
Abstract
Significant progress has been made in preventing severe COVID-19 disease through the development of vaccines. However, we still lack a validated baseline predictive biologic signature for the development of more severe disease in both outpatients and inpatients infected with SARS-CoV-2. The objective of this study was to develop and externally validate, via 5 international outpatient and inpatient trials and/or prospective cohort studies, a novel baseline proteomic signature, which predicts the development of moderate or severe (vs mild) disease in patients with COVID-19 from a proteomic analysis of 7000 + proteins. The secondary objective was exploratory, to identify (1) individual baseline protein levels and/or (2) protein level changes within the first 2 weeks of acute infection that are associated with the development of moderate/severe (vs mild) disease. For model development, samples collected from 2 randomized controlled trials were used. Plasma was isolated and the SomaLogic SomaScan platform was used to characterize protein levels for 7301 proteins of interest for all studies. We dichotomized 113 patients as having mild or moderate/severe COVID-19 disease. An elastic net approach was used to develop a predictive proteomic signature. For validation, we applied our signature to data from three independent prospective biomarker studies. We found 4110 proteins measured at baseline that significantly differed between patients with mild COVID-19 and those with moderate/severe COVID-19 after adjusting for multiple hypothesis testing. Baseline protein expression was associated with predicted disease severity with an error rate of 4.7% (AUC = 0.964). We also found that five proteins (Afamin, I-309, NKG2A, PRS57, LIPK) and patient age serve as a signature that separates patients with mild COVID-19 and patients with moderate/severe COVID-19 with an error rate of 1.77% (AUC = 0.9804). This panel was validated using data from 3 external studies with AUCs of 0.764 (Harvard University), 0.696 (University of Colorado), and 0.893 (Karolinska Institutet). In this study we developed and externally validated a baseline COVID-19 proteomic signature associated with disease severity for potential use in both outpatients and inpatients with COVID-19.
Similar content being viewed by others
Introduction
The 2019 novel coronavirus disease (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has persisted as one of the leading causes of death worldwide1,2. While significant progress has been made in preventing severe disease through the development of effective vaccines, we still lack a validated baseline predictive biologic signature for the development of severe disease in both outpatients and inpatients infected with COVID-19. Previous studies are limited by focusing on patients that required hospitalization or an emergency department (ED) visit and thus skewed towards sicker patients. Additionally, previous studies were limited by low sample sizes. The ALPS-COVID trials were 2 multi-center randomized controlled trials which evaluated the efficacy of losartan as a treatment in patients with COVID-193,4. In addition to populations present in other proteomic signature generation studies, the outpatient ALPS-COVID trial also enrolled symptomatic outpatients that did not require any ED or urgent care visits. Thus, the objective of this study was to develop and externally validate, via 3 additional international outpatient and inpatient trials, a novel baseline proteomic signature which predicted the development of moderate or severe (vs mild) disease in patients with COVID-19 from a proteomic analysis of 7000 + proteins. The secondary objective was exploratory, to identify (1) individual baseline protein levels and/or (2) protein level changes within the first 2 weeks of acute infection that are associated with the development of moderate/severe (vs mild) disease.
Methods
Proteomic signature development: patient population
This study was approved by the Advarra central institutional review board (Losartan for COVID-19 Outpatient trial: Advarra Pro00042760; Losartan for COVID-19 Inpatient trial: Advarra Pro00042757) and all participants provided written informed consent. All methods were performed in accordance with relevant guidelines and regulations. All data were fully de-identified prior to analysis. Clinical trial data is available from the corresponding author on reasonable request and also publicly available on Vivli-Registry ID NCT04312009 (https://vivli.org/resources/resources/).
Patients who participated provided blood samples for one of two multi-center, placebo-controlled randomized clinical trials to evaluate the efficacy of losartan in hospitalized and non-hospitalized patients with COVID-193,4. At each day of follow up, COVID-19 disease severity was recorded using a modification of the World Health Organization (WHO) ordinal scale: (0) Death, (1) Hospitalized, on invasive mechanical ventilation or ECMO, (2) Hospitalized on non-invasive mechanical ventilation or high flow devices, (3) Hospitalized requiring Oxygen, (4) Hospitalized, not on oxygen, (5) Not hospitalized. To handle missing day-15 outcomes, an a priori decision was made to assign all outpatients missing a day 15 outcome an outcome of 5. Inpatients who were missing a day 15 outcome had their last observed outcome carried forward.
Primary outcome
We constructed a binary outcome, “mild” vs. “moderate/severe,” to reflect the worst severity experienced by each individual. Patients were categorized as having “mild” disease if they had a WHO score of 5. “Moderate/severe disease” was defined as at least one WHO score of 4 or lower.
