
Abstract
Diabetes mellitus has a high prevalence rate and it has been deemed a severe chronic metabolic disorder with long-term complications. This research aimed to identify compounds that could potentially inhibit the vital metabolic enzyme α-glucosidase and thereby exert an anti-hyperglycemic effect. The main goal was to establish an effective approach to control diabetes. To proceed with this study, a series of novel coumarin-derived thiosemicarbazones 3a–3m was synthesized and examined using a variety of spectroscopic methods. Moreover, all the compounds were subjected to α-glucosidase inhibition bioassay to evaluate their antidiabetic potential. Fortunately, all the compounds exhibited several folds potent α-glucosidase inhibitory activities with IC50 values ranging from 2.33 to 22.11 µM, in comparison to the standard drug acarbose (IC50 = 873.34 ± 1.67 µM). The kinetic studies of compound 3c displayed concentration-dependent inhibition. Furthermore, the binding modes of these molecules were elucidated through a molecular docking strategy which depicted that the thiosemicarbazide moiety of these molecules plays a significant role in the interaction with different residues of the α-glucosidase enzyme. However, their conformational difference is responsible for their varied inhibitory potential. The molecular dynamics simulations suggested that the top-ranked compounds (3c, 3g and 3i) have a substantial effect on the protein dynamics which alter the protein function and have stable attachment in the protein active pocket. The findings suggest that these molecules have the potential to be investigated further as novel antidiabetic medications.
Similar content being viewed by others
Introduction
Diabetes mellitus (DM) has been recognized as one of the most widespread global health problems during the twenty-first century, with the prevalence surpassing 400M individuals globally1. According to the World Health Organization (WHO), diabetes is projected to become the seventh leading cause of death worldwide by 20302. Broadly this metabolic heterogeneous disorder has two types: (i). insulin-dependent DM (type 1) and (ii). insulin-independent DM (type 2)3. Amidst these distinctions, both types cause alterations in glucose transport in a variety of tissue systems, leading to hyperglycemia4. This disruption is associated with an increased production of α-glycosidase, an enzyme responsible for hydrolysis of oligo- and disaccharides into monosaccharides5. Several types of α-glucosidase inhibitors have been identified to control this situation like voglibose, acarbose, and miglitol6, however, unfortunately, these inhibitors have shown side effects like abdominal discomfort, flatulence, and diarrhea2. Such complexities pose immense pressure on the pharmaceutical industry to develop novel therapeutic drugs with low toxicity and improved efficiency7.
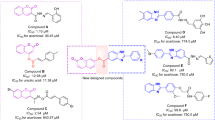
Based on our thorough research, a huge number of alternative proposed compounds for α-glucosidase inhibition have been reported to date8,9,10,11. Lately thiosemicarbazones with general formula R1R2C=N–NH(C=S)NHR12 and typical synthetic route involving condensation of thiosemicarbazide with aldehyde/ketone have been gaining popularity among medicinal chemists13. These scaffolds bear a range of biological activities like antibacterial14,15,16, antifungal17,18, antihelmintic19,20, antimalarial21,22, antiviral23,24, anticancer25,26, and antidiabetic potential27,28. Additionally, several research groups have documented the comprehensive account of the antidiabetic potential of thiosemicarbazones and coumarin based compounds Fig. 110,29,30,31,32,33.

Thiosemicarbazone and Coumarin based α-glucosidase inhibitors.
In continuation of our efforts to develop effective α-glucosidase inhibitors34,35,36,37 for combating the global health condition Diabetes Mellitus (DM) and to assess the efficacy of coumarin analogues, we have designed and synthesized several coumarin derived thiosemicarbazones in this study.
Chemistry
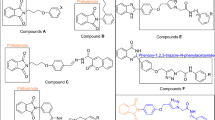
The preparation of coumarin derived thiosemicarbazones was carried out as in Fig. 2. Thiosemicarzides were generated by stirring the solution of phenyl isothiocyanate (5 mmol) in 20 mL of ethanol for 1 h, followed by dropwise addition of an ethanolic solution of hydrazine hydrate (5 mmol). The precipitates were filtered and washed with ethanol, and water and then dried over silica gel. Following this, thiosemicarbazone synthesis was commenced by dissolving coumarin aldehyde in EtOH at 90 °C with a few crystals of p-Toluenesulfonic acid (PTSA) as a catalyst. Once a clear solution is formed, respective thiosemicarbazide is introduced in the reaction mixture and set to reflux for 6–8 h. The progress of the reaction was continuously monitored through TLC. After the completion of the reaction, the reflux was stopped. Upon cooling, the coumarin derived thiosemicarbazone adduct was filtered and subsequently washed using cold MeOH.

Synthetic scheme of coumarin based thiosemicarbazones 3a–3m.
To determine the structures of compounds 3a–3m, spectral and elemental techniques were employed. In the infrared (IR) spectra C=S and C=N absorption bands appeared in the range of 1156–1228 cm−1 and 1590–1618 cm−1 respectively, confirming the presence of thiosemicarbazone derivatives. Likewise, C=O and NH, stretching vibrations correspondingly appeared in the region of 1699–1741 cm−1 and 3207–3371 cm−1. 1H NMR spectra added further structural confirmation to coumarin derived thiosemicarbazones through the observed chemical shifts for coumarin C–H and thiosemicarbazone N–H appearing within the range of 7.81–9.92 ppm and 8.74–11.19 ppm respectively. Finally, 13C NMR marked C=S and C=O signal at 173.52–179.38 ppm and 159.20–159.61 ppm respectively as the most downfield signals. Thus, all the analytical data provided consistent confirmation of the compound’s structures.
Results and discussion
In-vitro α-glucosidase inhibitory activity
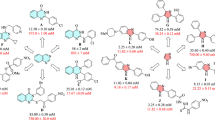
In the current studies, several coumarin-derived thiosemicarbazones were synthesized and their therapeutic capability by inhibiting α-glucosidase was evaluated which can control the postprandial hyperglycemia effect of dibetes. In this context, all the synthesized compounds 3a–3m were proceeded against α-glucosidase in-vitro bioassay, interestingly All of them exhibited several folds more antidiabetic potential (IC50 = 2.33–22.11 µM) as compared to the available marketed drug, acarbose (1C50 = 873.34 ± 1.67 µM). All the compounds have different R-substituents, which are responsible for the variation in the α-glucosidase inhibitory potential of these molecules. For instance, compound 3a, with phenyl moiety exhibited potent antidiabetic activity (IC50 = 10.0 ± 0.14 µM), while in compound 3g, substitution of β-phenyl increased its antidiabetic potential (IC50 = 2.33 ± 0.04 µM) as compared to 3a. The methoxy group substitutions at different positions were checked in compounds 3b and 3h. In compound 3b, 3-methoxyphenyl substituent resulted in potent inhibitory capability against α-glucosidase (IC50 = 6.10 ± 0.11 µM). While in compound 3h, the substitution of 4-methoxyphenyl further enhanced its α-glucosidase inhibition (IC50 = 3.40 ± 0.08 µM) as compared to 3b.
