-4,5,6,7-tetrol and Azelaic acid, among others, showed relatively high levels in ABA + NaCl treatment, whereas Eriodictyol, Glycerophospho-N-palmitoyl ethanolamine, Leucoside, among others, showed relatively low levels (Fig. 8) (Supplementary Table S10).

Volcano plot of different metabolites screened for different comparison groups (A,B). The ordinate log_t.test_p.value represents the P-value value of the-log10 for the t test, and the abscissa log2FC represents the log2 for the value of the FC of the difference multiple. Heatmap of the differential metabolites for the different comparison groups (C). The color from green to red indicates the expression abundance of metabolites from low to high phy.per.
All the differential metabolites were subjected to hierarchical clustering (Fig. 8C), we could obviously detected the differential accumulated between the two comparison groups.
Complex metabolic responses in organisms are generally influenced and regulated by multiple genes and proteins, ultimately leading to systemic changes in the metabolome. To identify the main pathways involved in the leaves of rice seedlings under salt stress that response to ABA, we mapped the DAMs to KEGG pathways and the metabolic pathway enrichment results were obtained (Fig. 9). 47 significant DAMs in ZCK vs. ZS were assigned to 28 KEGG pathways (Supplementary Table S11), including Alanine, aspartate and glutamate metabolism (00250), Butanoate metabolism (00650), Metabolic pathways (01100), Aminoacyl-tRNA biosynthesis (00970), Phenylalanine metabolism (00360), Glyoxylate and dicarboxylate metabolism (00630), Arginine and proline metabolism (00330), Carbon metabolism (01200), Biosynthesis of amino acids (01230) and 2-Oxocarboxylic acid metabolism (01210), among others. In ZS vs. ZSA, 63 significant DAMs were assigned to 16 KEGG pathways (Supplementary Table S12), including Metabolic pathways (01100), Valine, leucine and isoleucine biosynthesis (00290), Biosynthesis of amino acids (01230), Biosynthesis of secondary metabolites (01110), 2-Oxocarboxylic acid metabolism (01210), C5-Branched dibasic acid metabolism (00660), alpha-Linolenic acid metabolism (00592), Flavone and flavonol biosynthesis (00944) and Flavonoid biosynthesis (00941), among others.

Functional analysis of DAMs based on KEGG pathway annotations. Pathways with a Q-value ≤ 0.05 that were significantly enriched in DAMs between the control and NaCl group (A), NaCl and NaCl + ABA treatment group (B) after 72 h of foliar sprayed ABA, were analyzed with the KEGG database.
ABA regulated oxidation-related gene expression to protect rice leaves from salt stress
Antioxidant enzymes could protect plants against oxidative damage induced by stress. In our study, some redox-related genes were detected, including glutathione hydrolase, glutathione S-transferase, peroxidase and hydrogen peroxidase, which participate in the removal of ROS. In ZCK vs. ZS, 11 genes were up-regulated in ZS, in contrast, 30 genes (Os01g0372400, Os01g0374000, Os09g0367700, Os10g0527400, Os04g0651000, Os11g0112200, particularly) were up-regulated and 3 genes (Os03g0368900, Os01g0151400, Os01g0151500) were down-regulated in ZSA. 18 genes (10 genes down-regulated and 7 genes up-regulated in ZS, 17 genes up-regulated and 1 gene down-regulated in ZSA) were also assigned to hydrogen peroxide catabolic process (GO:0042744) (Supplementary Tables S5, 6 and S13).
Differential expression of starch and sucrose metabolism genes
Sucrose and starch are widely distributed in various plant tissues, and they are important transportation and storage substances of plant carbohydrate, especially their mutual transformation. All starch and sucrose genes expression levels were almost significantly down-regulated after salinity stress without ABA treatment, including five genes associated with glucan endo-1,3-beta-glucosidase 3, one trehalose 6-phosphate phosphatase and two probable alpha, alpha-trehalose-phosphate synthase [UDP-forming] 10. Conversely, ABA treatment prominently up-regulated these genes in the presence of salinity. Besides, we detected two sucrose synthase genes, sucrose synthase 7 and 5, which were obviously up-regulated in ZS vs. ZSA (Table 4), indicated that exogenous ABA enhanced the synthesis and accumulation of sucrose in photosynthesis process, which contributes to maintain normal growth under salt stress.
