
Abstract
Autism spectrum disorder (ASD) is a heterogenous multifactorial neurodevelopmental condition with a significant genetic susceptibility component. Thus, identifying genetic variations associated with ASD is a complex task. Whole-exome sequencing (WES) is an effective approach for detecting extremely rare protein-coding single-nucleotide variants (SNVs) and short insertions/deletions (INDELs). However, interpreting these variants' functional and clinical consequences requires integrating multifaceted genomic information. We compared the concordance and effectiveness of three bioinformatics tools in detecting ASD candidate variants (SNVs and short INDELs) from WES data of 220 ASD family trios registered in the National Autism Database of Israel. We studied only rare (< 1% population frequency) proband-specific variants. According to the American College of Medical Genetics (ACMG) guidelines, the pathogenicity of variants was evaluated by the InterVar and TAPES tools. In addition, likely gene-disrupting (LGD) variants were detected based on an in-house bioinformatics tool, Psi-Variant, that integrates results from seven in-silico prediction tools. Overall, 372 variants in 311 genes distributed in 168 probands were detected by these tools. The overlap between the tools was 64.1, 22.9, and 23.1% for InterVar–TAPES, InterVar–Psi-Variant, and TAPES–Psi-Variant, respectively. The intersection between InterVar and Psi-Variant (I ∩ P) was the most effective approach in detecting variants in known ASD genes (PPV = 0.274; OR = 7.09, 95% CI = 3.92–12.22), while the union of InterVar and Psi Variant (I U P) achieved the highest diagnostic yield (20.5%).Our results suggest that integrating different variant interpretation approaches in detecting ASD candidate variants from WES data is superior to each approach alone. The inclusion of additional criteria could further improve the detection of ASD candidate variants.
Similar content being viewed by others

Autism spectrum disorder (ASD) comprises a collection of heterogeneous neurodevelopmental disorders that share two behavioral characteristics—difficulties in social communication and restricted, repetitive behaviors and interests1,2. The etiology of ASD has a significant genetic component, as is evident from multiple twin and family studies3,4,5,6. Yet, over the years, very few genetic causes of ASD have been discovered; thus, today, despite extensive research, an understanding of the overall genetic architecture of ASD remains obscure6,7.
The emergence of next-generation sequencing (NGS) approaches in the past decade has transformed the genetic research of complex traits8. These NGS technologies have facilitated high-throughput DNA sequencing for large cohorts of patients, allowing the comparison of multiple variants that includes single-nucleotide variants (SNVs) and short insertions/deletions (INDELs) between large groups of patients9,10,11,12. In this realm, whole-exome sequencing (WES) is particularly suitable for studying the genetics of heterogenous traits such as ASD, as it focuses on a relatively limited number of protein-coding variants9,10,13,14,15,16,17,18.
However, understanding the functional consequences of coding variants is not a trivial task. In 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published standards and guidelines to generalize sequence variant interpretation and to address the issue of inconsistent interpretation across laboratories8. The resulting system for classifying variants recommends 28 criteria (16 for pathogenic and 12 for benign variants) and provides a set of scoring rules based on variant population allele frequency, variant functional annotation, variant familial segregation, etc.8,19; Variants are classified as pathogenic (P), likely pathogenic (LP), variants of uncertain significance (VUS), likely benign (LB) or benign (B). Subsequently, multiple in-silico tools were developed to implement these ACMG/AMP criteria for annotating the prospective pathogenicity of variants detected in WES studies.
While the ACMG/AMP scoring approach is highly effective for detecting de-novo highly penetrant mutations for rare Mendelian disorders, it is less suitable for detecting inherited partially penetrant variants20. Such variants, usually annotated as VUS in terms of the ACMP/AMP criteria, are expected to contribute significantly to the risk of developing neurodevelopmental conditions, including ASD9,17,18,21,22. Thus, relying solely on the ACMG/AMP criteria for variant annotation in WES studies of ASD may result in an under-representation of susceptibility variants, which will lead to a lower diagnostic yield for ASD. To overcome this potential limitation, we have developed “Psi-Variant,” a pipeline to detect different types of likely gene-disrupting (LGD) variants, including protein truncating and deleterious missense variants. We applied Psi-Variant – in comparison with InterVar and TAPES, two variant interpretation tools that use the ACMG/AMP criteria – to a large WES dataset of ASD to evaluate the concordance between these tools to detect variants and to assess their effectiveness in detecting ASD susceptibility variants.
