
Abstract
Bile acids and salts have been shown to play a role in liver carcinogenesis through DNA damage, inflammation, and tumor proliferation. However, the correlation between bile acid metabolism and hepatocellular carcinoma (HCC) prognosis remains unclear. This study aimed to identify a predictive signature of bile acid and bile salt metabolism-related long non-coding RNAs (lncRNAs) for HCC prognosis and treatment response. The study used HCC RNA-sequencing data and corresponding clinical and prognostic data from The Cancer Genome Atlas. A prognostic model consisting of five bile acid and bile salt metabolism-related lncRNAs was developed and evaluated in a training set, a validation set and an external set. The model demonstrated good performance in predicting HCC prognosis and was shown to be an independent biomarker for prognosis. Additionally, our study revealed a significant association between the signature and immune cell infiltration, as well as its predictive value for therapeutic responses to both immunotherapy and chemotherapy. Furthermore, three LncRNAs (LUCAT1, AL031985.3 and AC015908.3) expression levels in our signature were validated through qRT-PCR in a cohort of 50 pairs of HCC patient tumor samples and corresponding adjacent non-tumor samples, along with 10 samples of normal liver tissue adjacent to benign lesions. These findings suggest that this novel bile acid and bile salt metabolism-related lncRNA signature can independently predict the prognosis of patients with HCC and may be utilized as a potential predictor of response to treatment in this setting.
Similar content being viewed by others


Introduction
Liver cancer is one of the most common malignancies in humans and the second leading cause of cancer-related death worldwide1,2. Hepatocellular carcinoma (HCC) is the predominant form of liver cancer with a complex etiology and limited treatment options. As a result of the considerable efforts of researchers, various novel therapeutic methods are emerging. However, there is currently a lack of specific treatment. Recently, immunotherapy for advanced HCC has become a research hotspot. Unfortunately, the effectiveness of this treatment modality is limited3. Research on biomarkers of response or primary resistance to immunotherapies is essential for efficient treatment.
As endogenous metabolites of host-gut microflora co-metabolism, bile acids and their metabolites are attracting attention due to their carcinogenic potential. Bile acids, especially secondary bile acids, release arachidonic acid to promote the production of reactive oxygen species and induce DNA damage. Besides, bile acids and salts can also mediate inflammation, promote tumor proliferation and inhibit tumor cell apoptosis through a series of signal transduction pathways4,5,6. It has been shown that intestinal bacterial metabolite deoxycholic acid (DCA) promotes obesity-related liver cancer in mouse models7. In addition, the induction of inflammatory gene expression by DCA in hepatocytes was closely related to the occurrence and development of tumors8. Hang et al. suggested that bile acid metabolites directly regulate the balance between T-helper 17 and regulatory T cells to control host immune response without the involvement of intestinal flora9. Another study revealed that bile acids metabolized by intestinal flora can specifically regulate the number of natural killer T cells in the liver, thereby regulating the growth of liver tumors10. The above evidence clearly demonstrated that bile acids and salts play an important regulatory role directly or indirectly in shaping the immune system.
Long non-coding RNAs (lncRNAs) are non-coding transcripts with a length > 200 nucleotides, which regulate the expression of numerous cancer-related genes. An increasing number of studies have found that abnormally expressed lncRNAs are involved in regulating tumorigenesis-related biological functions, such as metastasis, immune response, and metabolic regulation11,12. In recent years, a growing body of evidence supports that many cellular actions of the bile acid/Farnesoid X receptor (FXR) pathway are mediated by lncRNAs, and lncRNAs are in turn powerful regulators of bile acid levels and FXR activities13. However, research on a bile acid and salt metabolism-related lncRNA prognostic signature in patients with HCC is currently lacking.
Results
Identification and visualization of differentially expressed bile acid and bile salt metabolism-related genes
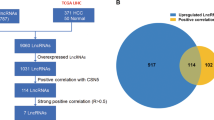
We obtained 23 bile acid and bile salt metabolism-related differentially expressed genes (DEGs), including 17 upregulated genes and and 6 downregulated genes (Fig. 1a, Supplementary Table S1). The heatmap shows the RNA expression levels of those DEGs (Fig. 1b). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to identify DEGs at the biologically functional level (Fig. 1c,d). In the biological process category, GO analysis showed that the DEGs were mainly enriched in the bile acid metabolic process, organic acid biosynthetic process, monocarboxylic acid biosynthetic process, etc. In the cellular components category, the DEGs were mainly enriched in peroxisome, peroxisomal part, peroxisomal membrane, microbody part, etc. In the molecular function category, the DEGs were mainly enriched in lipid transporter activity, oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, etc. KEGG pathway analyses indicated that bile acid and bile salt metabolism-related DEGs were mainly enriched in the primary bile acid biosynthesis, ATP-binding cassette transporters, peroxisome proliferator-activated receptor (PPAR) signaling pathway, etc.

GO and KEGG analyses of 23 bile acid and salt metabolism-related DEGs in HCC (a) Volcano plot of bile acid and bile salt metabolism-related genes in HCC. Red and green dots represent upregulated and downregulated genes, respectively. (b) Heatmap showing the RNA expression levels of 23 DEGs. (c) GO analysis. (d) KEGG analysis. ABC ATP-binding cassette, ATPase adenosine triphosphatase, BP biological process, CC cellular components, FC fold change, FDR false discovery rate, GO Gene Ontology, HCC hepatocellular carcinoma, KEGG Kyoto Encyclopedia of Genes and Genomes, MF molecular function, PPAR peroxisome proliferator-activated receptor, p.adjust adjusted p value.
Construction of a bile acid and bile salt metabolism-related lncRNA signature
A total of 498 bile acid and salt metabolism-related lncRNAs were obtained by Pearson's correlation analyses. To construct a prognostic model for HCC, univariate Cox regression was performed, and 57 lncRNAs related survival were selected. Subsequently, analysis with the LASSO algorithm was performed to identify stable markers from the survival-related lncRNAs (Fig. 2a,b). Finally, multivariate cox regression analysis was performed and a prognostic lncRNA model consisting of five lncRNAs (i.e., AL031722.1, AC015908.3, AL031985.3, LUCAT1 and PCCA-DT) was constructed. The risk score was calculated as follows: risk score = AL031722.1 expression * (− 0.246917061576047) + AC015908.3 expression * (− 0.178616640082393) + AL031985.3 expression * 0.317991474159641 + LUCAT1 expression * 0.110874567602666 + PCCA-DT expression * 0.0266570817050312. In addition, the hazard ratios of the five lncRNAs were calculated. AL031722.1 and AC015908.3 were protective factors for prognosis, whereas the other three lncRNAs were risk factors (Fig. 2c). Furthermore, the lncRNA-mRNA co-expression network (Fig. 2d) was constructed using the five lncRNAs identified through Pearson correlation analysis (|r| > 0.3 and p < 0.001). A Sankey diagram was used to visualize the network (Fig. 2e).