ABA regulated genes and metabolites involved in Flavonoid biosynthesis and JA biosynthesis to promote rice leaf development
A combined transcriptomic and metabolic analysis can better explain the transcriptional regulation of metabolic pathways. We simultaneously mapped differentially expressed genes and metabolites from the same treatment groups to KEGG pathways to elucidate the relationships between genes and metabolites. Between ZS and ZSA, differentially expressed genes and metabolites were assigned to Flavonoid biosynthesis (00941) and alpha-Linolenic acid metabolism (00592). In the Flavonoid biosynthesis (00941) pathway, we detected 16 genes (with 13 genes up-regulated and 3 genes down-regulated) and two metabolites (Eriodictyol and Luteolin) in ZSA, specially, the gene flavonoid 3'-monooxygenase CYP75B3-like(Os10g0320100) and the levels of Eriodictyol and Luteolinwere down-regulated (Supplementary Tables S7; S10 and Fig. S1; Fig. 10). Additionally, the genes probable 2-oxoglutarate-dependent dioxygenase At5g05600, flavanone 3-dioxygenase 2-like (Os03g0289800, Os04g0581000 and Os05g0127500) that catalyse an early step in the flavonoid biosynthesis pathway leading to the production of flavanols and anthocyanins, which were all up-regulated in ZSA, indicating that ABA may mitigate the effect caused by salinity via stimulating the flavonoid biosynthesis pathway.

Coordinate up and down-regulation of genes involved in Flavonoid biosynthesis. The transcripts in Flavonoid biosynthesis pathway were up-regulated in ZS vs. ZSA. Green rectangle boxes indicated down-regulated genes and the red indicated up-regulated.
In the alpha-Linolenic acid metabolism (00592) pathway, we detected 8 genes up-regulated, 2 genes (Os02g0194700, Os10g0562200) down-regulated in ZSA, as well as two metabolites (12-Oxo phytodienoic acid, 13(S)-HOTrE) levels were up-regulated (Fig. S2; Supplementary Tables S8 and S10), suggested that lipoxygenase 2.3, chloroplastic (EC:1.13.11.12) negatively regulated the 13(S)-HOTrE level, and the gene chloroplast envelope quinone oxidoreductase homolog (Os04g0372700) positively regulated the 12-Oxo phytodienoic acid level. Jasmonic acid (JA) are a class of fatty acid derivatives that are ubiquitous in plants. As signaling molecules, JA can not only effectively mediate plant defense responses to pathogens, herbivores and abiotic stresses, but also regulate plant growth and development, such as root elongation, carbohydrate accumulation and fruit ripening. In JA biosynthesis pathway (Fig. 11), we detected that phospholipase A1-Ibeta2, chloroplastic gene (Os10g0562200), which catalyzed the reaction of phosphatidylcholine into alpha-Linolenic acid, were down-regulated, as well as JA biosynthesis-related gene lipoxygenase 2.3, chloroplastic (Os02g0194700). However, we detected another six JA biosynthesis-related genes (putative 12-oxophytodienoate reductase 1–6, Os06g0215500, Os06g0215600, Os06g0215900, Os06g0216000, Os06g0216200, Os06g0216300) were up-regulated (Supplementary Table S8), indicating that ABA may mitigate the effect caused by salinity stress via stimulating the lipid metabolism and regulate JA biosynthesis pathway.

Coordinate the metabolites involved in JA biosynthesis pathway. The bright line and frame represent the synthetic pathway of JA in the metabolism patghway of α-Linolenic acid.
Discussion
High salinity causes impaired plant metabolism and physiological activities due to reduced osmotic potential, ion toxicity and nutritional imbalance, leading to organelle damage, disruption of photosynthesis, respiration, and protein synthesis8. Therefore, increased plant stress resistance and defense mechanisms is critical for sustainable crop production in salt-affected areas. An important role of ABA in a wide variety of plants is to improve the tolerance to stresses such as drought, salinity, cold, and heat9,10,11, which is considered an important signaling molecule that can trigger signaling and regulatory mechanisms to adverse stress. The molecular mechanisms underlying plant responses to ABA treatment under salinity are poorly understood. To characterize the enriched pathways and genes affected during salt and ABA treatment, we performed transcriptomic and metabolomic analyses in rice leaves. We identified DEGs and DAMs in salt-treated and ABA-treated rice leaves and investigated significantly enriched biological processes and pathways by GO and KEGG analysis. The present findings suggested that many phenylpropanoid biosynthesis-related genes, Flavonoid biosynthesis-related genes, and alpha-Linolenic acid metabolism-related genes involved in ABA signaling pathway in salt-stressed rice leaves. Similar results were obtained by Yang et al.3. Recently, more and more studies have pointed out that plant secondary metabolites involved in plant defense response to abiotic stress. Previous study has demonstrated that some angiosperms can accumulate flavonoids and flavonol glycosides, responding to UV stress by absorbing UV-B, as well as accumulate anthocyanins to combat drought, cold, high light, and other stresses12,13.