Proteomics data, quality control, preprocessing, filtering, and normalization
Blood samples were collected at baseline (at the time of study randomization) and at day 15 following randomization. Samples were collected in Ethylenediaminetetraacetic acid (EDTA) (96.5%) or citrated (3.5%) tubes and plasma was extracted as per trial protocol within 6 h of collection. Plasma was frozen at – 80 °C and batch analyzed at the end of the trial. The SomaLogic (Boulder, CO) SomaScan aptamer panel platform which measures 7000 + proteins was used5. The SomaLogic SomaScan aptamer panel reports abundances of aptamers which target specific proteins. Multiple aptamers may map to the same protein. All analyses were done on the SomaScan aptamers and results are reported based on the corresponding protein target.
We checked for the presence of a batch effect from the EDTA and citrated storage types using guided principal components analysis (gPCA)6. We applied gPCA to sample collection day and tube type. We combined the p-values across the days using Fisher’s method (p = 0.53). As pharmacodynamics studies did not identify a significant difference in renin–angiotensin–aldosterone system (RAAS) signaling between the losartan and placebo groups, we merged data from both outpatients and inpatients who were randomized to either cohort. We removed any proteins corresponding to non-human protein targets, leaving 7301 proteins in the dataset. We removed all quality control samples and 18 individuals for whom we did not have clinical data available. We also removed 8 samples whose median signal normalization scale factor at 20% mix fell below 0.26. This left 138 subjects for downstream analysis. Of these, 113 had baseline proteomic data and day 15 outcomes, and 73 had baseline and day 15 proteomic data as well as day 15 outcomes (Fig. S1).
We used an unsupervised approach to filter proteins in this dataset of 113 patients prior to analysis. We applied a log2 transformation to the protein abundances to normalize the data. We then calculated the variances of each log2-transformed protein measured at baseline, removing any whose variance fell below the 2.5th percentile (0.0325) and above the 97.5th percentile (1.63), resulting in removal of 366 proteins, leaving 6935 for the analysis. We used a Uniform Manifold Approximation and Projection (UMAP) plot7 on the baseline protein values to visualize any clustering.
Statistical analysis
We searched for candidate proteins and protein pathways that differentiate our primary outcome using resampling, Mann–Whitney U tests, logistic regression with an elastic net penalty, and logistic regression with an overlapping group lasso penalty. Prior to the multivariate analyses, all proteins were standardized to have mean 0 and standard deviation 1 to facilitate interpretation of the results.
Baseline analysis
We applied Mann–Whitney U tests8 to each protein to compare protein expression levels in patients with mild vs. moderate/severe COVID-19. We used a false discovery rate (FDR) correction to adjust for multiple hypothesis testing9. Proteins with an FDR-adjusted p-value ≤ 0.05 were considered statistically significant.
A multivariate approach to identify candidate proteins was used to analyze the dataset considering age, sex, BMI, and race as confounders. Resampling techniques were used for statistical rigor and robustness. We split the data into 50 training and testing sets with a 70:30 ratio, stratified according to the primary outcome of disease severity to ensure a similar proportion of patients by severity. For each training set, we fit a multivariable logistic regression model with an elastic net penalty10 to assess the conditional effect of each protein on disease severity and to perform variable selection (i.e. identify important biomarkers in predicting the log-odds of moderate/severe COVID-19). We averaged the coefficients from each fit to obtain proteins with consistently non-zero coefficients. For each testing dataset, the test prediction error and the area under the receiver operating characteristic curve (AUC) were obtained to assess model performance. We averaged these metrics across the 50 splits. The penalty parameter was fixed at 0.5 across all splits. We implemented the logistic regression model with elastic net using the glmnet package in R11.
Generation of a proteomic signature
To construct our proteomic signature, we identified top low- or moderately-correlated signatures at baseline with potential to predict the primary outcome when combined with clinical covariates. To do this, we combined results from the univariate and multivariate analyses above. Since the elastic net approach could result in strongly correlated proteins, we followed Brzyski et al.12 to select low- to moderately-correlated proteins. We began with the top 50 proteins with the largest conditional effect on the primary outcome (i.e. highest absolute coefficients from the elastic net model) given all the other proteins. From this list, we tagged the protein with the smallest p-value from the univariate analyses. Then, proteins that were highly correlated (Pearson’s r > 0.5) with this protein formed a cluster; the tagged protein is a representative of this cluster. We next tagged the protein with the smallest p-value from the univariate analysis that was not included in the first cluster to form the next cluster of proteins that were highly correlated with this protein. We repeated the process of tagging proteins and forming protein clusters until all proteins were in at least one of the clusters. Given our sample size of 113 and to ensure sufficient statistical power to detect differences in patients with mild vs. moderate/severe COVID-19, we only considered a final model with the top 5 signatures and/or clinical covariates.
External validation of proteomic signature
We validated the prediction estimates from our protein signature model using data from three independent prospective studies: data from Harvard’s Mass General Hospital in Boston, MA, USA (n = 384)13, two University of Colorado hospitals, Aurora, CO, USA (n = 99)14, and Karolinska University Hospital and Haga Outpatient Clinic, Stockholm, Sweden (n = 63)15. In particular, we obtained predicted estimates (i.e. predicted outcomes and log-odds) using the estimated model and the validation datasets. Each protein was standardized to have the same mean and variance in the discovery dataset. Since the dataset from Harvard University categorized the age variable, we created a model using the same 5 protein signatures and clinical covariates as described above while changing the age to the same categorical variable as defined in the dataset from Harvard. For the other two datasets, we kept age as a continuous variable. For prediction estimates, we assessed the AUC from the receiver operating characteristic (ROC) curve, and reported sensitivity, specificity, and F-1 estimates. These estimates were obtained from the optimal cutoff point on the ROC curve (i.e. Youden’s index). We also evaluated the 5-protein signature without clinical covariates.