Regression coefficient diagram based on the LASSO algorithm and lncRNA-mRNA network. (a) LASSO coefficient profiles of 57 bile acid and bile salt metabolism-related lncRNAs. (b) Cross-validation for the selection of tuning parameter in the LASSO regression. (c) Forest plots showing the relationships of each lncRNA subset with overall survival in the training group. The unadjusted hazard ratios are presented with 95% confidence intervals. (d) The co-expression network of prognostic bile acid and bile salt metabolism-related lncRNAs. (e) Sankey diagram of prognostic bile acid metabolism-related lncRNAs. LASSO least absolute shrinkage and selection operator, lncRNA long non-coding RNA.
The predictive power of the prognostic model in patients with HCC
To evaluate the power of this bile acid and bile salt metabolism-related lncRNAs classifier in predicting the survival of patients with HCC, ROC analysis was performed in the training, verification and whole sets. The results showed that the area under the curve of the risk score at 1, 3 and 5 years of prognosis time in the training set was 0.841, 0.648, and 0.718, respectively. These findings indicated high sensitivity and specificity for the prediction of survival (Fig. 3a). The prognostic accuracy of this risk prediction model was further tested using an internal verification set and a whole set, which revealed good predictive performance (Fig. 3b,c).

Internal validation of the predictive signature for OS based on the entire TCGA dataset (a–c) Time‐dependent ROC curves at 1, 3, and 5 years in training set, validation set and whole set. (d–f) The Kaplan–Meier survival curves of the bile acid and bile salt metabolism‐related lncRNA Signature in training set, validation set and whole set.
A prognostic curve and a scatter plot were used to exhibit the risk score and the survival status of each patient with HCC (Fig. 4a,b). The analysis showed that a high-risk score was associated with an increased mortality rate. In addition, Kaplan–Meier analysis was used to analyze the overall survival (OS) time in the low- and high-risk groups. Figure 3d–f showed that patients in the high-risk group had a worse prognosis than those in the low-risk group in the training, validation, and whole sets (all p < 0.05).

The correlation between the predictive signature and the prognosis of HCC patients. (a) Risk score curves based on the risk score of each patient with HCC. (b) Scatter plots displaying the survival status of each patient with HCC. (c) Forrest plot for the univariate Cox regression analysis of clinicopathological variables. (d) Forrest plot for the multivariate Cox regression analysis of clinicopathological variables. (e) The ROC curve of the risk score and clinicopathological variables. (f) Distribution heat map of clinicopathological variables in the low- and high-risk groups ***p < 0.001, **p < 0.01, *p < 0.05. AUC area under the curve, CI confidence interval, CTP Child–Turcotte–Pugh grade of liver function, HR hazard ratio, ROC receiver operating characteristic.
Cox regression analysis was performed to determine whether the risk score of the model is an independent prognostic factor for patients with HCC. Both univariate and multivariate Cox regression analyses showed that the risk score was significantly associated with the OS of patients with HCC (Fig. 4c,d). This observation indicated that the risk score was an independent factor for the prognosis of patients with HCC. We also compared several other clinical parameters to evaluate the important function of the risk score in predicting prognosis. Age, sex, stage, grade, and Child–Turcotte–Pugh (CTP) grade of liver function were examined as candidate predictive biomolecular indicators. The area under the curve of the risk score was 0.682, which was better than those of other clinicopathological variables in predicting the prognosis of patients with HCC (Fig. 4e). Figure 4f presents a heatmap displaying the distribution of clinicopathological variables in the high- and low-risk groups. The data reveal significant dissimilarities (all p < 0.05) between the two groups in terms of T stage, stage, grade, and age.
Construction and verification of the prognostic nomogram
To better evaluate the predictive power of this model for survival, we constructed a nomogram based on the age, sex, CTP grade of liver function, grade, stage, and risk score (Fig. 5a). In addition, calibration plots were produced to evaluate the consistence between the nomogram-predicted and actual 1-, 3-, and 5-year survival rates. The predicted lines were basically consistent with the reference lines, indicating that the constructed nomogram was credible (Fig. 5b–d).

Construction and verification of the nomogram. (a) A nomogram combining clinicopathological parameters and the risk score for estimating the probability of 1-, 3-, and 5-year survival of patients with HCC. (b–d) Calibration curves were used to investigate the deviation between the predicted and actual survival rates at 1, 3, and 5 years. CTP Child–Turcotte–Pugh grade of liver function, HCC hepatocellular carcinoma.
Validation of bile acid and bile salt metabolism-related risk model in external cohorts
Based on the risk model, we identified a differentially expressed gene signature A/B between the low-risk and high-risk groups within the TCGA-LIHC dataset. The Risk Gene Signature Score (RS score) was calculated based on the expression differences between gene signatures A and B in these high- and low-risk groups. In the TCGA-LIHC database, a significant correlation was observed between the risk score derived from bile acid metabolism-related lncRNAs and the RS score (as shown in Fig. 6a). Kaplan–Meier analysis further confirmed that high RS scores were associated with poor prognosis (Fig. 6b). These results collectively indicate that the RS score method represents a viable alternative risk score model.

Construction of the RS score and validation in the external data set GSE14520. (a) Spearman correlation analysis between RS score and risk score. (b) The Kaplan–Meier curve shows the OS of patients with different RS scores in the TCGA-LIHC database. (c) Distribution plots for the relationship between RS score and survival status (d) Scatter plots displaying the survival status of each patient with HCC (e) Heat maps for the gene signature A/B in the cohort (f) ROC curve for the RS score in the external cohort (g) Survival curves between high-and low-RS score groups in GSE14520.
In the GSE14250 dataset, we applied the same RS score calculation method to each patient. Patients were then divided into high-RS score and low-RS score groups based on the calculated median RS score. Survival analyses conducted on this external cohort consistently demonstrated that patients with a high RS score had an unfavorable prognosis (Fig. 6g). Additionally, ROC curve analysis illustrated the reliable prognostic predictive ability of the alternative model (Fig. 6f).
Gene set enrichment analysis (GSEA) in the low- and high-risk groups
GSEA was used to annotate the biological functions and determine the enrichment pathways in the low- and high-risk groups. In the high-risk group, there were 87 significantly enriched pathways (Supplementary Table S2), mainly related to proliferation, apoptosis, tumors, and other related pathways (Supplementary Fig. S1). In the low-risk group, there were 38 significantly enriched pathways (Supplementary Table S3), mainly related to metabolism pathways, such as primary bile acid biosynthesis, fatty acid metabolism, glycine serine and threonine metabolism, etc. (Supplementary Fig. S1).
The predictive signature and immune cell infiltration
To evaluate the association between our lncRNA signature and immune infiltration, we drew the correlation bubble plot of immune cells and risk score calculated using various algorithms (Supplementary Fig. S2). From the figure, it can be inferred that the scores of most immune cells were positively correlated with the risk score. In addition, the TIMER and CIBERSORT abs.mode algorithms were applied to evaluate the infiltration of immune cells in the low- and high-risk groups. The TIMER algorithm yielded six kinds of immune cells in total. The immune scores of B cells, CD4 + T cells, neutrophils, macrophages, and myeloid dendritic cells were higher in the high-risk group versus the low-risk group (Fig. 7a). The CIBERSORT abs.mode algorithm calculated the immune scores of 22 types of immune cells. It was found that numerous cell types (i.e., memory B cells, plasma B cells, activated memory CD4 + T cells, follicular helper T cells, regulatory T cells, activated NK cells, resting dendritic cells, M0 macrophages, M1 macrophages and M2 macrophages) had a higher immune score in the high-risk group versus the low-risk group (Fig. 7b). We also use ESTIMATE algorithm14 to evaluate the relationship of the risk score and the ratio of immune score in tumor samples. It was found that immune score was higher in the high-risk group (p = 0.037, Fig. 9b), which indicated that the high-risk group samples might had a higher ratio of immune cells.

