
Abstract
Treatment is challenging due to the heterogeneity of hepatocellular carcinoma (HCC). Chromatin regulators (CRs) are important in epigenetics and are closely associated with HCC. We obtained HCC-related expression data and relevant clinical data from The Cancer Genome Atlas (TCGA) databases. Then, we crossed the differentially expressed genes (DEGs), immune-related genes and CRs to obtain immune-related chromatin regulators differentially expressed genes (IRCR DEGs). Least absolute shrinkage and selection operator (LASSO) Cox regression analysis was performed to select the prognostic gene and construct a risk model for predicting prognosis in HCC, followed by a correlation analysis of risk scores with clinical characteristics. Finally, we also carried out immune microenvironment analysis and drug sensitivity analysis, the correlation between risk score and clinical characteristics was analyzed. In addition, we carried out immune microenvironment analysis and drug sensitivity analysis. Functional analysis suggested that IRCR DEGs was mainly enriched in chromatin-related biological processes. We identified and validated PPARGC1A, DUSP1, APOBEC3A, AIRE, HDAC11, HMGB2 and APOBEC3B as prognostic biomarkers for the risk model construction. The model was also related to immune cell infiltration, and the expression of CD48, CTLA4, HHLA2, TNFSF9 and TNFSF15 was higher in high-risk group. HCC patients in the high-risk group were more sensitive to Axitinib, Docetaxel, Erlotinib, and Metformin. In this study, we construct a prognostic model of immune-associated chromatin regulators, which provides new ideas and research directions for the accurate treatment of HCC.
Similar content being viewed by others
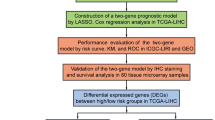


Introduction
Primary liver cancer is the sixth most common primary tumor and the fourth most common cause of cancer-related death worldwide1, HCC accounts for 80–90%2. Although early HCC can be cured by local ablation, surgical resection or liver transplantation, most HCC cases in the world are in the advanced stage. Targeted systemic therapy and immune checkpoint inhibitors (ICIs) have been proven to be effective treatment options for advanced HCC patients3,4, but some patients still do not benefit from them, which may be related to the complex tumor microenvironment (TME). TME is a micro-internal environment with immune inflammatory response, hypoxia and low pH composed of tumor cells and non-tumor cells. These characteristics affect the occurrence and development of tumor and treatment resistance. Among them, immune cells and a variety of factors released by them play an important role in TME5,6. Intensive investigation of immune cells and interactions within the TME is important to improve the efficacy of antitumor drugs7. Recently, the construction of immune-related risk models using bioinformatics techniques can accurately predict the prognosis of cancer patients and guide the efficacy of ICIs therapy8,9.
The heterogeneity of HCC is also related to genetics, epigenetics, proteomics, transcriptomics and metabolomics10,11,12. In recent years, epigenetics has attracted wide attention. Epigenetics does not change the gene sequence to achieve the regulation of gene expression, thus affecting tumorigenesis and all hallmarks of cancer13,14,15. The current studies suggest that epigenetic alterations contribute to promote tumor immune function16,17, and epigenetic therapeutics assist in enhancing the effect of immunotherapy18,19. For instance, the histone deacetylase inhibitor Belinostat improves the anti-tumor activity of CTLA-4 in a subcutaneous Hepa129 murine HCC model, reflecting the synergistic effect of combined therapy20. In HCC animal experiment with the EZH2 inhibitor DZNep and anti-PD-L1 antibody, combination treatment upregulated the expression of Th 1 chemokines and associated tumor antigens, promoted effector T cell infiltration and promoted antitumor immunity21. Chromatin regulators (CRs) were vital regulatory element in epigenetics22. CRs were mainly classified into three major categories according to roles in epigenetics: DNA methylators, histone modifiers, and chromatin remodelers23,24. Mutations in chromatin regulators, such as ARID1A, ARID1B, ARID2, MLL, and MLL3, may contribute to the occurrence and progress of HCC25. Chromatin remodeling factor ARID2 expression was negatively correlated with pathological grade and organ metastasis in HCC patients, and ARID2 knockout promotes metastasis in HCC mouse models26. Previous studies found that CRs drive epigenetic alterations play an important role in HCC, and also revealed their role patterns in HCC patients27,28. The study by Dai et al. identified three CR-related patterns and established the CRs phenotype-related gene signature to predict energy metabolism and cuproptosis activity in HCC29. Although the construction of polygenic prognostic models based on CRs and immune-related genes provides potential indicators for the response of ICIs. However, as far as we know, there is no study on the combined analysis of CRs and immune-related genes in HCC.
In this study, we investigated the expression profiles and functional enrichment of immune-related CRs in HCC. We successfully constructed a new prognostic model of HCC based on seven genes, PPARGC1A, DUSP1, APOBEC3A, AIRE, HDAC11, HMGB2, and APOBEC3B. Furthermore, we analyzed the correlation between risk score and clinical characteristics, and explored the correlation between the risk model and the immune microenvironment in HCC. Our results provide a new direction for revealing new biomarkers and new ideas for the accurate treatment of HCC.