Role of antioxidant systems in salinity stress
One of the molecular effects of compounds that enhance plant tolerance to abiotic stresses involves the activation of antioxidant processes. In the present study, we found some genes encoding key enzymes such as CAT, POD, S-transferase gene (GST), ascorbate peroxidase gene and glutathiredoxin (GRX), which were contributed to remove excess O2– and hydrogen peroxide and ensure plant survival14. Xie et al.15 found that 10 GSTs and 2 peroxidase genes were up-regulated in citrus roots, which participated in the salt resistance study of citrus roots. In the present study, we detected 7 and 10 peroxidase genes (including peroxidase 1, cationic peroxidase SPC4, peroxidase 70, etc.) were up-regulated and down-regulated, respectively under salt stress (Supplementary Table S6). However, most peroxidase genes were down-regulated in rice leaves under salt stress, which was opposite to some findings, this may be due to the fact that most of the peroxidase genes found in these studies belong to different members of the same gene family16. On the other hand, peroxidase belongs to a large gene family with more than 70 members in a plant species. Their expression levels and patterns were different in different tissues17. The down-regulated peroxidase genes may be due to that Huanghuazhan could reduce oxidative stress, or possibly because the up-regulated expression of these genes appears at post-transcriptional levels. Similar results were also obtained in the salt-sensitive rice variety IR2918 and IR6419. All of these results suggested that the antioxidant enzymes play an important role in multiple developmental processes. Additionally, we detected a blue copper protein (Os08g0482700), which is a ROS-scavenging signaling molecule and contributed to preventing the formation of a highly toxic OH-14, was up-regulated in ABA + NaCl treatment, all of these suggested that a strict regulation of the oxidative stress pathway to the salt stress response, and ABA could support the rice adaptation to salt stress via maintaining the redox status and improving salinity tolerance.
Role of flavonoids in salinity stress
Flavonoids are synthesized through the phenylpropanoid pathway to convert phenylalanine to 4-coumaroyl-CoA and eventually enter the flavonoid biosynthesis pathway. Our transcriptome results showed that the genes relate to phenylpropanoid pathway (00940): 4-coumarate-CoA ligase 2 (Os02g0697400) and 4-coumarate-CoA ligase 5 (Os08g0448000) were up-regulated under salt stress, which contributed to enhancing flavonoid biosynthesis (00941). Transcriptome analysis of the roots and stems of rice showed that salt stress significantly affected the metabolism of carbohydrates and amino acids, and induced the accumulation of secondary metabolites-related genes, phenolic salts, and flavonoids in roots20. Chandran et al.20 found that secondary metabolites-related genes such as flavonoid accumulation in rice roots could be induced under salt stress. In the present study, we detected 13 flavonoid biosynthesis-related genes (e.g. cytochrome P450 CYP73A100, probable 2-oxoglutarate-dependent dioxygenase, flavanone 3-dioxygenase 2-like, protein DMR6-LIKE OXYGENASE 1, etc.) were up-regulated in ABA + NaCl treatment, which is consistent with the result of qRT-PCR. Additionally, the up-regulation of cytochrome P450 CYP73A100 expression indicated that these proteins are required for the biosynthesis of several important compounds, such as hormones, defense-related products, and fatty acids21. Similar result was obtained in previous study22. Han et al.23 indicated that these up-regulated genes (cytochromeP450 76AD1, cytochromeP450 84A1, cytochromeP450 CYP73A100) may be involved in ABA signaling, implied that ABA signaling may be functionally important and exhibits a rapid response to Suaeda rigida early under salt stress. Exogenous ABA application prominently up-regulated cytochrome P450 CYP73A100, which may contribute to energy recovery, repair of cell membranes, and fatty acid synthesis.
It should be noted that we detected two important metabolites (Luteolin and Eriodictyol) were differentially accumulated. El-Shafey and AbdElgawad24 demonstrated that Luteolin could enhance the resistance of salt stress by enhancing the antioxidant systems in maize. In addition, the up-regulation of Flavonoid 3’hydroxylase (F3´H) expression in flowers of Taraxacum antungenseis consistent with the trend of Luteolin accumulation25. In the present study, the metabolic analysis showed that some DAMs were assigned to flavonoid biosynthesis (00941), and we detected that the gene flavonoid 3’-monooxygenase CYP75B3-like (Os10g0320100) and the levels of Eriodictyol and Luteolin were all down-regulated in ABA + NaCl treatment. Similar result was obtained by Song et al.26, who demonstrated that exogenous melatonin promoted Luteolin biosynthesis in pigeon pea through the CcPCL1 and CcF3´H-5 pathways under salt stress, and over-expression CcPCL1 and CcF3´H-5 in transgenic plants greatly improved resistance of salt stress by promoting Luteolin biosynthesis. Hence, we speculate that ABA enhanced rice tolerance to salt stress might mediate the accumulation of Luteolin and Eriodictyol through the gene flavonoid 3’-monooxygenase, CYP75B3-like (Os10g0320100), and this will be a focus of our future research.