Immune cell infiltration in the low- and high-risk groups. (a) The TIMER algorithm was used to calculate the infiltration of six kinds of immune cells. (b) The CIBERSORT abs.mode algorithm was used to calculate the infiltration of 22 kinds of immune cells (Wilcoxon rank-sum test, ns, not statistically significant; p ≥ 0.05; *p < 0.05; **p < 0.01; ***p < 0.001). TIMER tumor immune estimation resource.
Correlation between the predictive signature and immune checkpoint blockade (ICB) therapy-related molecules
To explore the role of the bile acid and salt metabolism-related lncRNA risk score model in ICB immunotherapy in patients with HCC, we analyzed the correlation between key targets of ICB therapy and this lncRNA signature. We compared the expression levels of 47 immune checkpoint genes (Supplementary Table S4) in the low- and high-risk groups. We found that the expression levels of 33 genes exhibited statistically significant differences; most of them were highly expressed in the high-risk group (Fig. 8g). To further clarify the relationship between our bile acid and salt metabolism-related lncRNA model and immune checkpoint genes, we analyzed six immune checkpoint genes. Spearman analysis was used to determine the correlation between the expression levels of these genes and the risk scores of the model (Fig. 8a–f). The results showed that the risk score was significantly positively correlated with PD-1 (r = 0.257, p < 0.001), CTLA4 (r = 0.320, p < 0.001), TIM-3 (r = 0.323, p < 0.001), TIGIT (r = 0.229, p < 0.001), CD276 (r = 0.462, p < 0.001), and CD47 (r = 0.278, p < 0.001).

Correlation between the expression of ICB-related genes and this bile acid and bile salt metabolism-related lncRNA signature. (a–f) Association between the risk score of this signature and the expression of six ICB-related genes (i.e., PD-1, CTLA4, HAVCR2, TIGIT, CD276 and CD47) (g) Comparison of 33 differentially expressed ICB-related genes in the low- and high-risk groups (Wilcoxon rank sum test, ns, not statistically significant; *p < 0.05; **p < 0.01; ***p < 0.001). CD47 cluster of differentiation 47, CD276 Cluster of differentiation 276, CTLA4 cytotoxic T-lymphocyte associated protein 4, ICB immune checkpoint blockade, lncRNA long non-coding RNA, PDCD1 programmed cell death 1, TIGIT T Cell immunoreceptor with Ig and ITIM domains.
Correlation between the predictive signature and therapy response in HCC
We further evaluated whether the bile acid and salt metabolism related signature could serve as an immunotherapy predictor for HCC patients based on the tumor immune dysfunction and exclusion (TIDE) algorithm. Higher TIDE prediction scores are associated with a greater likelihood of immune evasion, indicating that patients are less likely to benefit from ICI treatment. Interestingly, our study revealed significantly lower TIDE scores in high-risk group compared to the low-risk group (p < 0.01, Fig. 9d), implying that high-risk patients may derive more substantial benefits from ICI therapy. Furthermore, through an analysis of the IMvigor210 immunotherapy dataset, we observed that patients with high RS Scores (equivalent to a high-risk profile) exhibited a notably higher response rate to immunotherapy (p = 0.011). This finding further substantiates the notion that high-risk group patients stand to gain greater benefits from immunotherapy.

Differences in immune infiltration and immune therapy response between high- and low-risk groups. (a) Comparison of the Stromal scores between low- and high-risk group using the ESTIMATE algorithm. (b) Comparison of the Immune scores between low- and high-risk group. (c) Comparison of the Estimate scores between low- and high-risk group. (d) Comparison of the TIDE scores between low- and high-risk group. (e) Boxplot demonstrating the RS score difference between the response group and the non-response group in IMvigor210 dataset. (f) Bar plot displaying the relative frequency of different clinical response subgroups in the low or high RS score group in IMvigor210 dataset.
The "pRRophetic" package of the R project was used to predict the sensitivity of patients to different chemotherapeutic and targeted drugs using the risk score15,16 (Fig. 10). We found that low-risk group was associated with the low estimated IC50 of Docetaxel and Rapamycin, and high-risk group was associated with the low estimated IC50 of Bortezomib, Cisplatin, Doxorubicin, Gemcitabine, Mitomycin.C and paclitaxel (Fig. 10a–h). We also found the estimated IC50 of some targeted drugs like Erlotinib and Gefitinib were lower in low-risk group, and Tipifarnib was lower in high-risk group (Fig. 10i–k). However, there was no significant difference in estimated IC50 between high- and low-risk groups for Sorafenib (Fig. 10l). These findings suggested that the risk score can be used as a predictor of sensitivity to chemotherapy.

Comparison of the sensitivity of the low- and high-risk groups to chemotherapeutic and targeted drugs. (a–h) Estimated IC50 of chemotherapeutic drugs Bortezomib, Cisplatin, Docetaxel, Doxorubicin, Gemcitabine, Mitomycin.C, Paclitaxel and Rapamycin in the low- and high-risk groups. (i–l) Estimated IC50 of targeted drugs Erlotinib, Gefitinib, Tipifarnib and Sorafenib in the low- and high-risk groups. IC50 half-maximal inhibitory concentration.
qRT-PCR validation of liver cancer samples
We collected tissues and corresponding clinical prognostic information from 50 patients with HCC. Initially, the expression differences of three lncRNAs in tumor samples and adjacent non-tumor samples were compared. Through PCR analysis, we found that AC015908.3 was highly expressed in adjacent non-tumor tissues but exhibited low expression in tumor tissues (p < 0.001, Fig. 11c). However, the expression differences of LUCAT1 and AL031985.3 between tumor and adjacent non-tumor tissues were not found to be statistically significant (p values were 0.429 and 0.831, respectively, Fig. 11a,b). Subsequently, the expression of these three lncRNAs in tumor tissues was compared to that in normal liver tissues. It was found that both LUCAT1 and AL031985.3 were upregulated in tumor tissues compared to normal tissues (p values < 0.05 for both, Fig. 11e,f), while AC015908.3 exhibited higher expression in normal liver tissues and lower expression in tumor tissues (p < 0.04, Fig. 11g). To further investigate the clinical implications, stratified analyses were performed on LUCAT1 and AL031985.3. The high-expression group in tumor samples was defined as those with expression levels at least twice as high as in adjacent non-tumor samples, and the low-expression group as those with expression levels at half or less. Others were considered to have no significant change in expression. The LUCAT1 high-expression group comprised 14 cases, while the low-expression group included 18 cases. Similarly, the AL031985.3 high-expression group had 16 cases, and the low-expression group had 16 cases. The relationship between high and low-expression groups and clinical pathological features was explored. It was found that, for LUCAT1, there was no statistically significant difference in terms of overall survival (p = 0.057, Fig. 11h). However, a significantly higher cumulative recurrence rate was observed in the high-expression group compared to the low-expression group (p = 0.014, Fig. 11j). Additionally, a higher proportion of cases with moderately to poorly histological differentiation was found in the high-expression group compared to the low-expression group (p < 0.05, Fig. 11l). In the case of AL031985.3, there were no statistically significant differences in terms of overall survival or histological differentiation (Fig. 11j,l). However, a higher recurrence rate was observed in the high-expression group compared to the low-expression group (p = 0.049, Fig. 11k).