Exploratory analysis: protein pathways identification
We conducted two a priori designed exploratory analyses: (1) Identification of protein pathways most associated with the primary outcome and (2) association of protein level change (delta) during the first two weeks of infection and the primary outcome.
To identify protein pathways most associated with the primary outcome, we used an overlapping group lasso penalty16 in a logistic regression model. We used the ToppGene Suite17 to find pathways to which the proteins in our dataset belonged. Proteins were identified using their Ensembl ID. For each split for our primary analysis, we first fit the overlapping group lasso model on the training set considering 800 possible values for the penalty control parameter. We used fivefold cross validation to obtain the optimal penalty based on cross-validation error. In our primary analysis, we used each split’s optimal penalty and predicted severity on the test set and obtained the corresponding AUC and prediction error. We implemented the logistic regression model with group penalty using the grpregOverlap package in R and performed all analyses using R software version 4.0.0 (The R Foundation)18.
As an additional exploratory analysis, we were interested in the association of protein level change and moderate/severe disease. The change in proteins during the first two weeks of infection can provide valuable insight into host response to infection and inform an understanding of COVID-19 pathogenesis and potential therapeutic targets. We compared individuals with mild vs. moderate/severe COVID-19 disease using protein expression at baseline and the difference between baseline and day 15 protein expression (termed delta). For each protein and in each group, this was calculated as \(lo{g}_{2}Day 15 Expression - lo{g}_{2}Baseline Expression\).
Results
Proteomic signature development: study population
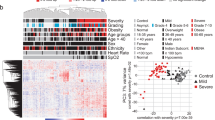
Table 1 summarizes the demographics of the participants used to generate the proteomic signature. The cohort of patients with moderate/severe COVID-19 (as compared with patients with mild COVID-19) were older in age (median: 58 [IQR: 48–66] vs median: 38.0 [IQR: 26.25–51] and had a higher BMI (median 32 [IQR 26.5–35.1] vs median 26.7 [IQR 24.2–31.6]). A larger percentage of individuals in the moderate/severe group (as compared with patients with mild COVID-19) identified as Black or African American (43.6% [n = 17] vs. 4.1% [n = 3]) or Latinx (17.9% [n = 7] vs 9.5% [n = 7]) and a lower percentage identified as white (28.2% [n = 11] vs. 78.4% [n = 58]). The majority of patients had WHO severity category 5 (not hospitalized) at baseline (Fig. 1A). Figure 1B, summarizes the top 2 components of baseline protein expression highlighting both separation between patients with moderate/severe vs mild COVID-19.

(A) Bar graph shows the counts for each WHO modified severity category within the moderate/severe group. (B) Uniform manifold approximation and projection plot (UMAP) using baseline protein expression shows clustering of patients with moderate/severe COVID-19 and mild COVID-19.
Identification of proteins with largest conditional effect on moderate/severe COVID-19 infection
Univariate analysis
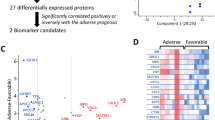
At baseline, 4110 out of 6935 proteins were significant at a FDR level of 0.05 comparing patients with mild vs. moderate/severe COVID-19. Figure 2 shows a volcano plot for the negative log10 p-value vs. fold change difference for the baseline proteins. 83 proteins are significant after FDR adjustment with a fold change difference magnitude greater than 1, and 17% of proteins (59/83) had higher median expression in moderate/severe cases. We summarize the top 10 baseline proteins in Table S1.

Volcano plot of -log10 (p-value) from Mann–Whitney U tests comparing each protein between moderate/severe and mild COVID-19 cases vs. the fold change difference in log2 protein expression between moderate/severe and mild COVID-19 cases. Proteins shown in dark blue are significant for COVID-19 severity according to the unadjusted p-value after log10 transformation, and proteins shown in light blue are significant according to the adjusted p-value after log10 transformation. Proteins selected for the 5-protein signature are labeled. Fold Difference is log2 protein for moderate/severe group—log2 protein for mild group. Values > 0 indicate the protein is elevated in the moderate/severe group. Values < 0 indicate the protein is elevated in the mild group. Values = 0 indicate the same protein levels in the two groups.
Multivariable analysis
Using logistic regression with an elastic net penalty to predict moderate/severe disease, 563 proteins at baseline were selected in at least one of the fifty training/test splits. Table 2 summarizes the top 10 proteins identified as increasing the risk for moderate/severe COVID-19. The average test error rate was 4.7% and the average test AUC was 0.964. The direction of the effects of each protein was consistent across the fifty training/test splits.