Methods
Study sample
Initially, the study sample comprised 250 children diagnosed with ASD who are registered in the National Autism Database of Israel (NADI)23,24 and whose parents gave consent for participation in this study. Based on our clinical records, none of the parents in the study has been diagnosed with ASD, intellectual disability, or any other type of neurodevelopmental disorder. Genomic DNA was extracted from saliva samples from participating children and their parents using Oragene®•DNA (OG-500/575) collection kits (DNA Genotek, Canada).
Whole exome sequencing
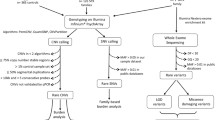
WES analysis was performed on the above-mentioned samples with Illumina HiSeq sequencers, followed by the Illumina Nextera exome capture kit at the Broad Institute as part of the Autism Sequencing Consortium, described previously11. Sequencing reads aligned to Genome Reference Consortium Human Build 38 and aggregated into BAM/CRAM files were analyzed using the Genome Analysis Toolkit (GATK)25 to generate a joint variant calling format (vcf) file for all subjects in the study. We excluded data for 30 probands from the raw vcf file due to incomplete pedigree information or low-quality WES data. Thus, WES data for 220 ASD trios were analyzed in this study (Fig. 1).

Analysis workflow for detecting LP/P/LGD variants from the WES data. InterVar and TAPES detected LP/P variants by implementing ACMG/AMP criteria. Psi-Variant detected LGD variants by utilizing in-house criteria.
Data analysis
The variant detection process in this study is outlined in Fig. 1. and explained below.
Data cleaning
The raw vcf file contained 1,935,632 variants. From this file, we removed variants with missing genotypes and/or variants in regions with low read coverage (≤ 20 reads) and/or with low genotype quality (GQ ≤ 50). In addition, we removed all common variants (i.e., those with a population minor allele frequency > 1% as per the Genome Aggregation Database (gnomAD))26,27 as well as those that did not pass the GATK’s “VQSR” and “ExcessHet” filters. Thereafter, we used an in-house machine learning (ML) algorithm to remove other potentially false-positive variants. The details of this ML algorithm and its efficiency in classifying true positive and false positive variants are summarized in the supplementary file S1. Finally, we used the pedigree structure of the families to identify proband-specific genotypes, including de-novo variants, recessively inherited variants, and X-linked variants (in males). Recessively inherited variants occur in the same loci of both copies of a gene in autosomes (where both the parents are carriers). Whereas one altered copy of the gene in chromosome X among males is defined as X-linked (males). We removed multiallelic variants from these genotypes and those classified as “de-novo” that appeared in more than two individuals in the sample. In this study, we haven’t considered compound heterozygote variants (in cis/trans).
Identifying ASD candidate variants
We searched for candidate ASD Variants using three complementary approaches. First, we applied InterVar19 and TAPES28, two commonly used publicly available command-line tools that use ACMG/AMP criteria8, to detect LP/P Variants. In addition, we assigned the ACMG/AMP PS2 criterion to all the de-novo Variants to detect additional LP/P Variants from the list of VUS. Since InterVar and TAPES are less sensitive tools for detecting recessive possible gene disrupting (LGD) variants20, we developed an integrated in-house tool, Psi-Variant, to detect LGD variants. The Psi-Variant workflow starts using Ensembl’s Variant Effect Predictor (VEP)26 to annotate the functional consequences for each variant in a multi-sample vcf file. Then, all frameshift indels, nonsense, and splice acceptor/donor variants are further analyzed by the LoFtool29 with scores of < 0.25 are annotated as intolerant variants. In addition, it applies six different in-silico tools to all missense substitutions to determine their likelihood to be “deleterious/damaging” using the following cutoffs: SIFT30 (< 0.05), PolyPhen-231 (≥ 0.15), CADD32 (> 20), REVEL33 (> 0.50), M_CAP34 (> 0.025) and MPC35 (≥ 2) as recommended by the dbNSFP database36.
Comparison between InterVar, TAPES, and Psi-variant
We compared the number of variants detected by the three tools and the percentages of variants detected by different combinations. Thereafter, we used the list of ASD genes (n = 1031) from the SFARI Gene database37 (accessed on 11 January 2022) as the gold standard to compute the odds ratio (OR) and positive predictive value (PPV) for detecting candidate ASD variants in SFARI genes. In addition, we assessed the diagnostic yield (%) for each tool combination by computing the proportion of children with detected candidate ASD variants in SFARI genes. Diagnostic yield (%) was computed as the proportion of ASD probands with at least one ASD candidate variant out of the total affected ASD probands with complete pedigree.