Validation of lncRNA expression by qRT-PCR in clinical samples and exploration of their association with clinical features. (a–c) Expression differences of LUCAT1, AL031985.3, and AC015908.3 in Hepatocellular Carcinoma (HCC) and corresponding adjacent non-tumor tissues. (d–f) Expression differences of LUCAT1, AL031985.3, and AC015908.3 in HCC and normal liver tissues. (h, i) Kaplan–Meier survival curves comparing high and low-expression groups of LUCAT1 and AL031985.3. (j, k) Cumulative incidence of recurrence curves for high and low-expression groups of LUCAT1 and AL031985.3. (l, m) Overlayed bar graphs displaying the differences in Histologic differentiation between high and low-expression groups of LUCAT1 and AL031985.3.
Comparisons between HCC and adjacent non-tumor tissues were assessed using the Wilcoxon matched-pairs signed rank test. Comparisons between HCC and normal liver tissues were conducted using the Wilcoxon rank-sum test. Survival and cumulative recurrence curves were compared using the log-rank test. Inter-group differences in histological frequencies were evaluated using Fisher's exact test.
Discussion
Bile acids are synthesized in the liver and transformed into bile salts by the association with Na+ or K+ ions before release into the gall bladder. These primary bile acids are metabolized into secondary bile acids by certain members of the normal gut microbiota. Bile salt metabolism is a subset of bile acid metabolism that specifically focuses on the modified and more water-soluble forms of bile acids, known as bile salts. Bile acids and salts play important roles in the maintenance of the healthy gut flora, balance of lipid and carbohydrate metabolism, insulin sensitivity, and innate immunity. Moreover, as signaling molecules, they also play a pivotal role in the development of various types of cancer17,18,19,20. Accumulating evidence indicates that hydrophobic bile acids (e.g., lithocholic acid, DCA and chenodeoxycholic acid) may contribute to the tumorigenesis and development of liver cancer21,22,23. Huang et al. identified six microbial markers, which are closely related to bile acid metabolism and the tumor immune microenvironment, by fecal 16sRNA sequencing of patients with HBV-RELATED HCC and healthy populations. These markers had good prediction potential for clinical outcomes of HBV-related HCC patients24. FXR activity is a major inhibitor of HCC carcinogenesis. Dysregulation of bile acid homeostasis in cirrhosis and non-alcoholic steatohepatitis leads to increased hepatic bile acid inflammation and reduced FXR signaling, which is a risk factor for the development of HCC2,25,26. The sodium taurocholate cotransporting polypeptide (NTCP), which is involved in bile acid transport, has been identified as a receptor for HBV, and its variant, S267F, has been strongly associated with a decrease in HBV-associated hepatocellular carcinoma27,28. Petrick et al. provided evidence that higher concentrations of bile acids-specifically, conjugated primary bile acids-are associated with increased HBV- and HCV-related HCC risk29. In recent years, bile acid-related lncRNAs have been increasingly recognized as essential in the pathogenesis of liver diseases, including liver injury, fatty liver, fibrosis, and hepatocarcinoma30,31,32,33. However, few studies have described the role of bile acid metabolism in the prognosis of HCC. A comprehensive bioinformatics analysis is needed to evaluate the role of bile acid regulators in predicting prognosis and their therapeutic potential in HCC.
In this study, we identified 23 bile acid metabolism-related DEGs of HCC. KEGG analysis showed that the DEGs were mainly enriched in bile acid synthesis, metabolism, transport, and the PPAR signaling pathway. Numerous studies have shown that abnormal regulation of the PPAR signaling pathway is involved in the development and progression of tumors34,35,36. However, further experiments are needed to verify whether bile acid metabolism-related genes are involved in tumorigenesis and development through the PPAR signaling pathway in HCC.
Through stepwise univariate Cox regression, LASSO regression and multivariate Cox regression analyses, we constructed a novel bile acid and bile salt metabolism-related five-lncRNA signature in a large-scale HCC cohort. The results demonstrated the sensitivity and specificity of the signature. Subsequently, Kaplan–Meier curves, time-dependent ROC curves, and Cox regression analysis were employed to confirm the predictive performance of this bile acid metabolism-related lncRNA risk score model, which can serve as an independent biomolecular indicator for the prediction of patient survival in HCC. Furthermore, the calibration curves of the nomogram validated that our novel risk score model performs better than traditional clinicopathological characteristics in predicting the prognosis of HCC.
GSEA analysis showed that pathways enriched in the high-risk group were mainly related to proliferation, apoptosis and tumors; these pathways included Cell cycle, Oocyte meiosis, p53 signaling pathway, mTOR signaling pathway and Bladder cancer. Pathways enriched in the low-risk group were primarily involved in synthetic and metabolic processes, such as primary bile acid biosynthesis, fatty acid metabolism, and glycine serine and threonine metabolism. This suggests that more cancer-driving pathways (e.g. tumor cell proliferation, growth, differentiation and metabolism) are involved in the high-risk group versus the low-risk group, thus resulting in a worse prognosis. Metabolic reprogramming is an important feature of neoplasms and mutually causal in the occurrence and development of tumors. Hence, cancer is a genetic disease as well as a metabolic disease37. From the enrichment of normal metabolic pathways in the low-risk group and the enrichment of cancer-driving pathways in the high-risk group, it can be hypothesized that the five lncRNAs identified in this study may play a role in changes in the metabolic pathway, leading to the progression of HCC.
ICB therapy has revolutionized the treatment of numerous malignancies. Nevertheless, its effectiveness is limited to a minority of patients with cancer. Hence, the discovery of predictive and prognostic biomarkers is urgently warranted. However, currently, there is no effective biomarker for predicting the response of patients with HCC to treatment with immune checkpoint inhibitors38. In our study, we explored the relationship between our predictive signature and immune cell infiltration. As an integral component of the tumor microenvironment, immune infiltrates contribute to tumor progression and response to immunotherapy39. In some solid tumors, high levels of immune infiltration involving macrophages and T cells are usually associated with better immune response compared with low levels of immune infiltration40. In addition to T cells which have been extensively studied, other immune cells of the innate and adaptive immune systems, including dendritic cells (DCs), macrophages, NK cells, and B cells, have also been shown to contribute to tumor progression and immunotherapy response41. Hollern et al. demonstrated that immune checkpoint therapy induces T follicular helper cell (TFH cells) activation of B cells to facilitate the anti-tumor response in mouse models of triple-negative breast cancer42. An extensive analysis of 70,000 cancer patients across various types identified immune factors such as CD8 + T cells, M1 macrophages, TLSs, TH1 cells, TFH cells, B cells, NK cells, and DCs that were associated with favorable prognosis for most cancers analyzed. On the contrary, regulatory T cells (Treg cells), M2 macrophages, T-helper 17 cells (TH17), T-helper 2 cells (TH2), and polymorphonuclear myeloid-derived suppressor cells (PMN MDSCs) predominantly signaled adverse prognosis43. However, various immune cells play a dual role in tumor initiation and progression. Immune cells such as macrophages, dendritic cells, NK cells, B cells, CD4 + T cells, and CD8 + T cells can bolster anti-tumor immune responses, shielding us against malignant cells. Nonetheless, they can also adopt a pro-tumorigenic role, thereby fostering tumor advancement and survival44.