Baseline protein signature
Combining the univariate and multivariate results, we identified 5 proteins with sufficient power to differentiate patients with mild vs. moderate/severe COVID-19. These proteins were Afamin (seq.18196.8), I-309 (seq.13687.5), NKG2A, PRS57, and LIPK. The Pearson correlation between pairs of proteins was low to moderate (r < 0.5) (Fig. S2). A logistic regression model of the outcome with these five proteins resulted in an AUC of 0.9548 and an error rate of 3.54%. The variance inflation factor (VIF) for these proteins ranged from 1.09 to 2.19 which indicated that the proteins were not highly linearly related. For the sake of brevity, we shall use the target names to refer to these protein signatures without further specification.
We further investigated inclusion of the clinical covariates age, gender, BMI, and treatment into this signature and age was ultimately included. BMI and sex were excluded due to convergence issues and high VIFs. Treatment was excluded due to a null effect from the inpatient and outpatient clinical trials. The addition of age to the 5 proteins increased the AUC (0.9804) and reduced the error rate (1.77%). The VIFs were also acceptable (< 5). Figure 3 shows a heatmap of log-transformed, scaled abundances and boxplots of the 5 proteins.

Heatmap and boxplots of the 5-protein signature. Proteins are log2-transformed and scaled. Heatmaps were generated using R Statistical Software v4.2.1.
External validation
Age distribution followed a bell-shaped curve across all 4 studies (Fig. S3a). Figure S3b–h present box-whisker plots for the identified signatures (and the aptamers under the same name) across the studies. Figure 4a shows notable variation in the distribution of each protein across all studies. The AUCs in the three studies are 0.764, 0.696, and 0.893 for the model with the 5 protein signatures and age using datasets from Harvard University, the University of Colorado, and Karolinska Institutet, respectively (Fig. 4b). A model with only the protein signature without any covariates resulted in the following AUCs for the three datasets, respectively: 0.722, 0.715, and 0.876. Sensitivity, specificity, and F-1 score at the cutoff point with optimal Youden index are also shown in Table 3.

(A) Boxplots showing distributions of each protein across the four datasets. (B) ROC curves for the model with protein signatures and age using each of the dataset when validating.
Exploratory analysis 1: identification of protein pathways associated with development of moderate/severe COVID-19 infection
From logistic regression with overlapping group lasso penalty models, we identified 54 baseline pathways selected in at least one of the fifty training/test splits. The average test error and average test AUC were 4.4% and 0.9875, respectively. These results were robust to the number of folds considered in obtaining the optimal penalty parameter when we added a ridge penalty in addition to the lasso penalty. The most aberrant pathway identified was dysfunction in the Extrinsic Pathway of Fibrin Clot Formation, specifically coagulation factors VII (lower in patients with moderate/severe disease) and IX (higher in patients with moderate/severe disease) (Fig. S4). In addition to the Extrinsic Pathway of Fibrin Clot Formation, the Scavenging by Class B Receptors pathway was also selected across all 50 data splits (Table 4). Ligand-receptor interactions was selected in 98% of the splits, and PI3K/AKT signaling was selected in 78% of the splits.
Exploratory analysis 2: change in protein levels associated with severe COVID-19 infection
Univariate analysis
In addition to investigating if baseline protein levels were associated with severity, we also studied if a delta, or change, in protein levels (baseline to day 15) was associated with disease severity by day 15. 1144 proteins were significant after FDR adjustment between baseline and day 15 when comparing mild vs. moderate/severe patients using Mann–Whitney U tests. Figure S5 shows a volcano plot for the negative log10 p-values vs. fold change difference for the protein deltas between baseline and day 15. Of note, CK-MB and CK-MM are located at the upper left quadrant, suggesting a highly significant p-value and relatively large log fold change, and lower levels of CK-MB and CK-MM in the moderate/severe group over 15 days. Retinol-binding protein 4 (RBP) and HSP-70 locate to the upper right quadrant, with higher levels observed in the moderate/severe group over 15 days. We summarize the top 10 protein deltas in Table 5 and show plots of these proteins in Fig. 5.

Line plots of the top 4 proteins identified in the delta analysis. CK-MM creatine kinase MM, CK-MB creatine kinase MB, RBP-4 retinol binding protein 4. The moderate/severe group has lower levels of CK-MM and CK-MB over 15 days. The moderate/severe group has higher levels of retinol-binding protein 4 and HSP70 over 15 days.
Discussion
The objective of this study was to develop and externally validate a baseline prognostic proteomic signature using data from patients that participated in 5 multi-center randomized controlled or prospective biomarker COVID-19 trials. To date, no previously developed prognostic proteomic signatures include data from both outpatients, emergency department, and inpatients with COVID-19. The deliverable of this project, is an internationally validated prognostic signature, which could be used in either the outpatient or inpatient setting. The secondary objective was exploratory, to identify (1) protein pathways and/or (2) protein level changes within the first 2 weeks of acute infection that are associated with the development of moderate/severe (vs mild) disease. During the secondary exploratory analysis we noted the following 3 findings worthy of future investigation: (1) baseline dysregulation of the extrinsic pathway of fibrin clot formation, ligand receptors, PI3K signaling, as well as visceral fat deposits and metabolic pathways, were most often associated with severe disease in patients with COVID-19, (2) a reduction in CK-MB/CK-MM during the initial 2 weeks of COVID-19 infection is associated with severe disease, and an (3) increasing HSP 70 and Retinol-binding protein 4 are associated with severe disease.