Software
Data storage, management, and analysis were conducted on a high-performing computer cluster in a Linux environment using Python version 3.5 and R Studio version 1.1.456. All the statistical analyses and data visualizations were incorporated into R Studio.
Ethics approval and consent to participate
Informed consent was obtained from all the families involved in the study.
Institutional review board statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Committee of Soroka University Medical Center (SOR-076-15; 17 April 2016).
Results
Determining the detection threshold for Psi-Variant
Psi-Variant integrates information from six in-silico tools that assess the functional consequence of missense variants. We examined the positive predictive value (PPV) and diagnostic yield for different numbers of in-silico tools annotating missense variants as “deleterious/damaging” (Fig. 2). There was a strong and statistically significant negative linear correlation (r = −0.98, p < 0.01) between the PPV and diagnostic yield for different detection thresholds with no noticeable optimal threshold (e.g., an increase in PPV without a comparable decrease in yield). Thus, we decided to use a conservative threshold of ≥ 5 tools for this study since it detected a similar number of variants as detected by both InterVar and TAPES (see below).

Performance of Psi-Variant using different detection thresholds. The positive predictive value (PPV; X-axis) and diagnostic yield (Y-axis) for different number of in-silico tools annotating missense variants as “deleterious/damaging”. Increasing the minimal number of detection tools increases the PPV but proportionally decreases the detection yield.
Detection of candidate variants by the different tools
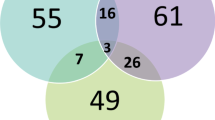
A total of 372 variants in 168 probands (highlighted in the supplementary Table S2) were detected by at least one of InterVar (n = 220), TAPES (n = 199), or Psi-Variant (n = 187) from a dataset of 2,035 high-quality, ultra-rare variants with proband-specific genotypes (Fig. 1). Of these, 55 variants (14.8%) were detected by all three tools. The highest concordance in detected variants was observed between InterVar and TAPES (64.3%), followed by TAPES and Psi-Variant (23.1%) and InterVar and Psi-Variant (22.9%).
The characteristics of the detected variants are shown in Table 1. As expected, missense variants comprised the majority of detected variants, with 58.8%, 53.5%, and 51.4% of the variants detected by TAPES, Psi-Variant, and InterVar, respectively. Notably, a higher rate of frameshift variants was detected by Psi-Variant than by InterVar and TAPES (31.5% vs. 17.7% and 11.1%, respectively).
Almost all (≥ 95%) variants detected by either InterVar or TAPES were de-novo variants, while de-novo variants comprised only 54.5% of the variants detected by Psi-Variant, which also detected a high portion of X-linked and autosomal recessive variants (17.6% and 27.8%, respectively). Examination of the distribution of the detected variants in genes associated with ASD according to the SFARI Gene database37 revealed a two-fold enrichment of variants distributed in ASD genes (for all detection tools) compared to their portion in the clean vcf file and even a higher enrichment of variants distributed in high-confidence ASD genes (P < 0.001).
Effectiveness of ASD candidate variants detection
To assess the effectiveness of the different tools in detecting ASD candidate variants, we calculated the PPV and the OR for detecting ASD genes (i.e., those listed in the SFARI Gene database37) for different combinations of utilization of the three tools. The results of these analyses are shown in Fig. 3. Utilization of any of the three tools resulted in a significant enrichment of ASD genes, with the highest enrichment being observed in variants detected by both InterVar and Psi-Variant (PPV = 0.178; OR = 4.10, 95% confidence interval (C.I.) = 2.77–5.90; and PPV = 0.176; OR = 4.02, 95% C.I. = 2.63–5.95) followed by TAPES (PPV = 0.158; OR = 3.53, 95% C.I. = 2.28–5.27). Notably, the best performance in detecting ASD candidate variants was obtained at the intersection of the detected variants between InterVar and Psi-Variant (I ∩ P) (PPV = 0.274; OR = 7.09, 95% CI = 3.92–12.22). The I ∩ P combination was also the most effective in detecting SNVs in high-confidence ASD genes (i.e., those with a score of 1 in the SFARI Gene database37 (Fig. 3A-B). However, the I ∩ P combination had a relatively low diagnostic yield of 7.3% for SFARI genes. On the other hand, the union of InterVar and Psi-Variant (I U P) achieved a diagnostic yield of 20.5% (Fig. 3C), but its effectiveness in detecting variants in SFARI genes was lower (PPV = 0.159; OR = 3.59, 95% C.I. = 2.57–4.91) (Fig. 3A-B).