In our study, most immune cells had a positive correlation with the risk score; five of the six types of immune cells identified by the TIMER algorithm were enriched and had infiltrated in the high-risk group. Based on the results obtained from the CIBERSORT abs.mode algorithm analysis, there were significant differences in immune cells (e.g. memory B cells, plasma B cells, activated memory CD4 + T cells, follicular helper T cells, regulatory T cells, activated NK cells, resting dendritic cells and macrophages) between the low- and high-risk groups. These cells exhibited higher levels of infiltration in the high-risk group versus the low-risk group. And the ESTIMATE algorithm revealed that immune score was higher in the high-risk group than low-risk group, which means high-risk patients had higher infiltration of immune cells than low-risk patients. Subsequently, the relationship between the bile acid and salt metabolism-related lncRNA risk model and 47 ICB-related genes was explored. We found that 33 genes were differentially expressed in the low- and high-risk groups; most of these genes were highly expressed in the high-risk group. Spearman correlation analysis showed that the risk score of our bile acid and bile salt metabolism related-lncRNAs model was significantly correlated with the ICB-related genes (i.e., PD-1, TIM-3, and CTLA4). We found that high risk scores were linked to high expression of immune checkpoints. This suggested that bile acid metabolism is important in the field of immuno-oncology. To ascertain the association of our model with the benefit of immunotherapy response, we independently validated the immune therapy response in high and low-risk groups using the TIDE database and the immunotherapy cohort IMvigor 210. The TIDE scores were lower in the high-risk group, indicating that there is a reduced likelihood of immune evasion in this group, suggesting that they may derive greater benefits from immunotherapy. Furthermore, due to the lack of expression data for the five lncRNAs in our model, we used the RS score as a substitute for the risk score and observed that higher RS scores were linked to improved immune responses in the IMvigor 210 cohort. In summary, it can be inferred that patients in the high-risk group may potentially benefit more from immunotherapy compared to those in the low-risk group. We also assessed the role of this model in predicting the response to conventional chemotherapeutic drugs used in HCC. By calculating the estimated IC50 values, we found that high-risk patients may be sensitive to cisplatin, mitomycin C and doxorubicin, but resistant to docetaxel. Based on our study, the use of immunotherapy or chemotherapeutic drugs can be customized to individual patients according to their risk score and sensitivity, thereby providing the basis for precise and personalized treatment in HCC. However, additional laboratory experiments and large-scale clinical trials are warranted in the future to confirm this hypothesis.
In our study, we conducted qRT-PCR to assess the expression levels of three lncRNAs included in our model in HCC tissues. We found that LUCAT1 and AL031985.3 exhibited higher expression levels in tumor tissues compared to normal liver tissues, while AC015908.3 showed lower expression in tumor tissues compared to normal liver tissues. These findings were consistent with the results obtained from mining the TCGA-LIHC database. Increasing evidence have showed that lncRNA lung cancer-associated transcript 1 (LUCAT1) participates in the regulation of proliferation, migration, invasion, and drug resistance of multiple tumors45,46,47,48. LUCAT1 exhibits high expression in various types of cancers. In breast cancer, elevated LUCAT1 expression is associated with poorer survival, larger tumor size, and later TNM staging46. In colorectal cancer, LUCAT1 promotes cell proliferation, apoptosis, migration, and invasion both in vitro and in vivo. Patients with higher LUCAT1 expression tend to have worse prognoses and poorer responses to chemotherapy47,48. Analyzing the TCGA-LIHC database, Jiao et al. found that increased LUCAT1 expression in liver cancer correlates with age, histological grade, T stage, and survival status49. Additionally, Zhu et al. identified elevated LUCAT1 expression in liver cancer samples through qRT-PCR, and in vitro experiments indicated a close association between high LUCAT1 expression and HCC cell proliferation and migration50. In our study, qRT-PCR analysis of human samples revealed that while the difference in LUCAT1 expression between cancer and adjacent tissues was not pronounced, tumor tissues exhibited higher LUCAT1 expression compared to normal liver tissues. Moreover, we found that high LUCAT1 expression was linked to higher tumor recurrence rates and histological characteristics of poorer differentiation. These factors suggest that elevated LUCAT1 expression is associated with poorer prognosis. However, in our own cohort, there was no significant difference in prognosis between high and low LUCAT1 expression, possibly due to our relatively small sample size. Further validation with a larger sample size will be necessary in the future.
Through bioinformatics analysis, we identified a bile acid and salt metabolism-related lncRNA signature associated with survival. This signature can also, to some extent, predict the response of patients to immunotherapy and chemotherapy. There are several limitations in this study. Firstly, this was a retrospective study based on data obtained from public databases. Hence, prospective cohort studies are needed to verify the accuracy of this model in the future. Secondly, these predictions were solely based on bioinformatic and comparative analyses with small scale, thus, further study with larger sample size in the real-world setting is warranted.
In conclusion, we performed a comprehensive analysis of the association between the bile acid metabolism-related lncRNA and survival of patients with HCC. A novel bile acid and bile salt metabolism-related prognostic risk signature was constructed. This signature could effectively serve as an independent prognostic biomarker and a potential predictive factor for the effectiveness of immunotherapy and chemotherapy in patients with HCC.
Materials and methods
Patients and datasets
We downloaded the fragments per kilobase of transcript per million mapped reads-standardized RNA-sequencing data and the corresponding clinical and prognostic data from TCGA portal (http://cancergenome.nih.gov) on January 4, 2022; the data included 374 tumor samples and 50 adjacent normal samples. Additionally, for external validation, we obtained a liver cancer dataset, GSE14520, from the Gene Expression Omnibus (GEO) database. To identify relevant gene sets related to bile acid and bile salt metabolism, we utilized the curated "HALLMARK_BILE_ACID_METABOLISM" signature from the Molecular Signatures Database (MSigDB), available at https://www.gsea-msigdb.org/gsea/msigdb. This hallmark gene set comprises 28 individual gene sets, encompassing a total of 112 genes primarily involved in the metabolism of bile acids and bile salts (Supplementary Tables S5, S6). "Hallmark" gene sets are meticulously curated to provide a more refined and consolidated representation of various biological processes or states. They offer a coherent and concise depiction of gene expression patterns associated with these processes, reducing redundancy and variability. Consequently, they serve as valuable inputs for gene set enrichment analysis, offering a more focused and streamlined alternative compared to the original gene sets. This approach enhances the interpretability and accuracy of our analysis, allowing us to investigate the specific role of bile acid metabolism-related genes in hepatocellular carcinoma (HCC) with greater precision51.