Proteomic profiling of sera can serve as a novel predictive biomarker early in the course of infection to predict with high accuracy (AUC 0.98 and error rate of 1.77%) if a patient will develop disease requiring hospitalization. Importantly, this biomarker outperforms published performance metrics of current clinical prognostic models19,20,21. To date, there have been over 13 published proteomic prognostic signatures with AUCs ranging from 0.8 to 0.9913,22,23,24,25,26. Similarly, previously described signatures have analyzed plasma (similar to our study) or serum using proximity extension assay (PEA) technology or mass spectrometry-based strategies, whereas in this study we used an aptamer-based proteomic platform. Of the previously published baseline proteomic signatures which sought to predict hospitalization or severe outcomes, only 5 included more than 50 patients. (1) Bauer et al.24 enrolled 97 patients that presented to the ED and identified a 5-protein signature ADM, IL-6, MCP-3, TRAIL-R2, and PD-L1 with AUROC 0.8–0.87); however, this was not externally validated. (2) Demichev et al.27 enrolled 139 hospitalized patients and developed a proteomic and clinical signature which could detect the need for mechanical ventilation with AUROC 0.99. This signature was then externally validated on a cohort of 99 hospitalized patients (AUROC 0.97). (3) Filbin et al.13 enrolled 306 patients that presented to the ED (23 were discharged home, and the rest admitted) and developed a predictive classifier which included the following proteins: IL-6, IL-1RL1, PTX3, IL-1RN, KRT-19, and TRIAP1 which was associated with an AUROC 0.85 to predict severe (mechanical ventilation or death) vs mild/moderate disease. (4) Al-Nesf et al.23 enrolled 100 patients that were admitted to the hospital and identified a 12-protein (MSTN, CLEC4C, PTX3, TNC, SMOC1, HGF, IL6, IL1RL1, AREG, KRT19, TNFRSF10B, IL18R1) and 7 clinical test signature which was associated with an AUROC 0.99. This signature was then externally validated using Filbin’s cohort with AUROC 0.84. (5) Finally, Perreau et al.26 enrolled 98 patients that were admitted to the hospital and identified a two protein signature (HGF and CXCL13) that predicted severe COVID-19 disease with AUROC 0.9–0.97.
It is important to point out that each of these studies were skewed towards patients that had symptoms severe enough requiring them to present to the ED. Thus it makes sense that the markers they identified which were predictive were also inflammatory cytokines which were likely upregulated at the time the patient presented to the ED and more associated with the development of severe disease and death. However, what is critically needed is a signature that can be used in mildly symptomatic outpatients at baseline and predict which patients will decompensate and require hospitalization. Our study sought to fill this void by leveraging a simultaneously enrolling outpatient biomarker trial in COVID-19. An additional strength of our study is the integration into a randomized controlled trial to ensure homogeneity of patients at study enrollment compared to other studies to date that suffer from heterogeneity in patient comorbidities or treatment received at the time of protein profiling.
External validation
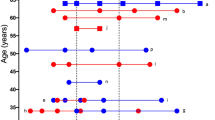
The AUCs using datasets from the Harvard University and University of Colorado studies are good, although not very high, compared to the AUC in the discovery dataset. Of the external datasets, the prediction of severe disease was lowest in the Colorado dataset (AUROC 0.69). The difference in prediction estimates between these studies might be attributed to the difference in the platforms for proteomic profiling used in the studies. We used the same platform as the Karolinska Institutet study, but our platform was different from the platforms used in the Harvard University and University of Colorado studies. The datasets from the Harvard and University of Colorado studies provide the protein Uniprot IDs but do not provide the Somascan aptamer sequence IDs. As a result, we can match the proteins in our signature using the Uniprot ID but not using the sequence ID so different aptamers may have corresponded to these proteins across studies. As elucidated in Fig. S3b–h, noticeable differences emerge in the distribution of distinct aptamers, which provide a possible explanation of why the performance of our model on the Karolinska dataset is better when compared to the Harvard and University of Colorado datasets. Further, the observed differences in protein levels and age distribution may explain the differences in prediction estimates across the three studies. Figure 4a highlights the distribution of proteins across each dataset. Here we note that the 5 protein levels were all lower in our dataset compared to levels from the Harvard University or the University of Colorado study. One hypothesis for our model's high external validation on the Karolinska data may be due to Afamin, the protein with the highest coefficient estimate in our model. In Fig. 4a, we observe a very similar distribution of Afamin between our dataset and that from Karolinska Institutet. We also noted that Afamin was the only protein from the five signatures that is differentially expressed consistently in all four datasets. Figure S3 highlights the age distribution across datasets. We note that the overall age is higher in the dataset from Karolinska Institutet than the other two datasets. When we categorized age, we observed that the general distribution of ages are similar across the studies. However, the proportions of individuals whose age is between 65 and 79 and whose age is over 80 from the Harvard University study are higher than the other studies. There were no patients with ages below 20 or greater than 80 years old from the Karolinska study, no participants below 20 in the Harvard University study, and no patients over 80 years old in the University of Colorado study.