Materials and methods
Data collection
The RNA sequencing (RNA-seq) data and relevant clinical data of HCC including 374 cancer samples and 50 para-cancer samples were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/)30. Another 232 Japanese population HCC samples were obtained from the ICGC portal (https://dcc.icgc.org/projects/LIRI-JP)31.
A total of 870 Chromatin regulators (CRs) were retrieved from previous topic research22. The lists of immune-related genes were downloaded from the InnateDB (https://www.innatedb.com/) and totaling 1040 human immune-related genes (Supplementary Table 1).
Identification of immune-related CRs differentially expressed genes (IRCR DEGs)
Based on the genes expression of cancer tissues and para-cancer tissues in the TCGA-HCC dataset, differentially expressed genes (DEGs) were obtained using the “limma” R package according to the criteria of |log2 FC (fold change)|> 1 and adjusted p values < 0.01. Then the “VennDiagram” R package was utilized to obtain IRCR DEGs for the above DEGs. In addition, we obtained the mutations of IRCR DEGs through Gene Set Cancer Analysis (GSCA) (http://bioinfo.life.hust.edu.cn/GSCA/#/)32.
Functional enrichment analyses and gene–gene interaction network
To analyze the identified IRCR DEGs, the Gene Ontology (GO)33 and Kyoto Encyclopedia of Genes and Genomes (KEGG)34 pathway enrichment analyses were performed and visualized using the “clusterProfiler”35 and “GOplot” R package36. An adjusted p value < 0.05 was considered the screening criterion for significantly enriched terms.
GeneMANIA37, a flexible plugin of Cytoscape, which was applied to identify the genes most relevant to the query gene set and to construct a composite gene–gene functional interaction network.
Construction of a prognostic model based on IRCRs
We performed lasso-penalized Cox regression analysis to construct the prognostic risk model through glmnet R package. Risk scores were calculated by the following formula:
All HCC patients were divided into high-risk group and low-risk group by the median risk score. Moreover, we plotted K-M survival curve to evaluate the discrepancy of OS between the two groups by the “Survminer” R package38, and time-related receiver operating characteristic (ROC) curves were applied to assess the accuracy of the risk model by the “survivalROC” (version 1.03) packages39. The ICGC LIRI-JP dataset was considered as a valid set for further external verification of the prognostic model.
Construction of nomogram model
We researched the relationship between IRCR-based signature and clinical characteristics in HCC. To verify whether the signature risk score could be used as an independent prognostic factor in HCC patients, univariate and multivariate Cox regression analyses were performed. A nomogram associated with outcome was built to investigate the probability of prognosis for HCC patients. The calibration curve was performed to assess the predictive utility of the nomogram.
Immune cell infiltration analysis
In order to determine the immune prognostic correlation of IRCR-based signature in HCC, we used CIBERSORT, CIBERSORT-ABS, QUANTISEQ, MCP-counter, XCELL, TIMER, and EPIC algorithms to evaluate the infiltration level of immune cells between high-risk group and low-risk group. Meanwhile, we explored the expression of several immune checkpoints to predict the effect of immune checkpoint blockade therapy. In addition, the TIMER database(https://cistrome.shinyapps.io/timer/)40 was used to identify the correlations between 7 IRCRs and six immune cells (B cells, CD4 + T cells, CD8 + T cells, neutrophils, macrophages, and dendritic cells) in HCC.
Drug sensitivity analysis
The half-maximal inhibitory concentration (IC50) of drugs were analysed by using the Genomics of Drug Sensitivity in Cancer (GDSC, http://www.cancerrxgene.org/)41 database, and the drugs sensitivity were predicted by using the “pRRophetic”42 R package.
Statistical methods
R (v.4.0.2) was used for statistical analysis and visualization. Differences between groups were compared using the Wilcoxon rank-sum test. P value < 0.05 was considered statistically significant (ns, p ≥ 0.05; *p < 0.05; **p < 0.01; ***p < 0.001).
Informed consent
All participating authors give their consent for this work to be published.
Results
Identification and mutation analysis of IRCR DEGs in HCC samples
We found 4274 DEGs between the HCC and normal samples, of which 3054 were upregulated and 1220 were downregulated in the HCC samples (Fig. 1A, Supplementary Table 2). 9 IRCR DEGs were obtained by overlapping 4274 DEGs, 870 CRs and 1040 human immune-related genes (Fig. 1B). The situation of single nucleotide variation (SNV) of 9 IRCR DEGs in HCC samples were summarized in Fig. 1C,D. The mutation frequency was 100% in the 9 samples. Among them, PPARGC1A, EHMT2, AIRE and PKN1 were the highest mutated genes with more than 15% mutation rates. Besides, the most common variant classification was missense mutation. In addition, we found that single nucleotide polymorphisms (SNP) play an important role in the above mutated genes, and there were six classes of base substitution and the most common class was C > T.

Identification and mutation profile analysis of IRCR DEGs from the TCGA-HCC cohort. (A) Volcano plot of 4274 differentially expressed genes. (B)The Venn diagram of DEGs, CRs, and immune-related genes were downloaded from the InnateDB. (C) Oncoplot displaying the situation of the SNV of IRCR DEGs in HCC samples from TCGA database. (D) The SNV classes of IRCR DEGs in TCGA-HCC cohort.