Fifty HCC tissues and corresponding paracancerous tissues in this study were obtained from Tianjin Third Central Hospital (Tianjin, China) after surgical resection, meanwhile, ten normal liver tissues adjacent to benign lesions (include hepatic cyst, liver cavernous hemangioma and angiomyolipoma of liver) were acquired in the same way, all samples were collected from 2015 to 2018, and were frozen and stored in – 80 °C for further analysis. All patients were followed until death or closure of data analysis on May 4, 2023.The clinical records of the patients were listed in Supplementary Table S7. All samples and relative study were approved by the Medical Ethics Committees of Tianjin Third central Hospital (Approval number: IRB2019-034-01).
Identification and functional enrichment analysis of differentially expressed bile acid metabolism-related genes (DEGs)
DEGs were selected using the R package “limma”, with a false discovery rate < 0.05 and |log2 fold change (FC) > 1| as screening criteria. A total of 23 DEGs were identified for further functional enrichment analysis (Supplementary Table S1). We performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses on these DEGs, and visualized the results with the “ggplot2” package.
Construction of the bile acid and bile salt metabolism-related lncRNA predictive signature
Pearson's correlation analyses were performed to determine the correlation between the expression levels of lncRNAs and bile acid and bile salt metabolism-related DEGs. LncRNAs with a Pearson's correlation coefficient |r| > 0.3 and p < 0.001 were selected for further analysis. Overall, we identified 498 lncRNAs. Subsequently, we used the screened bile acid metabolism-lncRNAs to construct a prognostic risk score model for HCC. Firstly, HCC patients with incomplete follow‐up information or less than 30 days of survival were excluded. Altogether, 343 cases of HCC were randomly assigned into the training (n = 172) and validation (n = 171) sets at the ratio of 1:1 using the R project “caret” package. In the training set, 498 bile acid metabolism-related lncRNAs were analyzed by univariate Cox regression analysis using the “survival” package of R project. The results with p value > 0.05 were filtered out. Secondly, the recognized lncRNAs were further screened and confirmed via the least absolute shrinkage and selection operator (LASSO) algorithm using the R project “glmnet” package; eight LncRNAs were chosen. Thirdly, a multivariate Cox regression model was applied to those bile acid metabolism‐related lncRNAs. Finally, five bile acid metabolism‐related lncRNAs and their corresponding coefficients were identified to construct the prognostic signature in HCC. The risk score was calculated using the following formula:
where, Coef is the coefficient value, and Expr is the expression value of selected bile acid metabolism-related lncRNAs. This formula was used to calculate the risk score for each patient with HCC. According to the median value of the risk score, patients were divided into low- and high-risk groups. Thereafter, Kaplan–Meier analysis and receiver operating characteristic (ROC) curve analysis were used to evaluate the prognosis in these groups. In addition, multivariate Cox regression analysis was performed to examine the usefulness of the signature as an independent biomolecular indicator for the prediction of survival.
To further validate the robustness of our model, we conducted external validation using the liver cancer dataset GSE14520 from the Gene Expression Omnibus (GEO) database. However, as the GEO dataset lacked expression data for the five lncRNAs included in our model, we referred to previous literature52,53 and employed the Gene Set Variation Analysis (GSVA) to establish an alternative scoring system. This system was used to delineate differential gene profiles between the high-risk and low-risk groups. Utilizing the risk model based on the five lncRNAs we previously identified, we determined distinct expression features, denoted as Gene Signature A and Gene Signature B, in the low-risk and high-risk groups within the TCGA-LIHC dataset, thereby further confirming the stability of our risk model. Gene Signature A comprises genes found to be highly expressed in the high-risk group, while Gene Signature B encompasses genes highly expressed in the low-risk group. By analyzing the differences between these gene signatures, as well as their correlation with Risk Scores (RS), this method serves as an alternative risk scoring approach. Subsequently, RS scores were computed for the external GEO cohort, and the Kaplan–Meier method was applied to compare the overall survival (OS) between high RS score and low RS score groups.
Nomogram construction
Various clinical traits, including age, sex, stage, grade, Child–Turcotte–Pugh (CTP) grade of liver function, and the risk score were incorporated to construct a prognostic nomogram that can predict the 1-, 3-, and 5-year survival of patients with HCC. Calibration curves were plotted to test whether the predicted survival rate was consistent with the actual survival rate.
Functional enrichment analysis of the bile acid and bile salt metabolism-related lncRNA predictive signature
We carried out gene set enrichment analysis (GSEA) to investigate the mechanisms significantly correlated with our bile acid metabolism-related lncRNA predictive signature. GSEA was used to determine the pathway genes that were enriched54. The GSEA 4.1.0 software (http://www.broad.mit.edu/gsea/) was used for the analysis. Nominal p values < 0.05 denoted statistically significant differences.
Estimation of tumor-infiltrating immune cells
TCGA immune cell infiltration data (e.g., CD8 + T cells, B cells, CD4 + T cells, dendritic cells, macrophages, and neutrophils) were obtained from the tumor immune estimation resource (TIMER) database (https://cistrome.shinyapps.io/timer/). Spearman correlation analysis was used to analyze the relationship between the types of immune infiltrating cells obtained through various algorithms (e.g., xCell, TIMER, CIBERSORT) and the risk score. A bubble graph was used to demonstrate the relationship. Subsequently, the low- and high-risk groups were compared for differences in immune cell infiltration. To assess the immune microenvironment in high and low-risk groups of HCC patients, the ESTIMATE algorithm14 which can use the RNA-seq transcriptome profiles of certain genes to quantitatively estimate the scores of stromal and immune cells in the TME was applied to calculate the stromal and immune scores in high- and low-risk groups.
Signature and immune checkpoint blockade (ICB)
According to previous research, the expression of immune checkpoint blockade therapy-correlated genes may be associated with the responsiveness to ICB therapy55. We employed six key genes linked to therapy with immune checkpoint inhibitors in HCC, namely programmed death 1 (PD-1, also known as PDCD1), T-cell immunoglobulin domain and mucin domain-containing molecule-3 (TIM-3, also known as HAVCR2), cytotoxic T-lymphocyte associated antigen 4 (CTLA4), T Cell immunoreceptor with Ig and ITIM domains (TIGIT), cluster of differentiation 47 (CD47), and cluster of differentiation 276 (CD276). Spearman correlation analysis was used to explore the correlation between the above six ICB-related genes and the risk score of our model. To further explore the potential role of our lncRNA risk model in ICB treatment of patients with HCC, the expression levels of 47 immune checkpoint blockage-related genes in low- and high-risk patients were measured56.
Role of the predictive signature in predicting response to clinical treatment
To assess the predictive capacity of our model regarding therapy response in HCC, we utilized the Tumor Immune Dysfunction and Exclusion (TIDE) algorithm, available at http://tide.dfci.harvard.edu/, to predict the therapeutic effectiveness of Immune Checkpoint Inhibitors (ICIs) between the high-risk and low-risk groups. Higher TIDE prediction scores indicate a greater likelihood of immune evasion, suggesting reduced responsiveness to ICI treatment. In a further exploration of our bile acid and salt metabolism related lncRNA risk model's predictive performance in the context of tumor immunotherapy, we employed the Risk Score (RS) as an alternative to the risk score to evaluate response in the anti-PDL1 cohort (IMvigor 210). Additionally, we leveraged the "pRRophetic" package within the R project to infer the sensitivity of different risk groups to various chemotherapeutic and targeted drugs. We calculated the half-maximal inhibitory concentration (IC50) for commonly used clinical HCC treatment drugs. The estimated IC50 values for the low-risk and high-risk groups were compared using the Wilcoxon rank-sum test.