Role of JA and α-linoleic acid pathway in salinity stress
Jasmonic acids (i.e. cis-OPDA, JA, JA-Ile, and methyl jasmonate) are the major phytohormone involved in the stress response27. In the present study, we detected six JA biosynthesis-related genes, putative OPR1 (Os06g0215500), OPR2 (Os06g0215600), OPR3 (Os06g0215900), OPR4 (Os06g0216000), OPR5 (Os06g0216200), OPR6 (Os06g0216300), which were all up-regulated in ABA treatment under salt stress, consistent with the confirmation of gene expression patterns by qRT-PCR, indicating that ABA may mediate the biosynthesis and signal transduction of JA plant hormones to promote the growth of rice under salt stress. Wang et al.6 showed that exogenous ABA significantly up-regulated genes related to lipid and fatty acid metabolism and cytoplasmic transport in the aerial parts of two genotypes of rice under salt stress, indicating that enhanced lipid metabolism may also contribute to improving salt tolerance in rice6. From the metabolic analysis, we detected the levels of two differentially metabolisms (12-OPDA and 13(S)-HOTrE) in the alpha-Linolenic acid metabolism (00592) pathway, which were regulated by lipoxygenase 2.3, chloroplastic (EC:1.13.11.12) and chloroplast envelope quinone oxidoreductase homolog (Os04g0372700), respectively, were significantly up-regulated in ABA + NaCl treatment, indicating that ABA may mitigate the effect caused by salinity stress via stimulating the lipid metabolism. On the other hand, some researches showed that JA synthesis-related genes were down-regulated in sweet Sorghum under salt stress28, or the negative expression of regulators of JA and ETH signaling was significantly up-regulated in S.alopecuroides root tissues, indicating that JA may have been a negative regulatory in S.alopecuroides roots in response to salt stress29. All of these results were also similar to our transcriptome analysis results. Additionally, the wheat OPR 1 gene (TaOPR1) has been shown to increase plant tolerance to high-salt stress by increasing MYC2 expression, thereby activating ABA-dependent signaling pathways30. All of these suggested that JA-biosynthesis related genes may play a key role in increasing salt tolerance, and this will be a focus of our future research.
Role of trehalose in salinity stress
Previous studies have shown that the trehalose pathway plays an important role in regulating the usage and distribution of sucrose, coordinating the source-reservoir relationships, and improving the effective utilization of carbohydrates31 and increased crop yield32. Overproduced trehalose has considerable potential to improve abiotic stress tolerance in transgenic rice plants, which showed higher levels of tolerance to salt, drought, and cold stress as compared with untransformed controls33. In higher plants, trehalose is catalyzed by two enzymes: trehalose-6-phosphate synthase (TPS) and trehalose-6-phosphate phosphatase (TPP). In a previous report, the expression levels of OsTPP1 and OsTPP2 were induced by exogenous ABA, which can improve the stress tolerance in rice34,35. Rahman et al.36 showed that OsTPP2, OsTPP3, OsTPP8 and OsTPP9 were up-regulated under salt, drought and cold treatments, while OsTPP1, OsTPP4, OsTPP5, OsTPP6, and OsTPP7 were down-regulated under salt stress. This clearly demonstrates their important role in abiotic stress tolerance. Additionally, GhTPS11 in cotton37, over-expressing IbMIPS138 in the sweet potato plants showed that ABA biosynthesis and signaling genes as well as trehalose synthesis-related genes were generally up-regulated. In our study, we detected that the expression levels of 3 TPS10 (Os05g0128900, Os05g0153500, Os08g0414700) and one TPP gene (Os12g0505800) associated with starch and sucrose metabolism pathway were down-regulated under salt stress, while ABA treatment significantly up-regulated these genes under salt stress. This may be due to that ABA is involved in protecting the plant from abiotic stress through osmoregulation, detoxification of ROS and stabilization of the quaternary structure of proteins39. Previous study showed that the cucumber genes were expressed in roots, stems and leaves, however, they were varied in both levels, indicating that the CsTPS genes were tissue-specific. In addition, CsTPS1 and CsTPS6 were found to be down-regulated upon salinity treatment, and reached the lowest expression levels at 24 h40. Thus, we speculate that the differentially expression levels of trehalose-related genes are related to different tissues. Vishal et al.41 detected that OsTPS8 may regulate suberin deposition and trehalose via ABA signaling, thus conferring salt tolerance in rice. These studies suggested a close relationship between trehalose and ABA in stress tolerance and growth and development.