Functional annotation of the IRCR DEGs
To explore the biological functions and potential mechanisms of the IRCR DEGs in the TCGA-HCC cohort, we performed GO and KEGG enrichment pathway analysis. A total of 94 Gene Ontology (GO) entries and 2 KEGG pathways were enriched in the 9 IRCRs (Supplementary Table 3). The results of biological process (BP) analysis showed that 9 IRCRs were remarkably involved in DNA methylation or demethylation, cytidine catabolic process, and cytidine deamination. Cellular component (CC) were located in P-body, cytoplasmic ribonucleoprotein granule, and ribonucleoprotein granule. Molecular function (MF) analysis suggested that cytidine deaminase activity, deoxycytidine deaminase activity, and hydrolase activity, acting on carbon–nitrogen (but not peptide) bonds were mainly enriched. From the KEGG pathway analysis, we found that these IRCRs were mainly associated with longevity regulating pathway and Viral life cycle-HIV-1 (Fig. 2A). Through the chord plot analysis of the top 15 biological processes, we found that APOBEC3A and APOBEC3B were mainly involved in the above biological processes, and revealed that they play an important role in chromatin-related biological processes (Fig. 2C).

Functional annotation of IRCR DEGs. (A) KEGG enrichment analysis and the top 4 GO enrichment significance items of IRCR DEGs sorted by adjusted p value in BP, CC and MF. (B) The gene–gene interaction network of IRCR DEGs were constructed using GeneMania. (C) Chord plot showed the distribution of the top 15 GO enrichment in BP.
Based on the 9 IRCR DEGs of identified and potential targets were obtained by shared protein domains, co-expression and pathway in the GeneMANIA, an entire network was constructed using Cytoscape(version 3.7.2). As shown in Fig. 3B, the results suggested that the 9 IRCR DEGs may interact with these 20 proteins, such as APOBEC1, APOBEC2, APOBEC3C, APOBEC4, ADAT2 and ADAT3, etc. (Fig. 2B).

Prognostic value of risk model in HCC patients. (A) Ten-time cross-validation for tuning parameter selection in the LASSO model.(B) LASSO coefficient profiles.(C) Distribution of survival status based on the median risk score. (D) Heatmap of 7 IRCR genes in HCC patients.(E) Kaplan–Meier survival analysis of HCC patients between high-risk group and low-risk group. (F) Time-independent receiver operating characteristic (ROC) analysis of risk scores predicting 1,2,3-year overall survival.
Construction of a prognostic model based on IRCRs
A risk model was constructed with 7 genes (PPARGC1A, DUSP1, APOBEC3A, AIRE, HDAC11, HMGB2, APOBEC3B) by using LASSO Cox regression analysis (Fig. 3A,B). The risk score was calculated by coefficients of 7 IRCRs as following formula: risk score = (-0.1335 × PPARGC1A expression) + ( 0.0302 × DUSP1 expression) + (0.1716 × APOBEC3A expression) + (0.3182 × AIRE expression) + (0.2219 × HDAC11 expression) + (0.1269 × HMGB2 expression) + (0.0219 × APOBEC3B expression) (Table 1). HCC patients were classified into two groups (high-risk group and low-risk group) according to the median risk score (Fig. 3C,D). The results of the KM curve showed that the prognosis of the high-risk group was significantly worse than that of the low-risk group (p < 0.001), which suggested that risk score was negatively correlated with prognosis (Fig. 3E). The time-dependent ROC analysis showed that the AUC values of 1, 2, and 3 years were 0.713, 0.66, and 0.674 respectively, indicating the accuracy of the model in predicting patient prognosis (Fig. 3F).
External validation of the prognostic model
We divided HCC patients into low-risk and high-risk groups in the ICGC cohort based on relevant coefficients of 7 IRCRs. The results showed the distribution of survival status of each HCC patient and the heatmap of 7 IRCRs in ICGC database (Fig. 4A,B). The results of Kaplan–Meier (p = 0.023) analysis showed consistency with the TCGA cohort (Fig. 4C). In addition, the ROC curve showed AUC values of 0.713 (1 year), 0.66 (2 years) and 0.674 (3 years) (Fig. 4D).

Validation of the prognostic model in ICGC cohort. (A) Distribution of survival status based on the median risk score. (B) Heatmap of 7 IRCR genes in ICGC LIRI-JP dataset. (C) Kaplan–Meier survival analysis of HCC patients in different risk groups. (D) Time-independent ROC analysis of risk scores predicting 1,2,3-year overall survival.
Correlation between the risk score and clinical characteristics
To analyze whether the prognostic model participated in the development and progression of HCC, we used the Chi-square test to compare the risk score in different clinical characteristics. The result (Fig. 5) showed that there were significant differences between high- and low-risk groups in pathological stage (p = 0.023) and T stage (p = 0.032). Moreover, we further analyzed the prognostic significance of the signature in subgroups. The result suggested that IRCR-based signature showed excellent performance in predicting outcome in age ≤ 65 (p = 0.009), age > 65 (p = 0.010), male (p = 0.002), female (p = 0.049), G1-G2 (p = 0.008), T1-T2 stage (p = 0.016), N0 (p < 0.001), M0 (p < 0.001) and Stage I-II (p = 0.019). While IRCR-based signature showed poor performance in predicting outcome in G3-G4 (p = 0.050), T3-T4 stage (p = 0.217) and Stage III-IV (p = 0.260) (Fig. 6).