The effect of chlorophenyl substitution in compounds 3c and 3k was evaluated to identify the favorable position of substituent which is responsible for their maximum inhibitory capability. Compound 3c with 2,4,5-trichlorophenyl group, displayed significant anti-α-glucosidase potential with IC50 of 2.57 ± 0.03 µM. On the other hand, in compound 3k, the substitution of 2,3-dichlorophenyl declined its α-glucosidase inhibitory potential (IC50 = 4.22 ± 0.10 µM) as compared to 3c. Similarly, the substitution of 2-fluorophenyl in compound 3l further decreased the α-glucosidase inhibitory potential of 3l with IC50 = 8.84 ± 0.16 µM. Similarly, we observed slight adverse effects of the substitution of benzyl and 4-tolyl groups in compounds 3d (IC50 = 12.55 ± 0.16 µM) and 3e (IC50 = 9.11 ± 0.20 µM), respectively. Likewise, the substitution of cyclohexyl in 3f. produced almost similar antidiabetic effects like 3c and 3d with an IC50 value of 11.20 ± 0.24 µM.
The effect of nitrophenyl, dimethylphenyl and methyl group substitutions was investigated in compounds 3i, 3j, and 3m, respectively to reveal their therapeutic use for the treatment of diabetes. In compound 3i, the substitution of 4-nitrophenyl displayed an overwhelming anti α-glucosidase activity (IC50 = 2.13 ± 0.04 µM) and made it the most potent antidiabetic agent in this series. While compounds 3j with 2,6-dimethylphenyl (IC50 = 22.11 ± 0.37 µM) and 3m with single methyl substitution (IC50 = 24.22 ± 0.31 µM) displayed almost similar α-glucosidase inhibitory potential.
This structure–activity relationship reveals that substitution of 4-nitrophenyl, β-phenyl, and 2,4,5-trichlorophenyl in 3i, 3g and 3c is most beneficial as compared to the addition of other moieties. Moreover, 3/4-methoxyphenyl in 3b/3h, and 2,3-dichlorophenyl in 3k also warrant further optimization to enhance the biological importance of 3b, 3h, and 3k. The results are summarized in Table 1.
Kinetic studies
The most potent compounds 3c, 3g and 3i were selected for kinetic studies to observe their mechanism of inhibition. In kinetic studies, compounds 3c, 3g and 3i were identified as the competitive inhibitor of α-glucosidase enzyme with ki values of 1.62 ± 0.041, 1.33 ± 0.0027 and 1.51 ± 0.0086 µM respectively. In such types of inhibition, the Vmax of the enzyme remains constant, while the Km value increases (Fig. 3). Lineweaver–Burk plots were used to determine the type of inhibition of the given compounds, in which the reciprocal of the reaction rate was plotted vs. the reciprocal of substrate concentrations to investigate their effect on the Km and Vmax of the α-glucosidase (Fig. 3). To determine the Ki values for all compounds evaluated for kinetics study secondary replot of Lineweaver–Burk plots were applied by taking the slope of each line in Lineweaver–Burk plots vs. different concentrations of the tested compounds. Further Dixon plots were used for the reconfirmation of Ki values (Fig. 3).

The inhibitory mode of 3c, 3g and 3i against α-glucosidase enzyme (A) Line weaver-Burk plot of reciprocal of rate of reaction (velocities) vs reciprocal of substrate p-nitro phenyl-α-d-glucopyranoside in the absence of (filled black square), and in the presence of 7.00 (filled black circle), 3.50 (open circle) and 1.75 µM (open square) of 3c, 3g and 3i. (B) Secondary replot of Line weaver-Burk plot between the slopes of each line on Line weaver-Burk plot against different concentrations of 3c, 3g and 3i. (C) Dixon plot of reciprocal of rate of reaction (velocities) vs different concentrations of 3c, 3g and 3i.
Docking studies
All the compounds showed excellent inhibitory potential against the α-glucosidase enzyme; therefore, these compounds were docked at the binding site of α-glucosidase. Docking results indicate that all these molecules adopt almost the same orientation with slight conformational differences. The thiosemicarbazone moiety of these molecules is inserted deep into the active site core where this moiety has a chance to interact with any one of the residues of the catalytic triad (Asp215, Glu277, and Asp352). While the chloro-substituted coumarin ring faces the entrance of the active site, the R group has a chance to rotate towards the loop residues (Tyr72, Phe159, Phe178, Gln182, and Arg442) which lines the catalytic triad.
The most active compound 3i, formed excellent interactions with Asp352 which is a part of the catalytic triad, in addition two crucial residues of the active site i.e., Gln353 and His112 also provide Hydrogen bonds (H-bond) to this molecule. The thiosemicarbazone moiety of 3i mediates H-bond with the side chains of Asp352 and Gln353, while the nitro group of 3i mediates H-bond with the side chain of His112. The thiosemicarbazone moiety of 3g, which is the second most active compound, was oriented towards Glu277 (of the catalytic triad) and forms H-bonds with Glu277, Arg213 and His351. While the phenyl ring (R group) of 3g interacts hydrophobically with Tyr72, and Phe178 and Phe178 provide π–π interaction to the R group. Due to the addition of bulky R groups in 3c (tri-chlorophenyl), 3h (methoxyphenyl) and 3k (di-chlorophenyl), their thiosemicarbazone moiety skipped interaction with catalytic triad residues, instead 3c, 3h and 3k formed H-bonds with Arg213/His351, Arg213/His112, and Arg213/His351, respectively.
Similarly, compounds 3b and 3l followed the interaction pattern of 3g, and their thiosemicarbazone moiety interacted with Glu277 and Arg213. In addition, compounds 3b and 3l also formed H-bonds with a water molecule and His351, respectively. We observed that compounds 3e and 3h have almost similar binding orientation and both these molecules interacts with His112 and a water molecule. Additionally, the R group of 3e also formed a hydrophobic (π–π) interaction with Phe303. Similarly, compounds 3a and 3m showed the same binding pattern, and these molecules bind with Asp352, and His351 through H-bonds. While the thiosemicarbazone functional group of 3f does not contribute to the binding interaction, and only its coumarin mediates H-bonding with the side chain of Gln353, and forms π-π interactions with Phe303. Likewise, the thiol group of 3d only formed an H-bond with His112. Like 3f, the coumarin oxygen of 3j also interacted with Gln353, while the carbazide nitrogen of this molecule formed an H-bond with the side chain of Asp352 like compounds 3a and 3m. The docking results indicate that the conformational difference can affect the binding pattern, as a result, the activity is affected. The docking scores and interactions are given in Table 2. The docking scores of the compounds are in the range of − 6.34 kcal/mol to − 3.21 kcal/mol, which reflects the good binding potential of these molecules with the active site of the enzyme. The binding modes of compounds are depicted in Fig. 4. The most active compounds (3i, 3g, and 3c) exhibited the highest docking scores, i.e., − 6.34 to − 6.34 kcal/mol, likewise, the moderate active molecules demonstrated docking scores in range of > − 5 to > − 3 kcal/mol, and two least active molecules in this series (3j and 3m) bear the least docking scores. According to the docking scores of compounds, the docking results correlate well with the in-vitro assay results. Furthermore, docking analysis reflects the importance of Asp352, Gln353, His112, Glu277, Arg213, His351, and Arg442 in the stabilization of compounds in the binding site of the enzymes, while Phe178, and Phe303 are crucial to providing hydrophobic interactions to these novel inhibitors.