Baseline proteomic pathways associated with the development of severe disease
In our study, the most aberrant pathway at baseline that was associated with that development of moderate/severe vs mild disease was dysfunction in the coagulation system, specifically coagulation factors VII (lower in patients with moderate/severe disease) and IX (higher in patients with moderate/severe disease) (Fig. S4). This suggests the specific aberration associated with the development of severe COVID-19 is an overactive intrinsic pathway early in the disease course28. We identified patients with moderate/severe disease had significantly reduced Factor VII at baseline, patients with moderate/severe disease also had increased tissue factor pathway inhibitor (TFPI), an inhibitor of the extrinsic pathway (Fig. S4). The intrinsic pathway can be initiated through contact activation (via FXII) or by FXI. Other cohorts23,29,30 have found severe illness more strongly associated with abnormal complement and inflammatory protein levels; however, in one of these studies, an elevated plasma level of Factor XII was found to be protective against severe disease30, which is congruent with our findings. The ligand-receptor complex pathway, specifically the sonic hedgehog (SHH)-CDON axis, was also frequently dysregulated at baseline between patients who ultimately developed moderate/severe disease and mild disease. In addition to its regulatory roles in embryonic development, the hedgehog family proteins continue to direct homeostasis and turnover of certain cell types in adults31,32,33,34, and dysregulation of SHH signaling has previously been implicated as a potential pathway driving loss of smell in COVID-1934,35. Our findings support other studies which have also identified aberrant PI3K/AKT signaling as one of the most associated pathways in patients that develop severe COVID-1936,37. The PI3K/AKT pathway is a potent activator of mTOR and NF-κB signals as well as platelet activity, and activation of PI3K may be effected via angiotensin II signaling38,39. Biophysical modeling has predicted that mTOR inhibition disrupts the SARS-CoV-2 viral life cycle40,41. Finally, we observed significant dysregulation in visceral fat deposit and metabolic pathways, a finding may provide biologic plausibility for the elevated risk of severe disease course associated with obesity and male sex as both are associated with increased visceral fat stores42.
Change in protein level associated with the development of severe disease
One of the a priori objectives of this study was to characterize the proteome change (or delta) from enrollment through day 15. Identifying temporal trends in biomarkers which are most associated with severe disease allows development of prognostic tools and provides targets to guide future therapeutics. Previous studies have demonstrated an association between elevations of multiple isoforms of CK-MB at initial evaluation and increased odds of severe COVID-19 disease43,44,45,46,47,48. However, whereas there is an overlap between entry CK-MB and CK-MM for mild and moderate/severe disease groups, the reduction in these biomarkers over 15 days was most strongly associated with moderate/severe disease. The overlap with the muscle pathology, post-ICU syndrome and ICU-acquired weakness seem to be vast49,50, and while mechanistic understanding the preponderance of autopsy studies point to an immune mediated myopathy which may be related to the interferon I response rather than hypoxic injury. As the degree of CK elevation is likely related to both a patient’s overall muscle mass and the state of their kidney function, the change in CK over time, rather than a binary ‘elevated’ or ‘normal,’ appears to provide better differentiation of disease outcomes. This is of great interest as CK isoforms is a common and readily available assay.
Limitations
This study is subject to the following limitations. First, patients for the proteomic signature development were included from two different randomized controlled trials. As each trial was run by the same team, the inclusion/exclusion criteria were nearly identical; the only difference was that one trial enrolled symptomatic outpatients with COVID-19. Second, this was a drug trial where patients were randomized to receive losartan vs. placebo; thus, it is possible when doing the delta analysis that patients were influenced by the study drug. However, it is important to note that in one of the trials that investigated pharmacokinetic and pharmacodynamic effects of losartan, losartan did not have significant downstream effects on angiotensin 2, angiotensin-[1-7], ACE or ACE2 within the first 15 days of treatment. Despite this, it is possible given losartan’s inhibition on the angiotensin 1 receptor that there were other pharmacodynamic effects not investigated in the initial trial. Third, the analysis took an agnostic approach with a well-curated but not selective patient cohort. We were interested in patients’ baseline proteomic profiles using samples collected at the time of their first healthcare encounter. We recognize there is variation from patient to patient when COVID-19 presentation arises and when a patient might seek out healthcare. Fourth, applying our signature in other sample cohorts provided insight into its performance, though evaluating these proteins with other assays is needed to address platform bias. Validation of the 5-protein signature and other key results with an orthogonal platform is a necessary future direction. Finally, our analysis did not consider samples from those without COVID-19 so it is unclear if our findings are exclusive to COVID-19 infection, and if not, to what extent our findings differ between those with and without COVID-19. A future analysis investigating the ability of this signature to discriminate between individuals with and without COVID-19 would be worthwhile.