Correlation between risk score and clinical characteristics.

Kaplan–Meier curves of OS differences stratified by age, gender, tumor grade, N stage, T stage, M stage, or pathologic stage between the high-risk group and low-risk group.
Construction of nomogram model
Univariate COX regression analysis showed that risk group and pathologic stage were significantly relevant to the survival of HCC patients (p < 0.001) (Fig. 7A). In multivariate COX regression analysis, the risk group and pathologic stage were still remarkably related to the survival of HCC patients (p < 0.01) (Fig. 7B). Which demonstrate that IRCR-based signature was an independent prognostic factor for HCC patients.

Forest plot and nomogram of the prognostic risk model. (A) Forest plot of univariate Cox regression analysis in HCC. (B) Forest plot of multivariate Cox regression analysis in HCC. (C)The nomogram for predicting 1-, 2-, and 3-year OS of HCC patients. (D) The calibration plots for predicting 1-, 2-, and 3-year OS.
To further forecast the survival of HCC patients, we structured a nomogram comprised of risk group, gender, tumor grade and pathologic stage. Nomography predicted the prognostic survival probability of HCC patients at 1, 2, 3 years (Fig. 7C). The calibration curve indicated that there was a good consistency between the actual survival probability and the predicted probability (Fig. 7D).
Immune infiltration analysis of the IRCR-based signature
According to the analyses of TIMER, CIBERSORT, CIBERSORTABS, XCELL, QUANTISEQ, EPIC, and MCP-counter, the relationship between the IRCR-based signature and immune infiltration was displayed in the heatmap (Fig. 8A). The result of XCELL indicated that the proportions of CD8 + naïve T cells, CD8 + central memory T cells, granulocyte-monocyte progenitor cells, hematopoietic stem cells, M2 macrophages, and Tregs were higher in the low-risk group, whereas myeloid dendritic cells, NK cells, Th1 cells and Th2 cells were higher in the high-risk group (Supplementary Fig. 1). We also investigated the correlation between risk groups and key immune checkpoints. The result showed that there was a difference in the expression of CD48, CTLA4, HHLA2, IDO2, TNFSF9, and TNFSF15 between the two groups. In addition, CD48, CTLA4, HHLA2, TNFSF9, and TNFSF15 were elevated in high-risk group, suggesting that the high-risk group are more likely to show immunosuppressive phenotype in tumor microenvironment (Fig. 8B).

Immune infiltration and immune checkpoints analysis. (A) Immune cells infiltration between high-risk group and low-risk group. (B) The relationship between the IRCR-based signature and immune checkpoints.
TIMER database was used to explore the relationship between immune cells and above 7 prognostic IRCRs. The results showed HMGB2 was positively associated with all immune cells. APOBEC3A and APOBEC3B were positively associated with multiple immune cells such as B cells, CD8 + T cells, macrophage, neutrophil, and dendritic cells. HDAC11 was positively associated with B cells, CD4 + T cells, macrophage, and neutrophil. DUSP1 was positively associated with neutrophil. (Supplementary Fig. 2).
Drug sensitivity analysis
We further investigated the differences in sensitivity of common chemotherapy drugs between the two groups in HCC patients. The results indicated that IC50 values of drugs including Axitinib, Docetaxel, Erlotinib, and Metformin were higher in the high-risk group than those of the low-risk group, which suggested that HCC patients in the high-risk group were much more sensitive to these drugs (Fig. 9). While IC50 values of Bleomycin, Bortezomib, Doxorubicin, Etoposide, and Gemcitabine were significantly lower in the low-risk group than those of the high-risk group, suggesting that HCC patients in the low-risk group were much more sensitive to these drugs.

Drug sensitivity analysis.
Comparison with other risk prognostic models in HCC
To evaluate the prognostic ability of our model for HCC, we compared other three prognostic models: the four-gene model43, the seven-CRs model44, and the four-immune-related-gene model45. For the TCGA-LIHC dataset, we used methods of externally validating our model to calculate the corresponding risk scores. The results showed that the AUC values for 1,3, and 5-year survival of the four-gene model were lower than our model, the AUC values for 1-year survival of the seven-CRs model and four-immune-related-gene model were slightly higher than our model, but the AUC values for five-year survival were lower (Fig. 10). These results suggested that our model was advantageous in predicting the long-term survival (5-year) of HCC patients.

The comparison of our prognostic model and other models. (A) Time-dependent ROC analysis for our prognostic model. (B) Time-dependent ROC analysis for four-gene model. (C) Time-dependent ROC analysis for seven-CRs model. (D) Time-dependent ROC analysis for the four-immune-related gene model.
Discussion
There are about 906,000 new cases and 830,000 deaths of primary liver cancer worldwide in 2020, severely threaten human health and life46. Hepatocellular carcinoma (HCC) is still the most common pathological type. Cancer immunotherapies has greatly changed the clinical treatment of HCC in recent years, but it remains one of the worst prognosis diseases due to the heterogeneity. Epigenetic alterations can influence the interactions between tumor cells and liver tumor microenvironment (TME), so the epigenetics study can enhance anti-tumor immunity and better combat HCC47. A growing number of studies have shown that CRs plays an important role in HCC. Therefore, the analysis of HCC sequencing data by combining CRs and immune-related genes could be beneficial in the search for new biomarkers to predict the response to immunotherapy, and provide potential therapeutic targets for the treatment of HCC.