Effectiveness of InterVar (I), TAPES (T), Psi-Variant (P), and their combinations in detecting candidate variants in ASD genes. (A) Positive predictive value (PPV) of detecting candidate variants in SFARI 1 and all SFARI genes. (B) Odds Ratios (ORs) of detecting candidate variants in SFARI 1 and all SFARI genes. (C) Diagnostic yield (%) achieved by the different tool combinations for detecting candidate variants in SFARI 1 and all SFARI genes.
Discussion
In this study, we assessed the concordance and effectiveness of three bioinformatics tools in the interpretation of variants detected in the WES of children with ASD. There was better agreement in variant detection between InterVar and TAPES than between Psi-Variant and each of these two tools, probably because both InterVar and TAPES are based on the ACMG/AMP guidelines8, while Psi-Variant uses the interpretation of seven in-silico tools in assessing the functional consequences of LGD variants. In addition, most (94%) of the variants detected by either InterVar or TAPES were de-novo variants, compared to only 54.5% of the variants detected by Psi-Variant. This difference may be attributed to the fact that ACMG/AMP guidelines are particularly designed to detect de-novo highly penetrant variants, while inherited variants (autosomal recessive and X-linked) are usually classified as VUS20. Of note, seven of these variants were detected by both InterVar and TAPES as de-novo variants in more than one proband from different families, thus raising the possibility for false positive findings. On the other hand, Psi-Variant detected more homozygote recessive variants than InterVar and TAPES (52 vs. 10 and 5 respectively) and more than estimated in a previously large study18 suggesting some of them may be false positive. However, it also could be partially explained by the ethnic mixture of the study sample, that included many children from consanguineous Bedouin families. Indeed, most of these recessive variants were found in Bedouin children. Importantly, such rare inherited variants have been found to be associated with a variety of neurodevelopmental conditions, including ASD9,17,18,21,22. Another major difference between these tools lies in the detection of in-frame insertions/deletions that comprised ~ 20% of the variants detected by either InterVar or TAPES, while such SNVs were discarded by Psi-Variant. We decided to exclude these variants from Psi-Variant because their clinical relevance has been demonstrated in several genetic disorders38,39 but not in ASD40,41,42.
Another important factor that could affect the concordance between the three tools is the annotation tools they use. Specifically, both InterVar and TAPES use AnnoVar43 for their variant annotation, while Psi-Variant uses Ensembl’s VEP26. It has already been shown that AnnoVar and VEP have a low concordance in the classification of LoF variants44. In addition, each tool, InterVar, TAPES, and Psi-Variant, utilizes a different set of in-silico tools for the classification of missense variants, with SIFT30 alone being shared by all three tools.
Today, there are no accepted guidelines for detecting ASD susceptibility variants from WES data. Many genetic labs use the ACMG/AMP guidelines8, leading to a relatively low diagnostic yield45,46. Our findings suggest that different combinations of bioinformatics tools for variant interpretation may improve the detection of ASD susceptibility variants. Furthermore, combining these tools provides more flexibility in selecting the desired proportion between the detection yield and false positives. Thus, future guidelines for the detection of ASD susceptibility variants should consider the integration of different variant interpretation criteria.
Of note, many of the variants detected by the integrative pipeline affect genes with no known association with ASD, according to the SFARI Gene database37. This finding highlights the capability of the integrative pipeline to detect novel ASD genes. In addition, the combination of these tools could be used for detection and prioritization of susceptibility genetic variants of other medical conditions and human traits. Obviously, the validity of our findings and the applicability of these tools for other conditions should be assessed in additional studies.
The results of this study should be considered under the following limitations. First, the effectiveness assessments of the different tools and their combinations were based on ASD genes from the SFARI Gene database37. While this is the most commonly used database for ASD genes and is continuously updated, it is based on data curated from the literature and may thus include genes falsely associated with ASD. Second, the variant detection analyses were performed on WES data of a cohort from the Israeli population, which may not necessarily be representative of the genetic architecture of ASD. Third, the tools used in this study were designed to detect only extremely rare variants with relatively large functional effects. Thus, a more effective approach for the detection of ASD susceptibility variants should also include the interpretation of other types of genomic variations, such as copy-number and compound heterozygote variants47,48,49,50,51,52, as well as other variants with milder functional effects17,53,54. Finally, it should be noted that there are many other approaches for variant interpretation from WES data. Thus, it is possible that combinations of other approaches will be more effective in the detection of ASD susceptibility variants from WES data than the approaches investigated in this study.