RNA extraction and real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from liver tissue using Trizol reagent (Takara, Dalian, China) following the manufacturer's instructions and referencing prior literature57. RNA concentration and quality were assessed using a NanoDrop ND-2000 spectrophotometer (Life Technologies, Grand Island, NY, USA). RNA concentration and quality were assessed using a NanoDrop ND-2000 spectrophotometer (Life Technologies, Grand Island, NY, USA). First-strand cDNA synthesis was performed using GoScript™ Reverse Transcriptase (Promega (Beijing) Biotech Co. Ltd). SYBR Green real-time PCR was conducted using TaKaRa Ex Taq® Hot Start Version (Takara, Beijing, China). Primer information can be found in Supplementary Table S8. The amplification conditions included an initial denaturation step at 95 °C for 3 min, followed by 40 cycles of denaturation at 94 °C for 45 s, annealing at 60 °C for 1 min. All real-time PCR experiments were conducted using the QuantStudio™5 Real-Time Fluorescence Quantitative PCR System (Thermo Fisher SCIENTIFIC). Each experiment was repeated three times for accuracy. To ensure reliable quantification, GAPDH mRNA expression levels were used as a reference for normalization. The amplification efficiencies for each LncRNA and GAPDH were consistent. Quantitative analysis was performed using the 2−ΔΔCT method.
Statistical analysis
All statistical analyses were performed with the R software (Version 4.0.5; R Foundation). GSEA (http://www.broadinstitute.org/gsea/index.jsp) was used to distinguish between two sets of functional annotations. All p values are two-side, and values < 0.05 denoted statistically significant differences.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Tianjin Third central Hospital (Approval number: IRB2019-034-01). All procedures involving human participants were in accordance with the ethical standards of national committee and the Helsinki declaration. Informed consent was obtained from all participants and/or their legal guardians.
Data availability
Collections of data from this study are available in public databases. The names of the repository/repositories are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2, 16018. https://doi.org/10.1038/nrdp.2016.18 (2016).
Al-Rugeebah, A., Alanazi, M. & Parine, N. R. MEG3: An oncogenic long non-coding RNA in different cancers. Pathol. Oncol. Res. 25, 859–874. https://doi.org/10.1007/s12253-019-00614-3 (2019).
El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502. https://doi.org/10.1016/S0140-6736(17)31046-2 (2017).
Quante, M. et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 21, 36–51. https://doi.org/10.1016/j.ccr.2011.12.004 (2012).
Bernstein, C. et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 85, 863–871. https://doi.org/10.1007/s00204-011-0648-7 (2011).
Lozano, E. et al. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.MCR-13-0503 (2014).
Yoshimoto, S. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. https://doi.org/10.1038/nature12347 (2013).
Allen, K., Jaeschke, H. & Copple, B. L. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 178, 175–186. https://doi.org/10.1016/j.ajpath.2010.11.026 (2011).
Hang, S. et al. Bile acid metabolites control T17 and T cell differentiation. Nature 576, 143–148. https://doi.org/10.1038/s41586-019-1785-z (2019).
Ma, C. et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360, 5931. https://doi.org/10.1126/science.aan5931 (2018).
Wei, L. et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol. Cancer 18, 147. https://doi.org/10.1186/s12943-019-1086-z (2019).
DiStefano, J. K. Long noncoding RNAs in the initiation, progression, and metastasis of hepatocellular carcinoma. Noncoding RNA Res. 2, 129–136. https://doi.org/10.1016/j.ncrna.2017.11.001 (2017).
Ma, Y., Harris, J., Li, P. & Cao, H. Long noncoding RNAs-a new dimension in the molecular architecture of the bile acid/FXR pathway. Mol. Cell Endocrinol. 525, 111191. https://doi.org/10.1016/j.mce.2021.111191 (2021).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. https://doi.org/10.1038/ncomms3612 (2013).
Geeleher, P., Cox, N. J. & Huang, R. S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 15, R47. https://doi.org/10.1186/gb-2014-15-3-r47 (2014).
Režen, T. et al. The role of bile acids in carcinogenesis. Cell. Mol. Life Sci. 79, 243. https://doi.org/10.1007/s00018-022-04278-2 (2022).
Kovács, P. et al. Lithocholic acid, a metabolite of the microbiome, increases oxidative stress in breast cancer. Cancers https://doi.org/10.3390/cancers11091255 (2019).
Mikó, E. et al. Microbiome-microbial metabolome-cancer cell interactions in breast cancer-familiar, but unexplored. Cells https://doi.org/10.3390/cells8040293 (2019).
Tsuei, J., Chau, T., Mills, D. & Wan, Y.-J.Y. Bile acid dysregulation, gut dysbiosis, and gastrointestinal cancer. Exp. Biol. Med. 239, 1489–1504. https://doi.org/10.1177/1535370214538743 (2014).
Kainuma, M., Takada, I., Makishima, M. & Sano, K. Farnesoid X receptor activation enhances transforming growth factor β-induced epithelial-mesenchymal transition in hepatocellular carcinoma cells. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19071898 (2018).
Hu, Y. et al. Bile acids regulate nuclear receptor (Nur77) expression and intracellular location to control proliferation and apoptosis. Mol. Cancer Res. 13, 281–292. https://doi.org/10.1158/1541-7786.MCR-14-0230 (2015).
Nguyen, P. T. et al. Senescent hepatic stellate cells caused by deoxycholic acid modulates malignant behavior of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 146, 3255–3268. https://doi.org/10.1007/s00432-020-03374-9 (2020).
Yang, C.-S. et al. Small heterodimer partner-targeting therapy inhibits systemic inflammatory responses through mitochondrial uncoupling protein 2. PLoS ONE 8, e63435. https://doi.org/10.1371/journal.pone.0063435 (2013).
Huang, H. et al. Integrated analysis of microbiome and host transcriptome reveals correlations between gut microbiota and clinical outcomes in HBV-related hepatocellular carcinoma. Genome Med. 12, 102. https://doi.org/10.1186/s13073-020-00796-5 (2020).
Zollner, G. et al. Hepatobiliary transporter expression in human hepatocellular carcinoma. Liver Int. 25, 367–379 (2005).
Halilbasic, E., Claudel, T. & Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 58, 155–168. https://doi.org/10.1016/j.jhep.2012.08.002 (2013).
An, P., Zeng, Z. & Winkler, C. A. The loss-of-function S267F variant in HBV receptor NTCP reduces human risk for HBV infection and disease progression. J. Infect. Dis. 218, 1404–1410. https://doi.org/10.1093/infdis/jiy355 (2018).