(A) The binding mode of all the compounds (presented in the cyan stick model) is shown in the active site of α-glucosidase. (B) The interactions of the most active compound (3i, green ball, and stick model) are shown with the active site residues (shown in yellow sticks). H-bonds are presented in black dotted lines.
Post-simulation analysis
Examining the protein dynamic stability
The RMSD of a simulation snapshot indicates how closely it matches a reference structure, which might be the starting simulation frame or a crystal structure. This parameter is used to examine the development of structural elements over time [70]. It is particularly useful in determining if a structure remains stable during the simulation time or deviates from its beginning coordinates. To assess the protein's stability during the simulation, we examined the RMSD plot of the Cα atoms (Fig. 5). This included comparing each frame in time to the simulation's initial frame. The RMSDs for the apo form of -glucosidase, the -glucosidase-maltose complex (3A4A positive control), and three -glucosidase -ligand complexes produced by molecular docking were calculated. The APO protein average RMSD calculated was 1.59 ± 0.003 Å. During the initial 60 ns, the RMSD of the protein gradually increases from 0.6 to 1.8 Å. The RMSD then remains stable for the rest of the simulation time. On the other hand, the ligand-bound protein 3A4A shows a significant increase in the mean RMSD (2.42 ± 0.006 Å). A rapid increase was observed in the first 40 ns simulation time (2 Å increase), where the protein went through structure confirmation from 41 to 70 ns simulation time. After 70 ns, the system got equilibrium and retained a stable RMSD with an average value of 2.7 Å. In the selected compounds complexes, 3c shows an average RMSD of 1.69 ± 0.006 Å, while the 3g and 3i mean RMSD calculated was 0.01 ± 0.001 Å and 1.43 ± 0.003 Å, respectively. The 3c system shows flexibility in the start 70 ns simulations where the RMSD value was increased gradually (0.9 Å to 2.4 Å). After that, the system gains equilibrium and has a stable RMSD value (2.5 Å mean RMSD). Compound 3g retains a stable behavior in the RMSD pattern till the end of the simulation, where the first 20 ns shows a slight increase in the RMSD values. The RMSD values of the 3i system increased from the start till 60 ns with a gradual increase pattern (1 Å increased). The system retains a stable RMSD by gaining equilibrium till the end of the simulation (1.8 Å mean RMSD). The RMSD results indicate that the ligand-attached systems showed a stable complex by retaining a stable RMSD for most of the simulation time. Thus, it signifies that the ligand and the target protein have a favorable interaction because the unbound protein may naturally adopt a conformation that favors binding with the small molecule in the complex. Further, the RMSD results indicate that there were no significant jumps in each system's RMSD profile, which signifies the simulations' output results.

Root mean square deviation analysis of the α-glucosidase in free state reference inhibitor attached (3A4A) and selected compounds (3c, 3g, and 3i) from 100 ns simulation trajectories.
Residues fluctuations measurement
Following that, we investigated whether protein residues or segments contributed to the observed changes in mobility shown in the RMSF data (Fig. 6). The RMSF value is critical for understanding how the side chains of residues change during MD simulation. The average RMSF calculated for the APO protein was 0.97 ± 0.02 Å. The high mobility regions observed in the APO protein were 276–298, 216–252, and 394–442 residues. The residues regions 276–298 and 216–252 show a loop confirmation, while the 394–442 region has a loop and H-19 to H22 helix regions. The average RMSF calculated for the reference system 3A4A was 1 ± 0.02 Å. The 394–442 residues in the 3A4A show a considerable fluctuation exceeding the 2 Å. The residues region 270–340 shows a confirmational shift from the free state protein, where the protein went structural confirmation. The average RMSF of the 3c system was 1.1 ± 0.02 Å, where the residues region 122–182 show a variable structural confirmation which exceeds the 2 Å value. The structural confirmation of the 122–182 residues region shows that most of the region is loop confirmation, which shows flexibility in the simulation. The 3g and 3i systems show mean RMSF values of 0.7 ± 0.01 Å and 0.8 ± 0.01 Å, respectively. The 3g system shows restricted movements in the regions that fluctuate high (above 2 Å) in the APO system, and the 3g shows fluctuation below 2 Å in the overall simulation. Similarly, the 3i system retains a stable RMSF where the 132–182 and 219–250 residues region shows high flexibility. The residues 376–312 region shows a restricted motion compared to the APO, where the APO shows fluctuation up to 4 Å. The RMSF results show that the protein adopts structural confirmation upon the ligand attachment with the protein active pocket. This indicates that the selected compounds bind tightly to the protein active site residues. The overall RMSF results of each system indicate that the proteins remain stable during the simulation run.

Root mean square fluctuation analysis to analyze the residues flexibility of the selected compound with a reference system 3A4A and free state in 100 ns simulation time.
Protein compactness analysis
The RG values calculated from MD modeling of unbound proteins and protein–ligand complexes may be regarded as measures of their overall compactness. It represents the root mean square distance between the component atoms' centers of mass. A lower RG number indicates a more compact and firmly folded structure, while a larger value indicates a more extended or flexible conformation. The value aids in determining how ligand binding affects the overall shape and flexibility of the protein by giving insight into the structural dynamics and stability of the protein or protein–ligand complex. Figure 7 shows the calculated RG profiles of simulated systems. The average Rg calculated for the APO protein was 24.62 ± 0.002 Å. In the first 60 ns simulation time, the Rg of the APO system increases gradually, and after 70 ns, the system gets a stable Rg having a compact protein confirmation. The reference ligand attached to system 3A4A shows a mean Rg value of 24.97 ± 0.002 Å. The 3A4A system shows flexibility in the first 50 ns simulation time where the Rg increases (0.9 Å increase). After that, the system retains the Rg with a mean value of 25.05 Å. A steady increase of 1.8 Å from the initial values to 70 ns simulation time. The system retains the Rg with a mean value of 24.9 Å till the end of the simulation. The average Rg values of the 3c and 3g systems are 24.60 ± 0.03 Å and 24.19 ± 0.001 Å, respectively. The 3g system retains a stable Rg in the simulation with minor bumps. The average Rg calculated for the 3i system reported was 24.44 ± 0.002 Å. The first 50 ns simulation time shows a constant increase in the Rg value (24.0 Å to 24.6 Å). After that, the structural confirmation of the system's Rg remains steady until the end of the simulation (average Rg 24.7 Å). As per the Rg results, the protein experienced conformational changes during the initial of the simulation to better constrain the ligand within the pocket, followed by establishing solid contact with the ligand and obtaining a stable Rg.

Protein compactness analysis of selected compounds free state (APO) and reference inhibited system 3A4A via Radius f gyration.