Conclusion
In this study we developed and externally validated a baseline COVID-19 proteomic signature associated with disease severity for potential use in both outpatients and inpatients with COVID-19.
Data availability
Clinical trial data is available from the corresponding author on reasonable request and also publicly available on Vivli-Registry ID NCT04312009 (https://vivli.org/resources/resources/).
References
Ahmad, F. B. & Anderson, R. N. The leading causes of death in the US for 2020. JAMA. 325(18), 1829–1830 (2021).
Woolf, S. H., Chapman, D. A. & Lee, J. H. COVID-19 as the leading cause of death in the United States. JAMA. 325(2), 123–124 (2021).
Puskarich, M. A. et al. A multi-center phase II randomized clinical trial of losartan on symptomatic outpatients with COVID-19. EClinicalMedicine. 37, 100957 (2021).
Puskarich, M. A. et al. Efficacy of losartan in hospitalized patients With COVID-19-induced lung injury: A randomized clinical trial. JAMA Netw. Open. 5(3), e222735 (2022).
Gold, L., Walker, J. J., Wilcox, S. K. & Williams, S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. Nat. Biotechnol. 29(5), 543–549 (2012).
Reese, S. E. et al. A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis. Bioinformatics. 29(22), 2877–2883 (2013).
McInnes, L., Healy, J., & Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction (2018). arXiv preprint arXiv:1802.03426.
Mann, H., & Whitney, D. On a test of whether one of two random variables is stochastically larger than the other. Vol 18 (Annals of Mathematical Statistics, 1947).
Benjamini, Y., & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Vol 57 (Royal Statistical Society, 1995).
Zou, H., & Hastie, T. Regularization and variable selection via the elastic net. Vol 67 (Royal Statistical Society, 2005).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Brzyski, D. et al. Controlling the Rate of GWAS False Discoveries. Genetics. 205(1), 61–75 (2017).
Filbin, M. R. et al. Longitudinal proteomic analysis of severe COVID-19 reveals survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell. Rep. Med. 2(5), 100287 (2021).
Sullivan, K. D. et al. The COVIDome Explorer researcher portal. Cell. Rep. 36(7), 109527 (2021).
Yu, M., Charles, A., Cagigi, A., et al. Delayed generation of functional virus-specific circulating T follicular helper cells correlates with severe COVID-19. medRxiv. 2022:2022.2007.2012.22277549.
Zeng, Y. & Breheny, P. Overlapping group logistic regression with applications to genetic pathway selection. Cancer Inf. 15, 179–187 (2016).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37(1), W305–W311 (2009).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/ (2021).
Lupei, M. I. et al. A 12-hospital prospective evaluation of a clinical decision support prognostic algorithm based on logistic regression as a form of machine learning to facilitate decision making for patients with suspected COVID-19. PLoS One. 17(1), e0262193 (2022).
Wynants, L. et al. Prediction models for diagnosis and prognosis of covid-19: Systematic review and critical appraisal. BMJ. 369, m1328 (2020).
Nuevo-Ortega, P. et al. Prognosis of COVID-19 pneumonia can be early predicted combining Age-adjusted Charlson Comorbidity Index, CRB score and baseline oxygen saturation. Sci. Rep. 12(1), 2367 (2022).
Costanzo, M. et al. COVIDomics: The proteomic and metabolomic signatures of COVID-19. Int. J. Mol. Sci. 23(5), 1 (2022).
Al-Nesf, M. A. Y. et al. Prognostic tools and candidate drugs based on plasma proteomics of patients with severe COVID-19 complications. Nat. Commun. 13(1), 946 (2022).
Bauer, W. et al. Plasma proteome fingerprints reveal distinctiveness and clinical outcome of SARS-CoV-2 infection. Viruses. 13(12), 1 (2021).
Guo, J. et al. Cytokine signature associated with disease severity in COVID-19. Front. Immunol. 12, 681516 (2021).
Perreau, M. et al. The cytokines HGF and CXCL13 predict the severity and the mortality in COVID-19 patients. Nat. Commun. 12(1), 4888 (2021).
Demichev, V. et al. A time-resolved proteomic and prognostic map of COVID-19. Cell Syst. 12(8), 780–794 (2021).
Fan, B. E. et al. COVID-19 associated coagulopathy in critically ill patients: A hypercoagulable state demonstrated by parameters of haemostasis and clot waveform analysis. J. Thromb. Thrombolysis. 51(3), 663–674 (2021).
Palmos, A. B. et al. Proteome-wide Mendelian randomization identifies causal links between blood proteins and severe COVID-19. PLoS Genet. 18(3), e1010042 (2022).
Gutmann, C. et al. SARS-CoV-2 RNAemia and proteomic trajectories inform prognostication in COVID-19 patients admitted to intensive care. Nat. Commun. 12(1), 3406 (2021).