In this study, we innovatively analysed CRs with immune-related genes in HCC, established a risk model associated with 7 IRCR and verified in ICGC LIRI-JP cohort. Univariate and multivariate COX analysis showed that the risk score based on 7 IRCR was an independent prognostic indicator for HCC patients. Compared with other prognostic models, our model is advantages in predicting long-term survival of HCC patients. In addition, we also analysed the relationship between the signature and immune cells infiltration in HCC.
GO analysis showed that IRCRs were mainly related to biological processes (BP), such as DNA methylation or demethylation, cytidine catabolic process, cytidine deamination, cytidine to uridine editing, and cytidine metabolic process. The result of KEGG pathway enrichment analyses indicated that IRCRs were mainly involved in the longevity regulating pathway and viral life cycle-HIV-1. Aging was a universal feature of organisms and tumorigenesis was also closely associated with cellular senescence, including HCC, and was regulated by longevity signaling pathways48,49. APOBEC3A and APOBEC3B were involved in most biological processes, and the relationship between the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family members was the most significant according to the gene- gene interaction network. APOBEC family was the main source of DNA modification of cancer genome, participated in immune response and antiviral response in human body with specific mutation pattern50.
Among the APOBEC protein family members, APOBEC3A and APOBEC3B are able to restrict the infection of multiple viruses, including parvovirus, hepatitis B virus (HBV), human papillomavirus, human immunodeficiency virus 1 (HIV-1) and carcinogenesis51,52,53,54. HBV is the main risk factor for HCC, but epigenetic factors are also involved in the underlying pathogenesis of HCC. APOBEC3A is an editing molecule of HBV DNA, APOBEC3A and APOBEC3B play crucial roles in inducing HBV DNA degradation55. Duowei found that APOBEC3B increased transcriptional expression through the non-classical NF-κB signal pathway, while the increased expression of APOBEC3B significantly increased CCL2 chemokine, thus recruiting myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) to participate in the development of HCC56. HMGB2, as a member of high-mobility group box(HMGB) proteins family, is involved in DNA replication, repair, transcription, differentiation, proliferation, cell signaling, inflammation, tumor migration, and cellular senescence57,58. It has been reported that HMGB2 gene knockout can induce cell senescence and inhibit the growth of tumor cells59. Cyclic cGMP-AMP synthase (cGAS) promotes inflammatory senescence-associated secretory phenotype (SASP) by recognizing cytoplasmic chromatin during cellular senescence. HMGB2 can retain the function of topoisomerase 1-DNA covalent cleavage complex (TOP1cc) in cytoplasmic chromatin. HMGB2-TOP1cc-cGAS axis functionally regulates SASP and immune checkpoint blocking response60. Dual-specificity protein phosphatase 1(DUSP1) expression declined in HCC tissue and was significantly associated with HCC progression and aggressiveness. DUSP1 down-regulation depends on promoter hypermethylation associated with loss of heterozygosity or ERK/SKP2/CKS1-dependent ubiquitination61. Hao revealed that DUSP1 expression correlated with the activation of p53, which in turn positively regulated DUSP1 transcription. If this destruction of the positive regulatory loop could contribute to HCC development and progress62. Autoimmune regulatory factor (AIRE) was a transcription factor mainly expressed in thymic medulla epithelial cells. AIRE expression was also found in other tissues outside the thymus63. Zhu showed that AIRE deficiency in mice led to increased immune response to melanoma and increased infiltration of CD4 + and CD8 + in tumor tissue, spleen and tumor draining lymph nodes64. HDAC11 was the sole class IV member of the histone deacetylases (HDAC) family and the smallest HDAC enzyme identified to date65. HDAC11 induced deacetylation of p53 transcription factor Egr-1 (early growth response 1), which prevented p53 transcription and promoted the development of HCC66. In a murine model, T cells lacking HDAC11 showed proinflammatory cytokine production and effector molecule expression67. PPARGC1A (peroxisome proliferator activated receptor gamma coactivator 1 alpha, PGC-1 α) was a transcriptional coactivator with important roles in mitochondrial biosynthesis, homeostasis, and energy metabolism68. PPARGC1A had oncogenic and tumor suppressive features, and high and low levels of PPARGC1A expression associated with the prognosis of different cancers. Compared with normal liver tissue, the expression of PPARGC1A in HCC tissue is downregulated and acted as a tumor inhibitory role in the occurrence and development of HCC69. Additionally, TIMER database showed that 7 IRCRs of the model were related to immune cells, which revealed that IRCRs might regulate HCC progression by influencing immune infiltration.
There were significant differences in immunotherapy among different patients, which was caused by the heterogeneity of immune environment in tumor microenvironment. Our results showed that the proportion of CD8 + T cells were higher in low-risk group, and the expressions of CD48, CTLA4, HHLA2, TNFSF9 and TNFSF15 in high-risk group were higher than those in the low-risk group, which suggested that the poor prognosis of HCC patients in high-risk group might be related to the immunosuppressive microenvironment. Moreover, HCC patients in high-risk group might benefit from checkpoint inhibitor immunotherapy. Also, we found that HCC patients in high-risk group might benefit from the treatments of Axitinib, Docetaxel, Erlotinib, and Metformin, while HCC patients in low-risk group might benefit from the treatments with Bleomycin, Bortezomib, Doxorubicin, Etoposide, and Gemcitabine.