Protein and ligand binding affinity estimation
The BFE was calculated using the most stable output MD trajectories (the final 2750 frames of each complex) of the three α-glucosidase-inhibitor structures and a reference 3A4A system using the MM-GBSA technique. The MM-GB/PBSA method uses a continuum solvent model to calculate the complex's binding free energy values. As a result, the BFE values determined in this research are related to the absolute or total free energy. The complexes with the lowest BFE value were the most stable. Table 3 contains the computed values. The results revealed variations in the BFE among the ligands. The reference compound 3A4A showed a stable complex with a total BFE (ΔGTOTAL) value of -12.8 ± 0.10 kcal/mol. Further analysis showed that contributions of electrostatic energy (ΔEELE) and gas phase free energy (ΔGgas) were the most significant factors in binding this ligand to the protein. Based on the results from the MMGBSA calculation, the 3g compound exhibits the highest BFE of − 24.7 ± 0.07 kcal/mol. The second highest binding energy was reported for the 3i compound of − 22.7 ± 0.07 kcal/mol, while the 3c system calculated binding energy is − 17.2 ± 0.06 kcal/mol. Consequently, the selected compounds (3c, 3g, and 3i) complexes are likely more stable than the reference compound 3A4A. The 3c system ΔEELE and ΔGgas components are the main contributors to the BFE, while the 3g and 3i system shows reduced in the ΔEELE and high ΔEVDW contribution. In conclusion, the study's results show that the selected compounds can serve as α-glucosidase inhibitors, making them viable candidates for building putative antidiabetic medicines.
Experimental
Material
All chemical reagents were purchased from Sigma-Aldrich Company, and no additional purification was performed. Commercially available oven-dried round bottom flasks were utilized for the reaction. The Heraeus Elemental Analyzer was used to execute elemental analyses. On an ABB Bomem FTIR instrument, KBr pellets were used to record infrared spectra in the range of 4000–500 cm−1. The Bruker DRX 500 with TMS as standard and DMSO-d6 solvent was used to record the 1H NMR and13C NMR spectra.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl)methylene)-N-phenylhydrazine-1-carbothioamide (3a)
Yellowish solid, Yield, 86%; IR υmax (cm−1): 1623 (C=N), 1655 (C=O), 3365 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.29 (1 H, s), 9.76 (1 H, s), 8.39 (1 H, s), 8.02 (1 H, dd, J 8.2, 1.3), 7.79–7.72 (1 H, m), 7.68 (2 H, d, J 7.6), 7.54–7.47 (2 H, m), 7.39 (2 H, dd, J 10.8, 5.0), 7.21 (1 H, t, J 7.4).13C NMR (101 MHz, DMSO) δ 176.10, 157.80, 151.68, 145.72, 138.93, 136.52, 134.26, 128.91, 126.45, 125.90, 125.77, 124.39, 119.32, 118.80, 117.07; Anal. Calcd. for C17H12ClN3O2S (357.81): C, 57.07; H, 3.38; N, 11.74; Found: C, 57.11; H, 3,33; N, 11.89.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl)methylene)-N-(3-methoxyphenyl)hydrazine-1-carbothioamide (3b)
Yellowish solid, Yield, 82%; IR υmax (cm−1): 1631 (C=N), 1666 (C=O), 3340 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.31 (1 H, s), 9.75 (1 H, s), 8.38 (1 H, s), 8.03 (1 H, dd, J 8.3, 1.4), 7.80–7.72 (1 H, m), 7.54–7.45 (3 H, m), 7.29 (1 H, t, J 8.1), 7.22–7.14 (1 H, m), 6.82–6.74 (1 H, m), 3.77 (3 H, s); 13C NMR (101 MHz, DMSO) δ 175.79, 159.70, 157.77, 151.69, 145.76, 140.04, 136.49, 134.28, 129.71, 126.46, 125.91, 119.32, 118.80, 117.07, 116.27, 111.14, 109.77, 55.65; Anal calcd for C18H14ClN3O3S (387.84); C, 55.74; H, 3.64; 10.83; Found: C, 55.91; H, 3.77; 10.97.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl)methylene)-N-(2,4,5-trichlorophenyl) hydrazine-1-carbothioamide (3c)
Off-white solid, Yield, 90%; IR υmax (cm−1): 1639 (C=N), 1664 (C=O), 3354 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.64 (1 H, s), 9.83 (1 H, s), 8.56 (1 H, s), 8.45 (1 H, s), 8.05 (1 H, dd, J 8.3, 1.5), 7.99 (1 H, s), 7.81–7.72 (1 H, m), 7.51 (2 H, ddd, J 5.9, 3.7, 1.3); 13C NMR (101 MHz, DMSO) δ 176.17, 157.82, 151.81, 146.14, 138.25, 136.19, 134.44, 130.79, 129.82, 128.86, 128.06, 127.87, 126.62, 125.92, 118.99, 118.81, 117.09; Anal calcd for C17H9Cl4N3O2S (461.14); C, 44.28; H, 1.97; N, 9.11; Found: C, 44.36; H, 2.01; N, 9.33.
(E)-N-benzyl-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene) hydrazine-1-carbothioamide (3d)
Light Yellow solid, Yield, 88%; IR υmax (cm−1): 1641 (C=N), 1654 (C=O), 3334 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.05 (1 H, s), 8.45 (1 H, t, J 6.1), 8.30 (1 H, d, J 0.7), 8.00 (1 H, dd, J 8.3, 1.4), 7.77–7.69 (1 H, m), 7.48 (2 H, ddd, J 4.4, 3.6, 1.9), 7.35 (4 H, t, J 5.5), 7.31–7.22 (1 H, m), 4.86 (2 H, d, J 6.1); 13C NMR (101 MHz, DMSO) δ 178.24, 157.99, 151.62, 145.07, 139.25, 136.34, 134.08, 128.89, 128.81, 128.64, 128.26, 127.94, 127.70, 127.45, 126.42, 125.84, 119.44, 118.83, 117.02, 47.25.