Conclusion
Salinity stress adversely affects the growth and development of rice, and ABA could increase its tolerance to salt stress. Compared with ZCK vs. ZS, DEGs specific to ZS vs. ZSA was significantly enriched in carbohydrate metabolism. Exogenous ABA obviously increased the expression of starch and sucrose genes (sucrose synthase 5 and sucrose synthase 7), which contributed to the synthesis and accumulation of sucrose in photosynthesis process under salt stress. Some redox-related genes including glutathione hydrolase, glutathione S-transferase, peroxidase and hydrogen peroxidase, as well as many differential metabolites (particularly Cyclamic acid) that belong to organic acid and derivatives or lipid and lipid-like molecules, were also up-regulated in ZS vs. ZSA when compared with ZCK vs. ZS, which mitigated oxidative damage. In addition, ABA could enhance rice leaves growth and development by regulating flavonoid biosynthesis and linoleic acid metabolism pathway. ABA decreased the level of Eriodictyol and Luteolin and up-regulated the expression of six JA biosynthesis-related genes. ABA also altered the expression of other genes related to glutathione metabolism, phenylpropanoid biosynthesis, flavonoid biosynthesis, photosynthesis-antenna proteins and alpha-Linolenic acid metabolism. In conclusion, the results in this study elucidated that exogenous ABA alleviated salinity toxicity and enhanced rice leaf development via mediating expression of genes in different pathways, however, candidate genes need to be further verified for their function.
Materials and methods
Plant growth and treatment
In the current study, seeds of inbred rice variety Huanghuazhan (Z) were used as the plant materials. The seeds were supplied by Guangdong Tianhong Seed Company Limited, Zhanjiang and the ABA solution was provided by Sichuan Lomon Fusheng Technology Co., Ltd. Healthy and uniform seeds were sorted manually and sterilize with 2.5% sodium hypochlorite solution for 15 min, then thoroughly rinse with distilled water. 75 seeds were germinated in the dark for 2 days at 30 ℃ using plastic pots (19.5 × 14.5 × 17.5 cm) containing 3 kg of substrates (Latosol: sand = 3:1, v/v). The relative humidity was maintained at 75% throughout the experiment. Thirty pots per experimental plot were arranged in a split plot scheme in a completely randomized block design with three replications, resulting in a total of 90 pots. After 8 d of germination, when rice seedlings grew to two leaves and one heart, 5 mg L−1 ABA were applied to leaves as foliar spray until the leaf surface was moist4. Salt treatment was initiated to the plants at the same time by adding NaCl at a rate of 25 mM increment per day, to reach a final concentration of 50 mM in the solution. The other treatments included: (i) control (ZCK): 0 mg L−1 ABA + 0 mM NaCl, (ii) (ZS): 0 mg L−1 ABA + 50 mM NaCl, (iii) (ZSA): 5 mg L−1 ABA + 50 mM NaCl. The leaves samples were collected after 24 h of all salt stress treatments with three biological replicates, including 4 pots of sample per biological replicate. And then immediately frozen in liquid nitrogen and stored at − 80 °C for further laboratory analysis.
RNA extraction and transcriptome sequencing
According to protocol the total RNA was extracted using Trizol reagent kit (Invitrogen, USA). RNA quality and concentration were assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Eukaryotic mRNA was enriched by Oligo (dT) beads, after RNAs fragmentation. The enriched mRNA was fragmented into short segments using fragmentation and reverse transcribed into cDNA with random primers. Second-strand cDNA were synthesized by DNA polymerase I, RNase H, dNTP and buffer. Rolling cycle amplification was used to duplicate DNA molecules in the PCR reaction. DNA nanoball (DNB) was generated which contain multiple copies of DNA. Sufficient quality DNBs loaded into patterned nanoarrays using high-intensity DNA nanochip technique and sequenced through combinatorial Probe-Anchor Synthesis (cPAS). Sequenced using Illumina HiSeq2500 by BGI-Shenzhen (Shenzhen, China).
Sequence assembly, annotation and classification
The sequencing data were processed by SOAPnuke42 by (1) Removing reads containing sequencing adapter; (2) Removing reads whose low-quality base ratio (base quality less than or equal to 15) is more than 20%; (3) Removing reads whose unknown base (‘N’ base) ratio is more than 5%, afterwards clean reads were obtained and stored in FASTQ format. The subsequent analysis and data mining were performed on Dr. Tom Multi-omics Data mining system (https://biosys.bgi.com). The clean reads were mapped to the reference genome using HISAT243. After that, Ericscript (v0.5.5)44 and rMATS (V3.2.5)45 were used to detect fusion genes and differential splicing genes (DSGs), respectively. Bowtie246 was applied to align the clean reads to the gene set, a database built by BGI (Beijing Genomic Institute in ShenZhen), in which known and novel, coding and noncoding transcripts were included. The Expression level of gene was calculated by RSEM (v1.3.1)47. The heatmap was drawn by pheatmap (v1.0.8)48 according to the gene expression difference in different samples. Essentially, differential expression analysis was performed using the DESeq2 (v1.4.5)49 or DEGseq50 or PoissonDis51 with Q value ≤ 0.05 (or FDR ≤ 0.001). To take insight to the change of phenotype, GO (http://www.geneontology.org/) and KEGG (https://www.kegg.jp/) enrichment analysis of annotated different expression gene was performed by Phy.per (https://en.wikipedia.org/wiki/Hypergeometric_distribution) based on Hypergeometric test. The significant levels of terms and pathways were corrected by Q value with a rigorous threshold (Q value ≤ 0.05)52.