Although the study could provide excellent aid for accurately treating HCC patients, there are still some shortcomings. First, although there is a large amount of high-throughput data stored in the TCGA database, the number of samples related to HCC is still insufficient. Second, the above results need to be further verified in vitro and in vivo. Therefore, further research is necessary to address the possible limitations in terms of results and conclusions.
Conclusion
We identified differentially expressed IRCRs and found that IRCRs are important for predicting the prognosis of HCC patients, and targeting IRCRs could be expected as an effective treatment for HCC. In conclusion, our prognostic model could provide new ideas and research directions for the accurate treatment of HCC.
Data availability
The datasets generated analysed during the current study are available in the UCSC Xena repository, we can download liver cancer related RNA sequencing and phenotype data from the link below, https://xenabrowser.net/datapages/?cohort=GDC%20TCGA%20Liver%20Cancer%20(LIHC)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443.
References
Llovet, J. M. et al. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 7, 6. https://doi.org/10.1038/s41572-020-00240-3 (2021).
Lawal, G. et al. The immunology of hepatocellular carcinoma. Vaccines 9, 1184. https://doi.org/10.3390/vaccines9101184 (2021).
Luo, X. Y., Wu, K. M. & He, X. X. Advances in drug development for hepatocellular carcinoma: Clinical trials and potential therapeutic targets. J. Exp. Clin. Cancer Res. 40, 172. https://doi.org/10.1186/s13046-021-01968-w (2021).
Su, T. H., Hsu, S. J. & Kao, J. H. Paradigm shift in the treatment options of hepatocellular carcinoma. Liver Int. 42, 2067–2079. https://doi.org/10.1111/liv.15052 (2022).
Budi, H. S. & Farhood, B. Targeting oral tumor microenvironment for effective therapy. Cancer Cell Int. 23, 101. https://doi.org/10.1186/s12935-023-02943-5 (2023).
Zhu, Y. H. et al. Immunosuppression, immune escape, and immunotherapy in pancreatic cancer: Focused on the tumor microenvironment. Cell Oncol. 46, 17–48. https://doi.org/10.1007/s13402-022-00741-1 (2023).
Mehraj, U. et al. The tumor microenvironment as driver of stemness and therapeutic resistance in breast cancer: New challenges and therapeutic opportunities. Cell Oncol. 44, 1209–1229. https://doi.org/10.1007/s13402-021-00634-9 (2021).
Xue, W. et al. A novel prognostic index of stomach adenocarcinoma based on immunogenomic landscape analysis and immunotherapy options. Exp. Mol. Pathol. 128, 104832. https://doi.org/10.1016/j.yexmp.2022.104832 (2022).
Xue, W. et al. miRNA-based signature associated with tumor mutational burden in colon adenocarcinoma. Front. Oncol. 11, 634841. https://doi.org/10.3389/fonc.2021.634841 (2021).
Cancer Genome Atlas Research Network, Electronic address, W. B. E. & Cancer Genome Atlas Research, N. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169, 1327–1341. https://doi.org/10.1016/j.cell.2017.05.046 (2017).
Hlady, R. A. & Robertson, K. D. Genetic and epigenetic heterogeneity in normal liver homeostasis and its implications for liver disease and hepatocellular cancer. Semin Liver Dis. 38, 41–50. https://doi.org/10.1055/s-0037-1621712 (2018).
Wu, Y., Liu, Z. & Xu, X. Molecular subtyping of hepatocellular carcinoma: A step toward precision medicine. Cancer Commun. 40, 681–693. https://doi.org/10.1002/cac2.12115 (2020).
Nebbioso, A., Tambaro, F. P., Dell’Aversana, C. & Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 14, e1007362. https://doi.org/10.1371/journal.pgen.1007362 (2018).
Pfister, S. X. & Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 16, 241–263. https://doi.org/10.1038/nrd.2016.256 (2017).
Flavahan, W. A., Gaskell, E. & Bernstein, B. E. Epigenetic plasticity and the hallmarks of cancer. Science 357, 6348. https://doi.org/10.1126/science.aal2380 (2017).
Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19. https://doi.org/10.1038/s41590-017-0006-x (2018).
Henning, A. N., Roychoudhuri, R. & Restifo, N. P. Epigenetic control of CD8(+) T cell differentiation. Nat. Rev. Immunol. 18, 340–356. https://doi.org/10.1038/nri.2017.146 (2018).
Dai, E. et al. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 20, 171. https://doi.org/10.1186/s12943-021-01464-x (2021).
Topper, M. J., Vaz, M., Marrone, K. A., Brahmer, J. R. & Baylin, S. B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 17, 75–90. https://doi.org/10.1038/s41571-019-0266-5 (2020).
Llopiz, D. et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 68, 379–393. https://doi.org/10.1007/s00262-018-2283-0 (2019).
