
Abstract
Preeclampsia (PE) is a significant threat to all pregnancies that is highly associated with maternal mortality and developmental disorders in infants. However, the etiopathogenesis of this condition remains unclear. This study aims to explore the regulatory roles of long noncoding RNAs (lncRNAs) and the mediated competing endogenous RNAs (ceRNA) in the etiopathogenesis of PE through analysis of lncRNA expression patterns in PE and healthy pregnant women (HPW), as well as the construction of lncRNA-mediated ceRNA regulatory networks using bioinformatics. A total of 896 significant differentially expressed lncRNAs, including 586 upregulated lncRNAs and 310 downregulated lncRNAs, were identified in comparison between PE and HPW. Analysis of these differential expressed lncRNAs revealed their predominant enrichment in molecular functions such as sphingosine-1-phosphate phosphatase activity, lipid phosphatase activity, phosphatidate phosphatase activity, thymidylate kinase activity, and UMP kinase activity. Moreover, these differential expressed lncRNAs were predominantly enriched in KEGG analyses such as fat digestion and absorption, lysine degradation, ether lipid metabolism, glycerolipid metabolism, and sphingolipid metabolism. Two ceRNA regulatory networks were constructed based on ceRNA score, including one that had 31 upregulated lncRNAs, 11 downregulated miRNAs, and 34 upregulated mRNAs, while the other contained 128 downregulated lncRNAs, 40 upregulated miRNAs, and 113 downregulated mRNAs. These results may provide a clue to explore the roles of lncRNAs in the etiopathogenesis of PE.
Similar content being viewed by others

Introduction
Preeclampsia (PE) is a serious condition that typically develops after 20 weeks of pregnancy1 and is characterized by high blood pressure and proteinuria2. It is a leading cause of maternal mortality3 and can also cause brain developmental disorders4, including intellectual disability, in infants, resulting in a significant burden on healthcare services5.
While the etiology of PE remains unclear, research has increasingly focused on several potential causes, including immunological intolerance6,7 and angiogenesis imbalance5, inflammatory cytokine disorders8 and immune maladjustment9. In addition, multi-omics approaches have identified potential metabolic disruptions in steroid hormones and imbalanced metabolism of androgens and estrogens in Placenta10,11 and a few lncRNA-mediated ceRNA regulatory mechanisms12, such as LncRNA-XIST regulation of KCNJ16 via miR-340-5p sponge13, CircFN1 modulation of ATF2 via miR-19a/b-3p sponge14, circ_0008726 regulation of RYBP via miR-345-3p sponge15, and Circ_0015382 acting as a miR-149-5p sponge to modulate the expression of TFPI2, associated with the onset and development of PE16. Increasing data for the etiopathogenesis of PE was from the cross-talk among circRNA, miRNA, and mRNAs. Recently, increasing reports demonstrated that dysregulated lnRNAs identified from trophoblast cells and placentas were predicted to be closely related to the pathogenesis of PE17,18,19,20. However, there is still only several reported data from the lncRNA-mediated ceRNA to describe the etiopathogenesis of PE from the patients using their peripheral blood19,20,21,22,23,24,25. In addition, biochemical tests of the PE showed that aminotransferases, creatinine, total bilirubin and Urea levels in were increased in the serum from pregnant women, and creatinine level in serum was predicted as the best diagnostic marker for PE26. A deleterious effect on renal and liver function happened in the PE followed by the changes of biochemical indexes26.
This study aimed to investigate the demographic characteristics, infant outcomes, serum biochemical indices, and expression profiles of lncRNAs in PE and build integrated bioinformatics-based lncRNA-mediated ceRNA regulatory networks to better understand the etiopathogenesis of PE.
Materials and methods
PE patients
Pregnant women with pre-eclampsia (PE) and healthy pregnant controls (HPW) were recruited in this study, with three participants in each group identified from the Maternity and Child Health Care Affiliated Hospital of Jiaxing University and the basic characteristics of the included subjects were shown in our previous report27. From the 30 pregnant women diagnosed with pre-eclampsia based on gestational hypertension and proteinuria and reported clinical features of PE, the demographic characteristics, infant outcomes, and serum biochemical indices, including glutamic-pyruvic transaminase (ALT) and glutamic oxalacetic transaminase (AST), were investigated. Ethics approval was obtained from the Ethics Committee of the Faculty of Medicine, and written informed consent was obtained from all participants. The study was approved by the research ethics committee for clinical studies at Suzhou Municipal Hospital, Jiangsu, China and all experiments were performed in accordance with relevant guidelines and regulations.
Total RNAs extraction
Total RNA was extracted from the fasting whole blood sample (3.0 mL) of each participant—the PE group and HPW group—with a Blood Cell Lysis Buffer kit (Tiangen, China). The purified leukocyte was used for extraction of total RNA with TRIzol reagent (Invitrogen, Carlsbad, USA) using standard protocols.
rRNA-depleted and RNA sequencing
The high-quality purified RNA was subjected to treatment with the TruSeq Stranded Total RNA with Ribo-Zero Gold kit (Illumina, RS-122-2301) to remove ribosomal RNA. The first strand cDNA was synthesized with the SuperScript II Reverse Transcriptase kit, followed by second strand cDNA synthesis with Second Strand Marking Master Mix. Purification of cDNA was performed using AMPure XP beads. The adenylated 3′ ends were then added with A-tailed Mix, and adapters were ligated with RNA Adapter Index. AMPure XP Beads were then used for further purification of the cDNAs, adenylated 3′ ends, and adapters and finally, to purify the DNA fragments. We built cDNA libraries with the TruSeq Stranded Total RNA with Ribo-Zero Gold kit; the HiSeqTM4000 sequencing platform was used to sequence the six cDNA libraries by Shanghai OE Biotech, Shanghai, China, and the sequencing reads are accessible in PRJNA665923.
Bioinformatics analysis
High-throughput sequencing reads were processed with Trimmomatic software28 to remove adapters. The high-quality clean reads were collected by removing low-quality data. Hisat229 was used to map clean reads to the reference sequence produced by the human reference genome (GRCh38.p12). The candidate long non-coding RNA (lncRNA) transcripts were selected to compare with the reference sequence using Cuffcompare software30. These transcripts with coding potential were screened out by using Coding Potential Calculator (CPC)31, Coding-Non-Coding Index (CNCI)32, Pfam33, and PLEK34 to obtain predicted lncRNA sequences. The counts were normalized using the DESeq (2012) R package, and the difference between the two groups was calculated using p-value and fold change values with the nbinomTest function. LncRNAs with p-values ≤ 0.05 and a fold change ≥ 2 were designated as differentially expressed. Geno ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) enrichment were conducted for the potential roles of these dysregulated lncRNAs. For the correction of p-values, the false discovery rate was calculated with the Benjamini and Hochberg multiple tests. LncRNA-mediated ceRNA networks were built using differentially expressed miRNAs and mRNAs with the ceRNA_score.
Statistical analysis
The statistical software GraphPad Prism for Windows version 5.0 (GraphPad Software, San Diego, CA, USA) was used to plot all graphs, and the student-T test was used for statistical analysis. Statistical significance was indicated with **** p < 0.0001 and * p < 0.05.
Results
Demographic characteristics and infant outcomes
A total of thirty patients with gestational hypertension and preeclampsia were strictly selected, and healthy pregnant women (HPW) were recruited as controls from hospitals. The age range of participants was 20–40 years. The delivery pregnancy week in the preeclampsia group was significantly lower than that of the control group (Fig. 1A). The fetal birth weight of the preeclampsia group was significantly lower than the control group (Fig. 1B). Compared to the control group, the levels of ALT and AST were significantly higher in the serum of the preeclampsia group (Fig. 1C, D). These results suggest that preeclampsia leads to infant developmental disorders and liver injury.