Validation of differential expression using qRT-PCR
To confirm the RNA-Seq results, 8 candidate DEGs were randomly validated by qRT-PCR. Based on the coding gene sequences, the primers of qRT-PCR were designed using primer premier 5.0 software, and β-actin genewas selected as the internal control53. The primers were designed and listed in Supplementary Table S1.The 2X SG Fast qPCR Master Mix (High Rox) (BBI, Canada, Inc) was used for the RT-qPCR assay and the RT-qPCR assays were carried out using the QuantStudio™ 1 Plus System (ABI/Thermo Fisher, USA). The relative targeted genes expression levels were calculated according to the 2–ΔΔCt method54. Each sample was performed in three replicates.
Extraction and quantification of metabolites
Metabolites were isolated from rice leaf tissues, with six replicates per treatment. After grinding and centrifugation, the supernatant was removed over the 0.22 μm filter membrane. The filtered samples were placed in the upper sample vial waiting for LC–MS analysis. A total of 20 μl from each sample was mixed into QC samples and used to assess the reproducibility and stability of the LC–MS analysis process. Sample analysis was performed on a Waters ACQUITY UPLC 2D (Waters, USA), coupled to a Q-Exactive mass spectrometer (Thermo Fisher Scientific, USA) with a heated electrosprayionization (HESI) source. Chromatographic separation was performed on a Hypersil GOLD aQ column (2.1 × 100 mm, 1.9 μm, Thermo Fisher Scientific, USA), with mobile phase A consisting 0.1% formic acid in water and mobile phase B consisting 0.1 formic acid in acetonitrile.The mass spectrometric settings for positive/negative ionization modes were as follows: spray voltage, 3.8/ − 3.2 kV; sheath gas flow rate, 40 arbitrary units (arb); aux gas flow rate, 10 arb; aux gas heater temperature, 350 °C; capillary temperature, 320 °C. The full scan range was 100–1500 m/z with a resolution of 70,000, and the automatic gain control (AGC) target for MS acquisitions was set to 1e6 with a maximum ion injection time of 100 ms. Top 3 precursors were selected for subsequent MSMS fragmentation with a maximum ion injection time of 50 ms and resolution of 30,000, the AGC was 2e5. The stepped normalized collision energy was set to 20, 40 and 60 eV.In order to provide more reliable experimental results, the samples were randomly sorted to reduce the system error. The QCs were injected at regular intervals (every 10 samples) throughout the analytical run to provide a set of data from which repeatability can be assessed.
Metabolomic data analysis
Data were analyzed by fold change and T test. Fold Change (FC) was obtained by variation fold analysis and p-value were corrected by FDR (False Discovery Rate) to obtain q-value. We used a combination of the p-values of T-test with the VIP value of the OPLS-DA model to filter differentially accumulated metabolites, with additional screening criteria (p < 0.05, VIP > 1).
Statistical analysis
The data were collated using WPS office software and SPSS 19.0 software for one-way ANOVA statistical analysis and the least significant difference (LSD) method for multiple comparisons, with significant differences defined at p < 0.05.
Ethics approval and consent to participate
The study was performed in accordance with relevant institutional, national, and international guidelines and legislation.
Data availability
The datasets generated and/or analysed during the current study are available in the SRA repository, BioProject Accession (PRJNA931648). https://www.ncbi.nlm.nih.gov/sra/PRJNA931648.
References
Kaur, H., Ozga, J. A. & Reinecke, D. M. Balancing of hormonal biosynthesis and catabolism pathways, a strategy to ameliorate the negative effects of heat stress on reproductive growth. Plant Cell Environ. 44, 1486–1503 (2021).
Wang, T. J. et al. JMJ17–WRKY40 and HY5–ABI5 modules regulate the expression of ABA-responsive genes in Arabidopsis. New Phytol. 230, 567–584 (2021).
Yang, W. et al. Transcriptome analysis reveals abscisic acid enhancing drought resistance by regulating genes related to flavonoid metabolism in pigeon pea. Environ. Exp. Bot. 191, 104627 (2021).
Chen, G. et al. Physiological mechanisms of ABA-induced salinity tolerance in leaves and roots of rice. Sci. Rep. 12, 1–26 (2022).