Hong, Y. K. et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol. 336, 66–74. https://doi.org/10.1016/j.cellimm.2018.12.010 (2019).
Lu, J. et al. FACER: comprehensive molecular and functional characterization of epigenetic chromatin regulators. Nucleic Acids Res. 46, 10019–10033. https://doi.org/10.1093/nar/gky679 (2018).
Plass, C. et al. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 14, 765–780. https://doi.org/10.1038/nrg3554 (2013).
Medvedeva, Y. A. et al. EpiFactors: A comprehensive database of human epigenetic factors and complexes. Database 2015, bav067. https://doi.org/10.1093/database/bav067 (2015).
Fujimoto, A. et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 44, 760–764. https://doi.org/10.1038/ng.2291 (2012).
Jiang, H. et al. Chromatin remodeling factor ARID2 suppresses hepatocellular carcinoma metastasis via DNMT1-Snail axis. Proc. Natl. Acad. Sci. U S A 117, 4770–4780. https://doi.org/10.1073/pnas.1914937117 (2020).
Liu, Z., Yang, H., Chen, Z. & Jing, C. A novel chromatin regulator-related immune checkpoint related gene prognostic signature and potential candidate drugs for endometrial cancer patients. Hereditas 159, 40. https://doi.org/10.1186/s41065-022-00253-w (2022).
Chen, J., Chen, X., Li, T., Wang, L. & Lin, G. Identification of chromatin organization-related gene signature for hepatocellular carcinoma prognosis and predicting immunotherapy response. Int. Immunopharmacol. 109, 108866. https://doi.org/10.1016/j.intimp.2022.108866 (2022).
Dai, Y. W. et al. Characterization of chromatin regulators identified prognosis and heterogeneity in hepatocellular carcinoma. Front. Oncol. 12, 1002781. https://doi.org/10.3389/fonc.2022.1002781 (2022).
Tomczak, K., Czerwinska, P. & Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 19, A68-77. https://doi.org/10.5114/wo.2014.47136 (2015).
Fujimoto, A. et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 48, 500–509. https://doi.org/10.1038/ng.3547 (2016).
Liu, C. J. et al. GSCA: An integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 24, 558. https://doi.org/10.1093/bib/bbac558 (2023).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Walter, W., Sanchez-Cabo, F. & Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. https://doi.org/10.1093/bioinformatics/btv300 (2015).
Montojo, J., Zuberi, K., Rodriguez, H., Bader, G. D. & Morris, Q. GeneMANIA: Fast gene network construction and function prediction for Cytoscape. F1000Res 3, 153. https://doi.org/10.12688/f1000research.4572.1 (2014).
Kassambara, A., Kosinski, M. & Biecek, P.survminer: Drawing Survival Curves using 'ggplot2'. R package version 0.4.9. https://CRAN.R-project.org/package=survminer (2021).
Heagerty, P. J., Lumley, T. & Pepe, M. S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 56, 337–344. https://doi.org/10.1111/j.0006-341x.2000.00337.x (2000).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 77, e108–e110. https://doi.org/10.1158/0008-5472.CAN-17-0307 (2017).
Yang, W. et al. Genomics of drug sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955-961. https://doi.org/10.1093/nar/gks1111 (2013).
Geeleher, P., Cox, N. & Huang, R. S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 9, e107468. https://doi.org/10.1371/journal.pone.0107468 (2014).
Zheng, Y. et al. Large-scale analysis reveals a novel risk score to predict overall survival in hepatocellular carcinoma. Cancer Manag. Res. 10, 6079–6096. https://doi.org/10.2147/CMAR.S181396 (2018).
Jia, X. & Zhang, G. Characterization of chromatin regulators in hepatocellular carcinoma to guide clinical therapy. Front. Genet. 13, 961018. https://doi.org/10.3389/fgene.2022.961018 (2022).
Zhang, F. et al. An immune-related gene signature predicting prognosis and immunotherapy response in hepatocellular carcinoma. Comb. Chem. High Throughput Screen 25, 2203–2216. https://doi.org/10.2174/1386207325666220304115006 (2022).
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. https://doi.org/10.3322/caac.21660 (2021).
Akce, M., El-Rayes, B. F. & Wajapeyee, N. Combinatorial targeting of immune checkpoints and epigenetic regulators for hepatocellular carcinoma therapy. Oncogene https://doi.org/10.1038/s41388-023-02646-1 (2023).
Lee, S. & Schmitt, C. A. The dynamic nature of senescence in cancer. Nat. Cell Biol. 21, 94–101. https://doi.org/10.1038/s41556-018-0249-2 (2019).
Moon, C. M., Shin, S. S., Heo, S. H. & Jeong, Y. Y. Metabolic alterations associated with early-stage hepatocellular carcinoma and their correlation with aging and enzymatic activity in patients with viral hepatitis-induced liver cirrhosis: A preliminary study. J. Clin. Med. 9, 765. https://doi.org/10.3390/jcm9030765 (2020).
Shi, R. et al. APOBEC-mediated mutagenesis is a favorable predictor of prognosis and immunotherapy for bladder cancer patients: Evidence from pan-cancer analysis and multiple databases. Theranostics 12, 4181–4199. https://doi.org/10.7150/thno.73235 (2022).