Demographic characteristics of PE patients and fetal birth weight. (A) Delivery pregnancy week between the PE group and the control HPW group. (B) Fetal birth weight of PE group and control HPW group. The level of ALT (C) and AST (D) in serum from PE group and control HPW group.
Potential lncRNAs exploration
Long non-coding RNAs (lncRNAs) without coding activity and longer than 200 nucleotides (nt) were selected. Four screening methods were utilized, primarily CPC14, CNCI15, Pfam16 and PLEK17, to remove transcripts with coding activity. A Venn diagram showed the screening yielded 4323 potential lncRNAs with an average length of 1259.63 nt (Fig. 2A), among which 2645 lncRNAs were over 200 nt and 1318 lncRNAs were more than 1000 nt. The length of the shortest lncRNA was only 201 nt (Fig. 2B).

The category of lncRNAs and length distribution. (A) Five types of the identified lncRNAs. (B) The identified lncRNAs length distribution.
Identification of differential expressed lncRNAs from PE vs. HPW
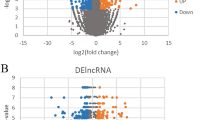
Principal component analysis (PCA) was used to understand the expression pattern of lncRNAs in preeclampsia (PE) vs. HPW and showed a total of 60.7% variance (PCA1 and PCA2), indicating that the samples were relatively concentrated in similar groups (Fig. 3A). After lncRNA count normalization with DESeq35, two parameters—p-value < 0.05 and an absolute value of the log2FC greater than 1 were used to screen for differentially expressed lncRNAs between the preeclampsia and HPW groups. A total of 896 differentially expressed lncRNAs were identified, of which 586 were upregulated and 310 were downregulated in the preeclampsia vs. HPW groups (Fig. 3B). These differentially expressed lncRNAs were visualized in a Volcano Plot (Fig. 3C). Unsupervised hierarchical clustering was performed, which clearly distinguished upregulated and downregulated lncRNAs between the PE and HPW groups (Fig. 3D).