Anal calcd for C18H14ClN3O2S (371.84); C, 58.14; H, 3.80; N, 11.30; Found: C, 58.24; H, 3.77; N, 11.52.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-(p-tolyl) hydrazine-1-carbothioamide (3e)
Yellow solid, Yield, 96%; IR υmax (cm−1): 1654 (C=N), 1655 (C=O), 3342 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.25 (1 H, s), 9.67 (1 H, s), 8.37 (1 H, s), 8.02 (1 H, dd, J 8.3, 1.4), 7.81–7.71 (1 H, m), 7.51 (4 H, ddd, J 6.3, 5.3, 3.4), 7.18 (2 H, d, J 8.2), 2.31 (3 H, s); 13C NMR (101 MHz, DMSO) δ 176.10, 157.80, 151.67, 145.62, 136.38, 135.03, 134.23, 129.51, 129.33, 126.44, 125.89, 124.46, 119.35, 118.81, 117.06, 21.03; Anal calcd for C18H14ClN3O2S (371.84); C, 58.14; H, 3.80; N, 11.30; Found: C, 58.29; H, 3.71; N, 11.60.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl)methylene)-N-cyclohexylhydrazine-1-carbothioamide (3f)
White solid, Yield, 84%; IR υmax (cm−1): 1651 (C=N), 1650 (C=O), 3335 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 11.95 (1 H, s), 8.29 (1 H, d, J 0.7), 8.04–7.99 (1 H, m), 7.77–7.71 (2 H, m), 7.53–7.46 (2 H, m), 4.22–4.07 (1 H, m), 1.91 (2 H, t, J 8.7), 1.67 (2 H, s), 1.56 (1 H, d, J 12.0), 1.37 (4 H, dd, J 18.8, 9.9), 1.27 (1 H, d, J 9.2); 13C NMR (101 MHz, DMSO) δ 176.52, 157.73, 151.59, 144.87, 135.56, 134.08, 126.35, 125.84, 119.43, 118.84, 117.02, 52.04, 32.16, 25.49, 24.58; Anal calcd for C17H18ClN3O2S (363.86); C, 56.12; H, 4.99; N, 11.55; Found: C, 56.19; H, 5.09; N, 11.67.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-phenethylhydrazine-1-carbothioamide (3g)
Yellow solid, Yield, 87%; IR υmax (cm−1): 1655 (C=N), 1641 (C=O), 3344 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 11.95 (1 H, s), 8.26 (1 H, s), 8.00 (1 H, dd, J 8.3, 1.1), 7.95 (1 H, t, J 5.6), 7.74 (1 H, td, J 8.0, 1.5), 7.51 (2 H, dd, J 11.9, 4.4), 7.38–7.21 (5 H, m), 3.83 (2 H, dd, J 13.3, 7.0), 2.92 (2 H, t, J 7.3); 13C NMR (101 MHz, DMSO) δ 177.70, 158.16, 151.59, 144.72, 139.43, 136.11, 134.04, 129.11, 128.98, 126.77, 126.42, 125.87, 119.35, 118.89, 117.04, 45.33, 34.92; Anal calcd for C19H16ClN3O2S (385.87); C, 59.14; H, 4.18; N, 10.89; Found: C, 59.19; H, 4.31; N, 10.99.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-(4-methoxyphenyl) hydrazine-1-carbothioamide (3h)
Light yellow solid, Yield, 80%; IR υmax (cm−1): 1645 (C=N), 1636 (C=O), 3321 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.22 (1 H, s), 9.60 (1 H, s), 8.37 (1 H, s), 8.03 (1 H, dd, J 8.3, 1.5), 7.80–7.72 (1 H, m), 7.56–7.46 (4 H, m), 6.96–6.90 (2 H, m), 3.77 (3 H, s); 13C NMR (101 MHz, DMSO) δ 176.50, 157.88, 157.42, 151.67, 145.56, 136.38, 134.21, 131.84, 126.61, 126.45, 125.90, 119.39, 118.82, 117.07, 114.03, 55.75; Anal calcd for C18H14ClN3O3S (387.84); C, 55.74; H, 3.64; N, 10.83; Found: C, 55.91; H, 3.52; N, 10.96.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-(4-nitrophenyl) hydrazine-1-carbothioamide (3i)
Yellow solid, Yield, 90%; IR υmax (cm−1): 1647 (C=N), 1655 (C=O), 3342 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 10.44 (1 H, s), 8.47 (1 H, s), 8.23–8.16 (3 H, m), 7.89–7.84 (2 H, m), 7.80–7.74 (1 H, m), 7.56–7.47 (2 H, m); 13C NMR (101 MHz, DMSO) δ 157.97, 153.66, 153.16, 149.78, 146.60, 142.03, 134.89, 134.58, 127.24, 125.52, 125.41, 121.15, 119.47, 118.80, 117.43, 99.99; Anal calcd for C17H11ClN4O4S (402.81); C, 50.69; H, 2.75; N, 13.91; Found: C, 50.77; H, 2.71; N, 13.96.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-(2,6-dimethylphenyl) hydrazine-1-carbothioamide (3j)
Yellow solid, Yield, 88%; IR υmax (cm−1): 1641 (C=N), 1625 (C=O), 3331 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.19 (1 H, s), 9.30 (1 H, s), 8.36 (1 H, s), 8.02 (1 H, dd, J 8.4, 1.5), 7.78–7.71 (1 H, m), 7.54–7.47 (2 H, m), 7.14–7.07 (3 H, m), 2.18 (6 H, s); 13C NMR (101 MHz, DMSO) δ 177.59, 158.31, 151.64, 145.17, 137.14, 136.77, 136.52, 134.08, 128.15, 127.58, 126.50, 125.88, 119.41, 118.94, 117.05, 18.46; Anal calcd for C19H16ClN3O2S (385.87); C, 59.14; H, 4.18; N, 10.89; Found: C, 59.21; H, 4.09; N, 10.99.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-(2,3-dichlorophenyl) hydrazine-1-carbothioamide (3k)
Off-white solid, Yield, 92%; IR υmax (cm−1): 1633 (C=N), 1635 (C=O), 3345 (N–H); 1H NMR (400 MHz, DMSO-d6) δ, ppm 12.54 (1 H, s), 9.82 (1 H, s), 8.44 (1 H, s), 8.05 (2 H, ddd, J 8.2, 3.8, 1.3), 7.81–7.72 (1 H, m), 7.58–7.48 (3 H, m), 7.42 (1 H, t, J 8.1); 13C NMR (101 MHz, DMSO) δ 176.60, 158.00, 151.77, 145.82, 138.13, 137.83, 134.35, 132.09, 128.14, 128.08, 127.61, 127.02, 126.59, 125.91, 119.09, 118.86, 117.09; Anal calcd for C17H10Cl3N3O2S (426.70); C, 47.85; H, 2.36; N, 9.85; Found: C, 47.90; H, 2.33; N, 9.99.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-(2-fluorophenyl) hydrazine-1-carbothioamide (3l)
White solid, Yield, 81%; IR υmax (cm−1): 1643 (C=N), 1636 (C=O), 3351 (N–H); 1H NMR (600 MHz, DMSO-d6) δ, ppm 12.44 (1 H, s), 9.61 (1 H, s), 8.40 (1 H, s), 8.09–8.00 (2 H, m), 7.74 (1 H, dd, J 11.4, 4.0), 7.49 (2 H, t, J 7.1), 7.29 (2 H, dd, J 19.9, 9.0), 7.22 (1 H, t, J 7.4); 13C NMR (151 MHz, DMSO) δ 176.31, 157.58, 156.36, 154.73, 151.27, 145.11, 136.87, 133.82, 127.22, 127.16, 126.95, 126.69, 126.62, 126.09, 125.45, 124.05, 118.71, 118.42, 116.62, 115.59, 115.46; Anal calcd for C17H11ClFN3O2S (375.80); C, 54.33; H, 2.95; N, 11.18; Found: C, 54.44; H, 2.83; N, 11.31.
(E)-2-((4-chloro-2-oxo-2H-chromen-3-yl) methylene)-N-methylhydrazine-1-carbothioamide (3m)
Light yellow solid, Yield, 87%; IR υmax (cm−1): 1652 (C=N), 1654 (C=O), 3332 (N–H); 1H NMR (600 MHz, DMSO-d6) δ, ppm 11.88 (1 H, s), 8.25 (1 H, s), 8.00 (2 H, d, J 8.0), 7.73 (1 H, t, J 7.5), 7.49 (2 H, t, J 7.1), 3.04 (3 H, d, J 4.5); 13C NMR (151 MHz, DMSO) δ 178.06, 157.67, 151.16, 144.47, 135.55, 133.58, 125.97, 125.41, 119.03, 118.42, 116.59, 31.12; Anal calcd for C12H10ClN3O2S (295.74); C, 48.74; H, 3.41; N, 14.21; Found: C, 48.87; H, 3.35; N, 14.31.