Conclusions
Our findings suggest that combination of different bioinformatics tools is more effective in the detection of ASD candidate variants from WES data than each of the examined tools alone. Future guidelines for the detection of ASD susceptibility variants should consider integrating different variant interpretation approaches to improve the effectiveness of ASD candidate variants detection from whole exome sequencing data.
Data availability
WES data were generated as part of the ASC and are available in dbGaP with study accession: phs000298.v4.p3. The generated results and codes are available in a GitHub public repository: https://github.com/AppWick-hub/Psi-Variant or available upon reasonable request to the corresponding author, Prof. Idan Menashe (idanmen@bgu.ac.il).
Abbreviations
- ACMG/AMP:
-
American college of medical genetics and genomics/association of molecular pathology
- ASD:
-
Autism spectrum disorder
- C.I.:
-
Confidence interval
- GATK:
-
Genome analysis toolkit
- LGD:
-
Likely gene disrupting
- LoF:
-
Loss of function
- LP:
-
Likely pathogenic
- ML:
-
Machine learning
- NADI:
-
National autism database in Israel
- NGS:
-
Next-generation sequencing
- OR:
-
Odds ratio
- P:
-
Pathogenic
- PPV:
-
Positive predictive value
- SNV:
-
Single nucleotide variants
- VEP:
-
Variant effect predictor
- Vcf:
-
Variant calling format
- VUS:
-
Variants of uncertain significance
- WES:
-
Whole exome sequencing
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th edn. American Psychiatric Publishing (2013).
Meng-Chuan Lai, Michael V Lombardo, S. B.-C. Autism. Lancet (2014).
Yoo, H. Genetics of autism spectrum disorder: current status and possible clinical applications. Exp. Neurobiol. 24, 257 (2015).
Lord, C. et al. Autism spectrum disorder. Lancet 392, 508–520 (2018).
Ronald, A. & Hoekstra, R. A. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am. J. Med. Gen., Part B: Neuropsychiatric Gen. 156, 255–274 (2011).
Hallmayer, J. et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68, 1095–1102 (2011).
Devlin, B. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–246 (2012).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Gen. Med. 17, 405–424 (2015).
Satterstrom, F. K. et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 22, 1961–1965 (2019).
Fu, J. M. et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 54, (2022).
Satterstrom, F. K. et al. Large-Scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism article large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell https://doi.org/10.1016/j.cell.2019.12.036 (2020).
Wu, D. et al. Large-scale whole-genome sequencing of three diverse asian populations in Singapore. Cell 179, 736-749.e15 (2019).
Satterstrom, F. K. et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism article large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of Autism. Cell https://doi.org/10.1016/j.cell.2019.12.03 (2020).
Feliciano, P. et al. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med 4, (2019).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Ishay, R. T. et al. Diagnostic Yield and Economic Implications of Whole-Exome Sequencing for ASD Diagnosis in Israel. (2022).
Wang, T., Zhao, P. A. & Eichler, E. E. Rare variants and the oligogenic architecture of autism. Trends in Gen. https://doi.org/10.1016/j.tig.2022.03.009 (2022).
Doan, R. N. et al. Recessive gene disruptions in autism spectrum disorder. Nat Genet 51, (2019).
Li, Q. & Wang, K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet 100, 267–280 (2017).
Houge, G. et al. Stepwise ABC system for classification of any type of genetic variant. Eur. J. Human Gen. 30, 150–159 (2022).
Wilfert, A. B. et al. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat Genet 53, 1125–1134 (2021).
Halvorsen, M. et al. Exome sequencing in obsessive–compulsive disorder reveals a burden of rare damaging coding variants. Nat. Neurosci. 24, 1071–1076 (2021).
Dinstein, I. et al. The national autism database of israel: a resource for studying autism risk factors, biomarkers, outcome measures, and treatment efficacy. J. Molecular Neurosci. 70, 1303–1312 (2020).
Meiri, G. et al. Brief Report: The Negev Hospital-University-Based (HUB) Autism Database. J Autism Dev Disord 47, 2918–2926 (2017).