Huang, Y. et al. Salt stress promotes abscisic acid accumulation to affect cell proliferation and expansion of primary roots in rice. Int. J. Mol. Sci. 22, 10892 (2021).
Wang, W. S. et al. Complex molecular mechanisms underlying seedling salt tolerance in rice revealed by comparative transcriptome and metabolomic profiling. J. Exp. Bot. 67, 405–419 (2016).
Zhou, D. et al. Pedigree-based analysis of derivation of genome segments of an elite rice reveals key regions during its breeding. Plant Biotechnol. J. 14, 638–648 (2016).
Flowers, T. J. Improving crop salt tolerance. J. Exp. Bot. 55, 307–319 (2004).
Chinnusamy, V., Gong, Z. & Zhu, J. K. Abscisic acid-mediated epigenetic processes in plant development and stress responses. J. Integr. Plant Biol. 50, 1187–1195 (2008).
Cutler, S. R., Rodriguez, P. L., Finkelstein, R. R. & Abrams, S. R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 61, 651–679 (2010).
Fujita, Y., Fujita, M., Shinozaki, K. & Yamaguchi-Shinozaki, K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 124, 509–525 (2011).
Allan, A. C., Hellens, R. P. & Laing, W. A. MYB transcription factors that colour our fruit. Trends Plant Sci. 13, 99–102 (2008).
Cheynier, V., Comte, G., Davies, K. M., Lattanzio, V. & Martens, S. Plant phenolics: Recent advances on their biosynthesis, genetics, and ecophysiology. Plant Physiol. Biochem. 72, 1–20 (2013).
Miller, G. A. D., Suzuki, N., Ciftci-Yilmaz, S. U. L. T. A. N. & Mittler, R. O. N. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 33, 453–467 (2010).
Xie, R. et al. Effect of salt-stress on gene expression in citrus roots revealed by RNA-seq. Funct. Integr. Genom. 18, 155–173 (2018).
Weeda, S. et al. Arabidopsis transcriptome analysis reveals key roles of melatonin in plant defense systems. PloS One 9, e93462 (2014).
Valério, L., De Meyer, M., Penel, C. & Dunand, C. Expression analysis of the Arabidopsis peroxidase multigenic family. Phytochemistry 65, 1331–1342 (2004).
Li, Y. F. et al. Comparative transcriptome and translatome analysis in contrasting rice genotypes reveals differential mRNA translation in salt-tolerant Pokkali under salt stress. BMC Genom. 19, 95–113 (2018).
Lakra, N., Kaur, C., Anwar, K., Singla-Pareek, S. L. & Pareek, A. Proteomics of contrasting rice genotypes: Identification of potential targets for raising crops for saline environment. Plant Cell Environ. 41, 947–969 (2018).
Chandran, A. K. N. et al. Transcriptome analysis of rice-seedling roots under soil–salt stress using RNA-Seq method. Plant Biotechnol. Rep. 13, 567–578 (2019).
Rawoof, A., Ramchiary, N. & Abdin, M. Z. A high-throughput RNA-Seq approach to elucidate the transcriptional response of Piriformospora indica to high salt stress. Sci. Rep. 11, 1–15 (2021).
Gahlot, S. et al. Isolation of genes conferring salt tolerance from Piriformospora indica by random overexpression in Escherichia coli. World J. Microb. Biotechnol. 31, 1195–1209 (2015).
Han, Z. J., Sun, Y., Zhang, M. & Zhai, J. T. Transcriptomic profile analysis of the halophyte Suaeda rigida response and tolerance under NaCl stress. Sci. Rep. 10, 1–10 (2020).
El-Shafey, N. M. & AbdElgawad, H. Luteolin, a bioactive flavone compound extracted from Cichorium endivia L. subsp. divaricatum alleviates the harmful effect of salinity on maize. Acta Physiol. Plant. 34, 2165–2177 (2012).
Li, L., Liu, Q., Liu, T., Cui, X. & Ning, W. Expression of putative luteolin biosynthesis genes and WRKY transcription factors in Taraxacum antungense kitag. Plant Cell Tissue Organ Culture 145, 649–665 (2021).
Song, Z. et al. Melatonin enhances stress tolerance in pigeon pea by promoting flavonoid enrichment, particularly luteolin in response to salt stress. J. Exp. Bot. 73, 5992–6008 (2022).
Gupta, A. et al. Global profiling of phytohormone dynamics during combined drought and pathogen stress in Arabidopsis thaliana reveals ABA and JA as major regulators. Sci. Rep. 7, 1–13 (2017).
Yang, Z. et al. Transcription profiles of genes related to hormonal regulations under salt stress in sweet sorghum. Plant Mol. Biol. Rep. 35, 586–599 (2017).
Zhu, Y. et al. Analysis of phytohormone signal transduction in Sophora alopecuroides under salt stress. Int. J. Mol. Sci. 22, 7313 (2021).