In-vitro α-glucosidase assay
The most recent investigations were carried out utilizing the techniques we previously discussed38. The enzyme and substrate were dissolved in phosphate buffer (pH 6.8) which also served as the reaction buffer for the experiments. A 96-well plate was used for the following ingredients: enzyme 2 U/2 mL, test samples (0.5 mM), 20 L/well, enzyme 20 L/well, and 135 L/well reaction buffer. The plate was incubated for 15 min at 37 °C. Changes in absorbance brought on by substrate breakdown were measured at 400 nm for 30 min after each well-received 25 L of 4-nitro phenyl α-D-glucopyranoside. Acarbose served as a positive control, and 7% DMSO served as a negative control.
Statistical analysis
The results of the recent research on the anti-diabetic impact of the studied materials presented were obtained using Excel and the SoftMax Pro suite.
The formula below was used to compute the percent inhibition of all drugs tested.
EZ-FIT (Perrella Scientific, Inc., USA) to calculate the IC50 for all antidiabetic agents under the study. All experiments were carried out in triplicate to prevent predicted errors, and variances in results are reported as Standard Error of Mean values (SEM).
Molecular docking
Docking of all the compounds was performed by Molecular Operating Environment (MOE version 2022.02)39,40. For the in-silico experiment, the X-ray crystal structure of α-glucosidase from an inbound form with α-D-glucopyranose was selected (PDB code 3A4A)38. The structures of compounds were first drawn on the Chem Draw program and saved into mol format, then each structure was imported into the MOE compound’s database, where the MOE WASH module was used to convert the structures 3D-format. During this conversion, Hydrogen atoms were added simultaneously on ligands, and partial charges (MMFF94x force field) were calculated. Later the structures of ligands were energy minimized with a gradient of 0.1kcal/mol/Å. The α-glucosidase enzyme file was modified for docking by adding hydrogen atoms and calculating charges (with AMBER 10:EHT force field) on residues through the QuickPrep module of MOE. Subsequently, docking was accomplished with default docking parameters of MOE, i.e., Triangle Matcher placement method and London dG scoring function. This docking procedure was initially validated by re-docking of co-crystallized ligand (isomaltose) in its original position in the PDB file, which perfectly bind at its cognate position determined by the X-ray crystallography with RMSD of 0.16 Å (Fig. S1, supporting information). In the end, thirty conformations of each ligand were saved for further analysis.
Molecular dynamics simulations
In addition to providing energetic insights into protein–ligand interactions, molecular dynamics (MD) simulation can give various dynamic structural information regarding biomacromolecules. These discoveries might lead to more effective strategies for creating protein–ligand interactions. To properly guide the drug development process, a solid knowledge of protein–ligand interactions is crucial41. The conformational dynamics of numerous protein systems were investigated in this work using MD simulation. These systems included apo α-glucosidase, three highly rated α-glucosidase-inhibitor complexes (3c, 3g, and 3i), and a reference complex (α-glucosidase-alpha-d-glucopyranose complex, PDB ID: 3A4A). The AMBER222 software with an explicit solvent model was used during the simulation. The simulation was initiated using the AMBER22 LEaP module, and the residue-specific ff19SB force field was employed. As a result, coordinate and topology files were built for the parameters of protein residues42. An OPC (optimal point charge) water model with a 12 Å buffer was used to solvate each system in a truncated octahedral box. A 1 Å grid of monovalent OPC ions (Na+ and Cl−) with a concentration of 0.1 M was utilized to determine the system's neutrality43. The complexes of α-glucosidase small inhibitors structure were adjusted using the General Amber Force Field-2 (GAFF2). Small-molecule AM1-BCC charges were calculated to refine the geometry44. Hydrogen atoms that were missing were added using the LEaP module, and AMBER22's Parmchk2 tool was used to produce the necessary force field parameters for the small compounds in the complexes45. Hydrogen atom locations were constrained using the SHAKE algorithm46 during the MD simulation, allowing a time step of 2 fs. Long-range electrostatic interactions were calculated using the particle-mesh Ewald method (PMEMD)8 with a threshold of 8 Å. GPUs were used to simulate maximum parallel scalability. The complex systems underwent two minimization cycles following MD initialization to refine the structure. In the first iteration of optimization, constraints were placed on the protein residues, and the system was subjected to 50,000 steps of steepest descent minimization. The conjugate gradient algorithm was then in the second minimization with a step size of 25,00047. The systems were heated gradually using a Langevin thermostat46. Weak restraints were utilized to keep the protein in place as the system was heated from 0.1 to 300 K throughout 400 picoseconds. The heating relied on an NVT ensemble, where N is the number of particles, V is the volume of the simulation box, and T is the temperature. The harmonic oscillator's kinetic energy was regulated by the 2.0 ps−1 collision frequency with the Langevin thermostat48. The system's density was then modified the same way as the heating phase in a 400 ps time scale. After the system reached 300 K, a 2000 ps equilibration operation was performed in a constraint-free NPT ensemble (where N is the number of particles, P is the pressure, and T is the temperature). An isotropic position scaling approach was employed to maintain a consistent pressure during the equilibration phase. This made it possible to attain a relaxation time of 2 picoseconds. The free state, the benchmark complex (alpha-D-glucopyranose-glucosidase complex), and the best three docked protein-inhibitor complexes were all subjected to a 100 ns production MD simulation in an NPT ensemble. The temperature was maintained by a Langevin thermostat, and the electrostatic interactions were computed using a cutoff distance of 8 Å. Each system's output trajectory was collected after10 ps simulation time step.
Evaluation of post-dynamic trajectory
Investigation of protein stability
The CPPTRAJ module in AMBER22 was used to evaluate the 100 ns trajectory data produced by the simulation of each system49. Root Mean Square Deviation (RMSD) profiles were generated for each system based on the Cα atoms of the protein, using data from 11,000 frames of trajectories. The RMSD was computed using the first trajectory frame as the coordinate reference. The average RMSD was calculated for each trajectory to examine how the RMSD changed over time.
Investigating residue fluctuations and compactness
When calculating the distances that each amino acid position shifted from its initial position, the Root Mean Square Fluctuation (RMSF) was used49. Protein residue flexibility is shown by the RMSF values calculated. During a 100 ns simulation, the RG of the protein was determined to determine how compact the protein was before and after ligand binding. RG uses the equation from50 to determine the positions of the protein's atoms concerning its mass centroid. We calculated the amount of pucker in the five-membered ring using the method developed by Altona and Sundaralingam13. In addition, cyclic averages in Rg calculations are depicted for periodic torsions.
Binding free energy calculations
Computational methods have been developed to determine the binding free energy (BFE) of protein–ligand complexes in recent years. There has been much research on the efficacy and applicability of these techniques51,52,53 to different protein–ligand systems. Free energy is broken down into constituent energy contributors that result from different interactions, providing remarkable accuracy at a computationally affordable price. THE BFE between protein and ligand complexes was calculated using AMBER22's Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method14,17,18. The BFE was computed using the latest 2750 frames (25 ns) of the roduction trajectry for each complex. The idea topology of the systems was generated using the mbondi3 radii and a solvent probe with a 2 Å radius. We calculated the protein–ligand complexe' BFE (ΔGbind) using the following equation54
To calculate the binding free energy (BFE), first, we subtract the free energies of the free ligand (ΔGL) and the free protein (ΔGR) from the free energy of the protein–ligand complex (ΔGR+L). The free energy component (G) in the equation may be decomposed into many energetic contributions using the Molecular Mechanics/Gauss-Boltzmann Surface Area (MM/GBSA) and Molecular Mechanics/Poison Boltzmann Surface Area (MM/PBSA) methods (Eq. 4).