McKenna, A. et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297 (2010).
McLaren, W. et al. The Ensembl variant effect predictor. Gen. Biol. 17, 1–14 (2016).
Chen, S. et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv https://doi.org/10.1101/2022.03.20.485034 (2022).
Xavier, A., Scott, R. J. & Talseth-Palmer, B. A. TAPES: a tool for assessment and prioritisation in exome studies. PLoS Comput. Biol. 15, 1–9 (2019).
Fadista, J., Oskolkov, N., Hansson, O. & Groop, L. LoFtool: A gene intolerance score based on loss-of-function variants in 60 706 individuals. Bioinformatics 33, 471–474 (2017).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Current Protocols in Human Genetics vol. 2 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47, D886–D894 (2019).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99, 877–885 (2016).
Jagadeesh, K. A. et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 48, 1581–1586 (2016).
Samocha, K. E. et al. (2017) Regional missense constraint improves variant deleteriousness prediction. bioRxiv doi:https://doi.org/10.1101/148353.
Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med 12, 1–8 (2020).
Abrahams, B. S. et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 2–4 (2013).
Sergouniotis, P. I. et al. The role of small in-frame insertions/deletions in inherited eye disorders and how structural modelling can help estimate their pathogenicity. Orphanet J. Rare Dis. 11, 1–8 (2016).
Sallah, S. R. et al. Assessing the pathogenicity of in-frame CACNA1F indel variants using structural modeling. J. Molecular Diagnostics 24, 1232–1239 (2022).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012).
Dong, S. et al. De novo insertions and deletions of predominantly paternal origin are associated with autism spectrum disorder. Cell Rep 9, 16–23 (2014).
Kopp, N., Amarillo, I., Martinez-Agosto, J. & Quintero-Rivera, F. Pathogenic paternally inherited NLGN4X deletion in a female with autism spectrum disorder: clinical, cytogenetic, and molecular characterization. Am J Med Genet A 185, 894–900 (2021).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, 1–7 (2010).
McCarthy, D. J. et al. Choice of transcripts and software has a large effect on variant annotation. Genome Med 6, (2014).
Trost, B. et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 185, 4409-4427.e18 (2022).
Tammimies, K. et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA – J. Am. Med. Assoc. 314, 595–903 (2015).
Husson, T. et al. Rare genetic susceptibility variants assessment in autism spectrum disorder: detection rate and practical use. Transl Psychiatry 10, (2020).
Turner, T. N. et al. Genomic patterns of de novo mutation in simplex Autism. Cell 171, 710-722.e12 (2017).
Leppa, V. M. M. et al. Rare inherited and de novo cnvs reveal complex contributions to ASD risk in multiplex families. Am J Hum Genet 99, 540–554 (2016).
Krumm, N. et al. Excess of rare, inherited truncating mutations in autism. Nat Genet 47, 582–588 (2015).
Lin, B. D. et al. The role of rare compound heterozygous events in autism spectrum disorder. doi: https://doi.org/10.1038/s41398-020-00866-7.
Tuncay, I. O. et al. The genetics of autism spectrum disorder in an East African familial cohort. Cell Genom. 3, 100322 (2023).
Du, Y. et al. Nonrandom occurrence of multiple de novo coding variants in a proband indicates the existence of an oligogenic model in autism. Gen. Med. 22, 170–180 (2020).
Guo, H. et al. Genome sequencing identifies multiple deleterious variants in autism patients with more severe phenotypes. Gen. Med. 21, 1611–1620 (2019).
Acknowledgements
We thank the families who participated in this research, without their contributions, genetic studies would be impossible.
Funding
This study was supported by a grant from the Israel Science Foundation (1092/21).
Author information
Authors and Affiliations
Contributions
Conceptualization: A.S. and I.M.; methodology: A.S. and I.M.; software: A.S. and L.L.; validation: A.S. and I.M.; formal analysis: A.S.; resources: H.G., G.M., A.M., Y.S., A.A., and I.D.; data curation: A.S.; writing—original draft preparation: A.S. and I.M.; writing—review and editing: I.M., and A.S.; supervision: I.M.; project administration: I.M.; funding acquisition: I.M. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shil, A., Levin, L., Golan, H. et al. Comparison of three bioinformatics tools in the detection of ASD candidate variants from whole exome sequencing data. Sci Rep 13, 18853 (2023). https://doi.org/10.1038/s41598-023-46258-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46258-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