Dong, W. et al. Wheat oxophytodienoate reductase gene TaOPR1 confers salinity tolerance via enhancement of abscisic acid signaling and reactive oxygen species scavenging. Plant Physiol. 161, 1217–1228 (2013).
Schluepmann, H., Pellny, T., van Dijken, A., Smeekens, S. & Paul, M. Trehalose 6-phosphate is indispensable for carbohydrate utilization and growth in Arabidopsis thaliana. Proc. Natl. Acad. Sci. 100, 6849–6854 (2003).
Paul, M. J., Watson, A. & Griffiths, C. A. Trehalose 6-phosphate signalling and impact on crop yield. Biochem. Soc. Trans. 48, 2127–2137 (2020).
Garg, A. K. et al. Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proc. Natl. Acad. Sci. 99, 15898–15903 (2002).
Ge, L. F. et al. Overexpression of the trehalose-6-phosphate phosphatase gene OsTPP1 confers stress tolerance in rice and results in the activation of stress responsive genes. Planta 228, 191–201 (2008).
Shima, S., Matsui, H., Tahara, S. & Imai, R. Biochemical characterization of rice trehalose-6-phosphate phosphatases supports distinctive functions of these plant enzymes. FEBS J. 274, 1192–1201 (2007).
Rahman, M. M., Rahman, M. M., Eom, J. S. & Jeon, J. S. Genome-wide identification, expression profiling and promoter analysis of trehalose-6-phosphate phosphatase gene family in rice. J. Plant Biol. 64, 55–71 (2021).
Wang, C. L., Zhang, S. C., Qi, S. D., Zheng, C. C. & Wu, C. A. Delayed germination of Arabidopsis seeds under chilling stress by overexpressing an abiotic stress inducible GhTPS11. Gene 575, 206–212 (2016).
Zhai, H. et al. A myo-inositol-1-phosphate synthase gene, Ib MIPS 1, enhances salt and drought tolerance and stem nematode resistance in transgenic sweet potato. Plant Biotechnol. J. 14, 592–602 (2016).
Bohnert, H. J. & Jensen, R. G. Strategies for engineering water-stress tolerance in plants. Trends Biotechnol. 14, 89–97 (1996).
Dan, Y., Niu, Y., Wang, C., Yan, M. & Liao, W. Genome-wide identification and expression analysis of the trehalose-6-phosphate synthase (TPS) gene family in cucumber (Cucumis sativus L.). Peer J. 9, e11398 (2021).
Vishal, B., Krishnamurthy, P. & RamamoorthyR, K. P. P. OsTPS 8 controls yield-related traits and confers salt stress tolerance in rice by enhancing suberin deposition. New Phytol. 221, 1369–1386 (2019).
Li, R., Li, Y., Kristiansen, K. & Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 24, 713–714 (2008).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Benelli, M. et al. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics 28, 3232–3239 (2012).
Shen, S. et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. 111, E5593–E5601 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 1–16 (2011).
Kolde R, & Kolde M R. Package ‘pheatmap’. R package, 1 (2008).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Audic, S. & Claverie, J. M. The significance of digital gene expression profiles. Genome Res. 7, 986–995 (1997).
Storey, J.D., Bass A.J., Dabney, A., Robinson, D. q value: Q-value estimation for false discovery rate control. R Package 2.6. 0. (2021).
Kim, B. R., Nam, H. Y., Kim, S. U., Kim, S. I. & Chang, Y. J. Normalization of reverse transcription quantitative-PCR with housekeeping genes in rice. Biotechnol. Lett. 25, 1869–1872 (2003).
Livak, K. J. & SchmittgenT, D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
We would like to acknowledgement the financial supports from Special projects in key areas of ordinary colleges of Educational Commission of Guangdong Province (2021ZDZX4027); Innovation Team Project of ordinary colleges of Educational Commission of Guangdong Province (2021KCXTD011); “Ling Hang” Program of Zhanjiang Innovation and Entrepreneurship Team Introduction (2020LHJH01); Research start-up project of Guangdong Ocean University (R20046); Research start-up project of Guangdong Ocean University (060302052012); “Innovation and Strong School Project” of Guangdong Ocean University in 2020 (230420006). Guangdong Tianhong Seed Company Limited, China, for provide rice seeds.
Author information
Authors and Affiliations
Contributions
D.Z. & N.F. Funding acquisition; project administration; designed the experiments. C.H. & G.C. Carried out the experiments; analyzed the data; wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, C., Chen, G., Zheng, D. et al. Transcriptomic and metabolomic analyses reveal that ABA increases the salt tolerance of rice significantly correlated with jasmonic acid biosynthesis and flavonoid biosynthesis. Sci Rep 13, 20365 (2023). https://doi.org/10.1038/s41598-023-47657-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-47657-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