Overall bond energy, or Ebond, is the sum of a molecule's bond energy and the angle and dihedral energies. Evdw represents Van der Waals's contribution, whereas electrostatic energy is represented by the sign Eelec. Polar (GGB) and non-polar (GSA) components comprise the solvation energy. The non-polar component is estimated in SASA through the LCPO method, which represents a linear pairwise overlap combination. However, the GB model determines the polar component55. The energy was calculated in kcal/mol, whereas Å2 was used to estimate the protein's surface area. Absolute temperature T and solute entropy SS were calculated. We calculated the free energy of four distinct complex systems using the MM/PB(GB)SA model. The α-glucosidase-alpha-D-glucopyranose complex (3A4A) and three α-glucosidase-inhibitor complexes were among these systems.
Data examination
The structural drawings were constructed using the software programs MOE2022.0221 and Blender22. OriginPro, a graphing application, was used56.
Conclusion
Several multifactorial diseases are associated with diabetes mellitus and its complications, necessitating advancements in drug discovery for the prevention and treatment of diabetes-based issues. In the present study, a series of coumarin-based drug-like molecules were designed as drug candidates for diabetes mellitus. Their antidiabetic potential was assessed by evaluating their ability to block the α-glucosidase enzyme function, and all the compounds displayed excellent α-glucosidase inhibitory potential with IC50 values ranging from 2.33 to 22.11 µM. The concentration-dependent inhibition of compound 3c, 3g and 3i was studied by Kinetic studies. Furthermore, the binding pattern of these molecules at the atomic level was examined through molecular docking, which revealed that thiosemicarbazide moiety plays a crucial role in binding these molecules to the catalytic and active site residues of the enzyme. However, the conformational changes in these molecules are primarily responsible for their diverse bioactivity. The molecular dynamic simulations explored the firm attachment of the compounds to the protein’s active site and showed an impact on the protein structural confirmation.
Data availability
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
Esfahani, N. A. et al. Design and synthesis of phenoxymethybenzoimidazole incorporating different aryl thiazole-triazole acetamide derivatives as α-glycosidase inhibitors. Mol. Divers. 26(4), 1995–2009 (2022).
Ali, M. et al. Synthesis, in vitro α-glucosidase inhibitory activity, and in silico study of (E)-thiosemicarbazones and (E)-2-(2-(arylmethylene)hydrazinyl)-4-arylthiazole derivatives. Mol. Divers. 22(4), 841–861 (2018).
Gulcin, I. et al. Antidiabetic and antiparasitic potentials: Inhibition effects of some natural antioxidant compounds on α-glycosidase, α-amylase, and human glutathione S-transferase enzymes. Int. J. Biol. Macromol. 119, 741–746 (2018).
Stringer, D. M., Zahradka, P. & Taylor, C. G. Glucose transporters: Cellular links to hyperglycemia in insulin resistance and diabetes. Nutr. Rev. 73(3), 140–154 (2015).
Pedrood, K. et al. Design, synthesis, characterization, enzymatic inhibition evaluations, and docking study of novel quinazolinone derivatives. Int. J. Biol. Macromol 170, 1–12 (2021).
Yue, L. M. et al. Computational prediction integrating the inhibition kinetics of gallotannin on α-glucosidase. Int. J. Biol. Macromol 103, 829–838 (2017).
Ibrar, A., Shehzadi, S. A., Saeed, F. & Khan, I. Developing hybrid molecule therapeutics for diverse enzyme inhibitory action: Active role of coumarin-based structural leads in drug discovery. Bioorg. Med. Chem. 26(13), 3731–3762 (2018).
Wu, P.-P., Zhang, K., Lu, Y.-J., He, P. & Zhao, S.-Q. In vitro and in vivo evaluation of the antidiabetic activity of ursolic acid derivatives. Eur. J. Med. Chem. 80, 502–508 (2014).
Fujieda, H. et al. Discovery of a potent glucokinase activator with a favorable liver and pancreas distribution pattern for the treatment of type 2 diabetes mellitus. Eur. J. Med. Chem. 156, 269–294 (2018).
Bakherad, Z. et al. In silico and in vitro studies of thiosemicarbazone-indole hybrid compounds as potent α-glycosidase inhibitors. Comput. Biol. Chem. 97, 107642 (2022).
Kaur, R., Kumar, R., Dogra, N. & Yadav, A. K. Design, synthesis, biological evaluations, and in silico studies of sulfonate ester derivatives of 2-(2-benzylidenehydrazono) thiazolidin-4-one as potential α-glucosidase inhibitors. J. Mol. Struct. 1247, 131266 (2022).
Glinma, B. et al. Study of benzoin 4-phenyl-3-thiosemicarbazone and benzil Bis(4-phenyl-3-thiosemicarbazone): Synthesis and their antiparasitic activity on trypanosome. JCBPS 10(4), 408–418 (2020).
Aygun, O. et al. Monomeric octahedral bismuth(III) benzaldehyde-N1-alkyl thiosemicarbazones: Synthesis, characterization and biological properties. Polyhedron 215, 115683 (2022).
Sheikhy, M., Jalilian, A., Novinrooz, A. & Motamedi-Sedeh, F. Synthesis and in vitro antibacterial evaluation of some thiosemicarbazides and thiosemicarbazones. J. Biomed. Sci. Eng. 5, 39–42 (2012).
Toan, V. N., Thanh, N. D., Tri, N. M. & Huong, N. T. T. Synthesis and biological screening of thiosemicarbazones of substituted 3-acetylcoumarins having d-glucose moiety. Bioorganic Med. Chem. Lett. 30, 127664 (2020).
Kumar, S. & Choudhary, M. Synthetic aromatic organic compounds bearing 4,4-dimethyl-3-thiosemicarbazide moiety: Theoretical and experimental approach. Polycycl. Aromat. Compoud. 43, 1–23 (2022).
Siwek, A., Stefańska, J., Dzitko, K. & Ruszczak, A. Antifungal effect of 4-arylthiosemicarbazides against Candida species. Search for molecular basis of antifungal activity of thiosemicarbazide derivatives. J. Mol. Model. 18(9), 4159–4170 (2012).
Hamed, M. Y., Aly, A. F., Abdullah, N. H. & Ismail, M. F. Synthesis, characterization and antifungal evaluation of novel pyridazin-3(2H)-one derivatives. Polycycl. Aromat. Compoud. 43(3), 2356–2375 (2023).
Dziduch, K., Kołodziej, P., Paneth, A., Bogucka-Kocka, A. & Wujec, M. Synthesis and anthelmintic activity of new thiosemicarbazide derivatives—A preliminary study. Molecules 25(12), 2770 (2020).
Prajapati, N. P. & Patel, H. D. Novel thiosemicarbazone derivatives and their metal complexes: Recent development. Synth. Commun. 49, 2767–2804 (2019).
Summers, K. L. A structural chemistry perspective on the antimalarial properties of thiosemicarbazone metal complexes. Mini Rev. Med. Chem. 19(7), 569–590 (2019).