Bhardwaj, G. et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2(2), 172–180 (2001).
Ahn, S. & Joyner, A. L. In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature. 437(7060), 894–897 (2005).
Paladini, R. D., Saleh, J., Qian, C., Xu, G. X. & Rubin, L. L. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J. Invest. Dermatol. 125(4), 638–646 (2005).
Castillo-Azofeifa, D. et al. SOX2 regulation by hedgehog signaling controls adult lingual epithelium homeostasis. Development. 145(14), 1 (2018).
Henkin, R. I. How does Covid-19 infection affect smell?. Am. J. Otolaryngol. 42(3), 102912 (2021).
Sun, F. et al. Capivasertib restricts SARS-CoV-2 cellular entry: A potential clinical application for COVID-19. Int. J. Biol. Sci. 17(9), 2348–2355 (2021).
Shin, J. et al. SARS-CoV-2 infection impairs the insulin/IGF signaling pathway in the lung, liver, adipose tissue, and pancreatic cells via IRF1. Metabolism. 133, 155236 (2022).
Cheng, W. H. et al. Renin activates PI3K-Akt-eNOS signalling through the angiotensin AT(1) and Mas receptors to modulate central blood pressure control in the nucleus tractus solitarii. Br. J. Pharmacol. 166(7), 2024–2035 (2012).
Saward, L. & Zahradka, P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ. Res. 81(2), 249–257 (1997).
Karam, B. S. et al. mTOR inhibition in COVID-19: A commentary and review of efficacy in RNA viruses. J. Med. Virol. 93(4), 1843–1846 (2021).
Castle, B. T., Dock, C., Hemmat, M., et al. Biophysical modeling of the SARS-CoV-2 viral cycle reveals ideal antiviral targets. bioRxiv. 2020:2020.2005.2022.111237.
Favre, G. et al. Visceral fat is associated to the severity of COVID-19. Metabolism. 115, 154440 (2021).
Shi, L., Wang, Y., Wang, Y., Duan, G. & Yang, H. Meta-analysis of relation of creatine kinase-MB to risk of mortality in coronavirus disease 2019 patients. Am. J. Cardiol. 130, 163–165 (2020).
Zinellu, A., Sotgia, S., Fois, A. G. & Mangoni, A. A. Serum CK-MB, COVID-19 severity and mortality: An updated systematic review and meta-analysis with meta-regression. Adv. Med. Sci. 66(2), 304–314 (2021).
Zhu, Z. et al. Cardiac biomarkers, cardiac injury, and comorbidities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Immun. Inflamm. Dis. 9(4), 1071–1100 (2021).
Wungu, C. D. K. et al. Meta-analysis of cardiac markers for predictive factors on severity and mortality of COVID-19. Int. J. Infect. Dis. 105, 551–559 (2021).
Akbar, M. R. et al. The prognostic value of elevated creatine kinase to predict poor outcome in patients with COVID-19—A systematic review and meta-analysis. Diabetes Metab. Syndr. 15(2), 529–534 (2021).
Orsucci, D. et al. Increased creatine kinase may predict a worse COVID-19 outcome. J. Clin. Med. 10(8), 1 (2021).
Gil, S. et al. Muscle strength and muscle mass as predictors of hospital length of stay in patients with moderate to severe COVID-19: A prospective observational study. J. Cachexia Sarcopenia Muscle. 12(6), 1871–1878 (2021).
Soares, M. N. et al. Skeletal muscle alterations in patients with acute Covid-19 and post-acute sequelae of Covid-19. J. Cachexia Sarcopenia Musc. 13(1), 11–22 (2022).
Acknowledgements
The authors would like to acknowledge the University of Minnesota Acute Care Research Coordinator Group for the conduct of the ALPS-COVID trials. The authors would also like to acknowledge SomaLogic for the performance of the SomaScan assay on serum samples.
Funding
This article was funded by Gates Foundation and National Institute of Health (1R35GM142695).
Author information
Authors and Affiliations
Contributions
Concept and design: all authors. Acquisition, analysis, or interpretation of data: C.J.T., M.P., A.S.-S. Drafting of the manuscript: all authors. Critical revision of the manuscript for important intellectual content: all authors. Statistical analysis: S.C.P., S.S., K.Y., E.L., S.E.S.
Corresponding author
Ethics declarations
Competing interests
Christopher Tignanelli and Michael Puskarich are the principal investigators of the ALPS-COVID trial. Trial support was provided by the Bill and Melinda Gates Foundation and the Minnesota Partnership for Biotechnology and Medical Genomics. Sandra Safo was partly supported by grant number 1R35GM142695 from the National Institute Of General Medical Sciences of the National Institutes of Health. All other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Castro-Pearson, S., Samorodnitsky, S., Yang, K. et al. Development of a proteomic signature associated with severe disease for patients with COVID-19 using data from 5 multicenter, randomized, controlled, and prospective studies. Sci Rep 13, 20315 (2023). https://doi.org/10.1038/s41598-023-46343-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46343-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