Hu, H. H. et al. The rs2296651 (S267F) variant on NTCP (SLC10A1) is inversely associated with chronic hepatitis B and progression to cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis B. Gut 65, 1514–1521. https://doi.org/10.1136/gutjnl-2015-310686 (2016).
Petrick, J. L. et al. Prediagnostic concentrations of circulating bile acids and hepatocellular carcinoma risk: REVEAL-HBV and HCV studies. Int. J. Cancer 147, 2743–2753. https://doi.org/10.1002/ijc.33051 (2020).
Li, X. et al. Cholangiocyte-derived exosomal lncRNA H19 promotes macrophage activation and hepatic inflammation under cholestatic conditions. Cells https://doi.org/10.3390/cells9010190 (2020).
Lan, X. et al. The human novel gene LNC-HC inhibits hepatocellular carcinoma cell proliferation by sequestering hsa-miR-183-5p. Mol. Ther. Nucleic Acids 20, 468–479. https://doi.org/10.1016/j.omtn.2020.03.008 (2020).
Lan, X. et al. Long noncoding RNA lnc-HC regulates PPARγ-mediated hepatic lipid metabolism through miR-130b-3p. Mol. Ther. Nucleic Acids 18, 954–965. https://doi.org/10.1016/j.omtn.2019.10.018 (2019).
Lv, S., Wang, W., Wang, H., Zhu, Y. & Lei, C. PPARγ activation serves as therapeutic strategy against bladder cancer via inhibiting PI3K-Akt signaling pathway. BMC Cancer 19, 204. https://doi.org/10.1186/s12885-019-5426-6 (2019).
Zou, Y. et al. Polyunsaturated fatty acids from astrocytes activate PPARγ signaling in cancer cells to promote brain metastasis. Cancer Discov. 9, 1720–1735. https://doi.org/10.1158/2159-8290.CD-19-0270 (2019).
Chang, W. H. & Lai, A. G. The pan-cancer mutational landscape of the PPAR pathway reveals universal patterns of dysregulated metabolism and interactions with tumor immunity and hypoxia. Ann. N. Y. Acad. Sci. 1448, 65–82. https://doi.org/10.1111/nyas.14170 (2019).
Li, Z., Wu, T., Zheng, B. & Chen, L. Individualized precision treatment: Targeting TAM in HCC. Cancer Lett. 458, 86–91. https://doi.org/10.1016/j.canlet.2019.05.019 (2019).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: The next generation. Cell 144, 646–674. https://doi.org/10.1016/j.cell.2011.02.013 (2011).
Sangro, B. et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 73, 1460–1469. https://doi.org/10.1016/j.jhep.2020.07.026 (2020).
Balkwill, F. R., Capasso, M. & Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 125, 5591–5596. https://doi.org/10.1242/jcs.116392 (2012).
Tobin, J. W. D. et al. Progression of disease within 24 months in follicular lymphoma is associated with reduced intratumoral immune infiltration. J. Clin. Oncol. 37, 3300–3309. https://doi.org/10.1200/JCO.18.02365 (2019).
Zhang, Y. & Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 17, 807–821. https://doi.org/10.1038/s41423-020-0488-6 (2020).
Hollern, D. P. et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell https://doi.org/10.1016/j.cell.2019.10.028 (2019).
Bruni, D., Angell, H. K. & Galon, J. The immune contexture and immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680. https://doi.org/10.1038/s41568-020-0285-7 (2020).
Peña-Romero, A. C. & Orenes-Piñero, E. Dual effect of immune cells within tumour microenvironment: Pro- and anti-tumour effects and their triggers. Cancers 14, 1681. https://doi.org/10.3390/cancers14071681 (2022).
Xing, C., Sun, S. G., Yue, Z. Q. & Bai, F. Role of lncRNA LUCAT1 in cancer. Biomed. Pharmacother. 134, 111158. https://doi.org/10.1016/j.biopha.2020.111158 (2021).
Zheng, A. et al. Long non-coding RNA LUCAT1/miR-5582-3p/TCF7L2 axis regulates breast cancer stemness via Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 38, 305. https://doi.org/10.1186/s13046-019-1315-8 (2019).
Zhou, Q. et al. LUCAT1 promotes colorectal cancer tumorigenesis by targeting the ribosomal protein L40-MDM2-p53 pathway through binding with UBA52. Cancer Sci. 110, 1194–1207. https://doi.org/10.1111/cas.13951 (2019).
Huan, L. et al. Hypoxia induced LUCAT1/PTBP1 axis modulates cancer cell viability and chemotherapy response. Mol. Cancer 19, 11. https://doi.org/10.1186/s12943-019-1122-z (2020).
Jiao, Y., Li, Y., Ji, B., Cai, H. & Liu, Y. Clinical value of lncRNA LUCAT1 expression in liver cancer and its potential pathways. J. Gastrointestin. Liver Dis. 28, 439–447. https://doi.org/10.15403/jgld-356 (2019).
Zhu, J. et al. Construction of a novel LncRNA signature related to genomic instability to predict the prognosis and immune activity of patients with hepatocellular carcinoma. Front. Immunol. 13, 856186. https://doi.org/10.3389/fimmu.2022.856186 (2022).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Zhao, Z. et al. The DNA damage repair-related lncRNAs signature predicts the prognosis and immunotherapy response in gastric cancer. Front. Immunol. 14, 1117255. https://doi.org/10.3389/fimmu.2023.1117255 (2023).
Xiao, S., Liu, X., Yuan, L. & Wang, F. A ferroptosis-related lncRNAs signature predicts prognosis and therapeutic response of gastric cancer. Front. Cell Dev. Biol. 9, 736682. https://doi.org/10.3389/fcell.2021.736682 (2021).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 (2005).
Goodman, A., Patel, S. P. & Kurzrock, R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol. 14, 203–220. https://doi.org/10.1038/nrclinonc.2016.168 (2017).
Hu, J. et al. Differential expression of the gene in pan-cancer and its related mechanism. Front. Cell Dev. Biol. 9, 700661. https://doi.org/10.3389/fcell.2021.700661 (2021).
Zhang, K. et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFβ and notch pathways. Nat. Commun. 8, 144. https://doi.org/10.1038/s41467-017-00204-4 (2017).
Funding
This research was sponsored by Tianjin Municipal Health Science and Technology Project (Nos. TJWJ2022XK029, TJWJ2021ZD003), Tianjin Key Medical Discipline (Specialty) Construction Project (No. TJYXZDXK-034A), and Natural Science Foundation of Tianjin Science and Technology Bureau (No. 21JCZDJC01050).
Author information
Authors and Affiliations
Contributions
H.T. and X.H.L. conceived and designed the study. C.H., L.J. and X.B.G. were responsible for analysis of the research data and trial operations. G.Y.T. directed the projects and designed the experiments. S.J.X. was responsible for the collection and analysis of liver cancer patient samples and relevant clinical information. C.H. drafted the manuscript. H.T., X.B.G., L.J. and Y.Z.J. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cui, H., Lian, J., Xu, B. et al. Identification of a bile acid and bile salt metabolism-related lncRNA signature for predicting prognosis and treatment response in hepatocellular carcinoma. Sci Rep 13, 19512 (2023). https://doi.org/10.1038/s41598-023-46805-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46805-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