Savir, S. et al. Synthesis, cytotoxicity and antimalarial activities of thiosemicarbazones and their nickel (II) complexes. J. Mol. Struct. 1211, 128090 (2020).
Abbas, S. Y. et al. Design, synthesis and antiviral evaluation of new N-(4)-(benzo[d][1,3]-dioxol-5-yl)thiosemicarbazone derivatives. Egypt. J. Chem. 64(7), 3791–3800 (2021).
Negutsa, E., Balan, G., Gulya, A. & Bulimestru, I. Antimicrobial and antifungal activity of Cu(II) and Bi(III) complexes based on aminopolycarboxylate ions and 2-formyl and 2-acetylpyridine thiosemicarbazones. One Health Risk Manag. 2(4-S), 52 (2021).
Kalaiarasi, G. et al. New organoruthenium(II) complexes containing N, X-donor (X = O, S) heterocyclic chelators: Synthesis, spectral characterization, in vitro cytotoxicity and apoptosis investigation. Inorganica Chim. Acta 535, 120863 (2022).
Yadav, P. N. et al. Exploration of anticancer potency of N(4) thiomorpholinyl isatin/5-haloisatin thiosemicarbazones on coordination to Cu2+ ion. Inorg. Chem. Commun. 143, 109767 (2022).
Shehzad, M. T. et al. Exploring antidiabetic potential of adamantyl-thiosemicarbazones via aldose reductase (ALR2) inhibition. Bioorg. Chem. 92, 103244 (2019).
Kumar, L. V., Sunitha, S. & Nath, G. R. Antioxidant, antidiabetic, and anticancer studies of nickel complex of vanillin-4-methyl-4-phenyl-3-thiosemicarbazone. Mater. Today Proc. 41(3), 669–675 (2021).
Ullah, H. et al. Benzimidazole bearing thiosemicarbazone derivatives act as potent α-amylase and α-glucosidase inhibitors; synthesis, bioactivity screening, and molecular docking study. Molecules 27, 6921 (2022).
Basri, R. et al. Synthesis, biological evaluation, and molecular modeling of 3-formyl-6-isopropylchromone derived thiosemicarbazones as α-glucosidase inhibitors. Bioorg. Chem. 139, 106739 (2023).
Hu, C.-M. et al. Synthesis and evaluation of coumarin-chalcone derivatives as α-glucosidase inhibitors. Front. Chem. https://doi.org/10.3389/fchem.2022.926543 (2022).
Zhang, X. et al. Synthesis and biological evaluation of coumarin derivatives containing oxime ester as α-glucosidase inhibitors. Arab. J. Chem. 15(9), 104072 (2022).
Ichale, R., Kanhed, A. & Vora, A. Coumarin linked thiazole derivatives as potential α-glucosidase inhibitors to treat diabetes mellitus. Mol. Divers. https://doi.org/10.1007/s11030-023-10652-4 (2023).
Ahmad, M. U. et al. Synthesis of benzimidazole based hydrazones as non-sugar based α-glucosidase inhibitors: Structure-activity relation and molecular docking. Drug Dev. Res. 82, 1033–1043 (2021).
Qamar-un-Nisa Tariq, S. et al. Xanthenone-based hydrazones as potent α-glucosidase inhibitors: Synthesis, solid state self-assembly, and in silico studies. Bioorg. Chem. 84, 372–383 (2019).
Hashmi, S. et al. Probing 4-(diethylamino)-salicylaldehyde-based thiosemicarbazones as multi-target directed ligands against cholinesterases, carbonic anhydrases, and α-glycosidase enzymes. Bioorg. Chem. 107, 104554 (2020).
Khan, S. et al. Probing 2-acetylbenzofuran hydrazones and their metal complexes as α-glucosidase inhibitors. Bioorg. Chem. 102, 104082 (2020).
Alam, A. et al. Bio-oriented synthesis of novel (s)-flurbiprofen clubbed hydrazone Schiff’s bases for diabetic management: In vitro and in silico studies. J. Pharm. 15(6), 672 (2022).
Pasha, A. et al. Synthesis of new diphenyl urea-clubbed imine analogs and its implications in diabetic management through in vitro and in silico approaches. Sci. Rep. https://doi.org/10.1038/s41598-023-28828-1 (2023).
Molecular Operating Environment (MOE), 2022.02 Chemical Computing Group ULC, 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7, Canada (2023).
Yamamoto, K., Miyake, H., Kusunoki, M. & Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 277(20), 4205–4214 (2010).
Liu, X. et al. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 13(1), 23–37 (2018).
Tian, C. et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16(1), 528–552 (2020).
Sengupta, A., Li, Z., Song, L. F., Li, P. & Merz, K. M. Jr. Parameterization of monovalent ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB water models. J. Chem. Inf. Model. 61, 869–880 (2021).
Wang, B. & Merz, K. M. A fast QM/MM (quantum mechanical/molecular mechanical) approach to calculate nuclear magnetic resonance chemical shifts for macromolecules. J. Chem. Theory Comput. 2, 209–215 (2006).
Wang, J., Wang, W., Kollman, P. A. & Case, D. A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 222, U403. https://www.webofscience.com/wos/woscc/full-record/WOS:000170690002032 (2001).
Kräutler, V., Van Gunsteren, W. F. & Hünenberger, P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22(5), 501–508 (2001).
Press, W. H., Flannery, B. P., Teukolsky, S. A., Vetterling, W. T. & Kramer, P. B. Numerical recipes: The art of scientific computing. AIP. (1987).
Sindhikara, D. J., Kim, S., Voter, A. F. & Roitberg, A. E. Bad seeds sprout perilous dynamics: Stochastic thermostat induced trajectory synchronization in biomolecules. J. Chem. Theory Comput. 5(6), 1624–1631 (2009).
Roe, D. R. & Cheatham, T. E. III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Kollman, P. A. et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33(12), 889–897 (2000).
Wang, W., Donini, O., Reyes, C. M. & Kollman, P. A. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. 30, 211–243 (2001).
Wang, J., Hou, T. & Xu, X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput. Aided Drug Des 2, 287–306 (2006).
Onufriev, A., Bashford, D. & Case, D. A. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins Struct. Funct. Genet 55(2), 383–394 (2004).
Weiser, J., Shenkin, P. S. & Still, W. C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 20(2), 217–230 (1999).
OriginLab Corporation. (2021).
Acknowledgements
The authors express their appreciation to the Deanship of Scientific Research at King Khalid University, Saudi Arabia, for funding this work through research group program under grant number (RGP. 2/232/44). Also, the current work was supported by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R419), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
S.B.Z. did synthesis; S.U., S.A.H., and A.K. did biological assays; N.U.H. and J.H. made graphs and docking discussion; A.Y.B. and A.F.E. provided facilities for analysis and funds acquisition; A.A.H. and Z.S. interpreted spectra, developed idea and wrote the manuscript idea; M.W. did IFD, MM-GBSA and Molecular Dynamic Simulation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zahra, S.B., Ullah, S., Halim, S.A. et al. Synthesis of novel coumarin-based thiosemicarbazones and their implications in diabetic management via in-vitro and in-silico approaches. Sci Rep 13, 18014 (2023). https://doi.org/10.1038/s41598-023-44837-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44837-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