Prasetyo, A. A. et al. The APOBEC3B deletion polymorphism is associated with prevalence of hepatitis B virus, hepatitis C virus, Torque Teno virus, and Toxoplasma gondii co-infection among HIV-infected individuals. J. Clin. Virol. 70, 67–71. https://doi.org/10.1016/j.jcv.2015.07.009 (2015).
Warren, C. J. et al. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 89, 688–702. https://doi.org/10.1128/JVI.02383-14 (2015).
Nakaya, Y., Stavrou, S., Blouch, K., Tattersall, P. & Ross, S. R. In vivo examination of mouse APOBEC3- and human APOBEC3A- and APOBEC3G-mediated restriction of parvovirus and herpesvirus infection in mouse models. J. Virol. 90, 8005–8012. https://doi.org/10.1128/JVI.00973-16 (2016).
Gansmo, L. B. et al. APOBEC3A/B deletion polymorphism and cancer risk. Carcinogenesis 39, 118–124. https://doi.org/10.1093/carcin/bgx131 (2018).
Lucifora, J. et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343, 1221–1228. https://doi.org/10.1126/science.1243462 (2014).
Wang, D. et al. APOBEC3B interaction with PRC2 modulates microenvironment to promote HCC progression. Gut 68, 1846–1857. https://doi.org/10.1136/gutjnl-2018-317601 (2019).
Kucirek, M., Bagherpoor, A. J., Jaros, J., Hampl, A. & Stros, M. HMGB2 is a negative regulator of telomerase activity in human embryonic stem and progenitor cells. FASEB J. 33, 14307–14324. https://doi.org/10.1096/fj.201901465RRR (2019).
Zirkel, A. et al. HMGB2 loss upon senescence entry disrupts genomic organization and induces CTCF clustering across cell types. Mol. Cell 70, 730–744. https://doi.org/10.1016/j.molcel.2018.03.030 (2018).
Aird, K. M. et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 215, 325–334. https://doi.org/10.1083/jcb.201608026 (2016).
Zhao, B. et al. Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat. Commun. 11, 908. https://doi.org/10.1038/s41467-020-14652-y (2020).
Calvisi, D. F. et al. Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 68, 4192–4200. https://doi.org/10.1158/0008-5472.CAN-07-6157 (2008).
Hao, P. P. et al. Disruption of a regulatory loop between DUSP1 and p53 contributes to hepatocellular carcinoma development and progression. J. Hepatol. 62, 1278–1286. https://doi.org/10.1016/j.jhep.2014.12.033 (2015).
Zhao, B., Chang, L., Fu, H., Sun, G. & Yang, W. The role of autoimmune regulator (AIRE) in peripheral tolerance. J. Immunol. Res. 2018, 3930750. https://doi.org/10.1155/2018/3930750 (2018).
Zhu, M. L., Nagavalli, A. & Su, M. A. Aire deficiency promotes TRP-1-specific immune rejection of melanoma. Cancer Res. 73, 2104–2116. https://doi.org/10.1158/0008-5472.CAN-12-3781 (2013).
Chen, J. et al. Erratum to: Functional analysis of histone deacetylase 11 (HDAC11). Methods Mol. Biol. 1436, E1. https://doi.org/10.1007/978-1-4939-3667-0_22 (2016).
Gong, D., Zeng, Z., Yi, F. & Wu, J. Inhibition of histone deacetylase 11 promotes human liver cancer cell apoptosis. Am. J. Transl. Res. 11, 983–990 (2019).
Woods, D. M. et al. T cells lacking HDAC11 have increased effector functions and mediate enhanced alloreactivity in a murine model. Blood 130, 146–155. https://doi.org/10.1182/blood-2016-08-731505 (2017).
Yun, C. W., Han, Y. S. & Lee, S. H. PGC-1alpha controls mitochondrial biogenesis in drug-resistant colorectal cancer cells by regulating endoplasmic reticulum stress. Int. J. Mol. Sci. 20, 1707. https://doi.org/10.3390/ijms20071707 (2019).
Bost, F. & Kaminski, L. The metabolic modulator PGC-1alpha in cancer. Am. J. Cancer Res. 9, 198–211 (2019).
Acknowledgements
We would like to acknowledge the financial support from Shandong Traditional Chinese Medicine Science and Technology Project (No. 2020M139) and the Scientific Research Project of Shandong College of traditional Chinese Medicine (No. 2021FY02). We thank the TCGA database for sharing the data.
Funding
This work was supported by Grants from the Development Plan of Shandong Traditional Chinese Medicine Science and Technology Project (No. 2020M139), and the Scientific Research Project of Shandong College of traditional Chinese Medicine (No. 2021FY02).
Author information
Authors and Affiliations
Contributions
J.W.C., X.W.H. and Y.N.D. conceived and designed the study. J.W.C. and X.W.H. performed data analysis. J.W.C., X.W.H. and M.M.Z. wrote the manuscript. YND reviewed the manuscript. All the authors finally approved the submitted version and agreed on the journal to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chai, Jw., Hu, Xw., Zhang, Mm. et al. Seven chromatin regulators as immune cell infiltration characteristics, potential diagnostic biomarkers and drugs prediction in hepatocellular carcinoma. Sci Rep 13, 18643 (2023). https://doi.org/10.1038/s41598-023-46107-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46107-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