Identification of differential expressed lncRNAs from PE vs. HPW. (A) Principal component analysis (PCA). (B) The differential expressed lncRNAs were calculated by p-value ≤ 0.05 and fold change ≥ 2. (C) The differential expressed lncRNAs were shown in the Volcano plot. (D) The differential expressed lncRNAs were shown with hierarchical clustering.
GO enrichment analysis of differential expressed lncRNAs
GO enrichment analysis divided the differential expressed lncRNAs into biological process, cellular component, and molecular function. The top 30 GO terms of the GO enrichment analysis for the upregulated lncRNAs with their derived genes were phospholipid dephosphorylation, negative regulation of heat generation, negative regulation of multicellular organismal metabolic processes, positive regulation of adrenergic receptor signaling pathways, negative regulation of locomotion involved in locomotory behavior, dUDP, dTDP, and dTTP biosynthetic processes, nervous system processes, and fat pad development in the biological process. Meanwhile, sphingosine-1-phosphate phosphatase activity, lipid phosphatase activity, beta-3 adrenergic receptor binding, thymidylate kinase activity, UMP kinase activity, phosphatidate phosphatase activity, cytidylate kinase activity, histone-lysine N-methyltransferase activity, nucleoside diphosphate kinase activity, and ubiquitin-ubiquitin ligase activity were enriched in the molecular function (Fig. 4A). The top 30 GO terms of the GO annotation analysis for the downregulated lncRNAs with their derived genes were posttranslational protein targeting to the endoplasmic reticulum membrane, positive regulation of long-term synaptic depression, regulation of microtubule-based processes, negative regulation of excitatory postsynaptic potential, establishment or maintenance of cell polarity protein secretion, cerebellar granule cell differentiation, heterophilic cell–cell adhesion via plasma membrane cell adhesion molecules, positive regulation of synapse assembly, and phosphatidylinositol biosynthetic process in the biological process. Meanwhile, protein serine/threonine kinase activator activity, protein tyrosine kinase activity, protein complex binding, protein C-terminus binding, transporter activity, ATPase activity, serine-type peptidase activity, histone-lysine N-methyltransferase activity, ATP binding, and microtubule binding were enriched in the molecular function (Fig. 4B).

GO enrichment analysis of differential expressed lncRNAs. GO enrichment analysis was performed on differential expressed lncRNAs according to GO functional annotation information of lncRNA-derived genes. According to the GO enrichment analysis, it was mainly divided into three types including biological process, cellular component, and molecular function. (A) The 30 GO terms of upregulated lncRNAs with their derived genes. (B) The 30 GO terms of downregulated lncRNAs with their derived genes.
KEGG enrichment of differential expressed lncRNAs
A hypergeometric distribution test was utilized to analyze the significance of differential lncRNA enrichment in each pathway entry for KEGG analysis. The upregulated lncRNAs were enriched in pathways like fat digestion and absorption, ether lipid metabolism, glycerolipid metabolism, sphingolipid metabolism, choline metabolism in cancer, lysine degradation, NF-kappa B signaling pathway, glycerophospholipid metabolism, phospholipase D signaling pathway, Fc gamma R-mediated phagocytosis, prostate cancer, ferroptosis, fluid shear stress, and atherosclerosis, SNARE interactions in vesicular transport, measles, RNA degradation, protein processing in endoplasmic reticulum, autophagy—animal, lysosome, and cytokine-cytokine receptor interaction (Fig. 5A). The downregulated lncRNAs were enriched in pathways like ubiquitin-mediated proteolysis, thyroid cancer, Huntington's disease, microRNAs in cancer, FoxO signaling pathway, RNA transport, Fc epsilon RI signaling pathway, pyrimidine metabolism, long-term potentiation, lysine degradation, inositol phosphate metabolism, acute myeloid leukemia, Fc gamma R-mediated phagocytosis, melanoma, thermogenesis, long-term depression, prostate cancer, thyroid hormone synthesis, hepatocellular carcinoma, and renal cell carcinoma (Fig. 5B).

KEGG enrichment of differential expressed lncRNAs. (A) The KEGG analysis enriched upregulated lncRNAs in the related pathways. (B) The KEGG analysis enriched downregulated lncRNAs in the related pathways.
Construction of ceRNA networks
To explore the potential regulatory roles of differentially expressed lncRNAs in preeclampsia, long non-coding RNA competing endogenous RNA (lncRNA ceRNA) networks were built. Differential miRNAs and mRNAs were retrieved from the database, and the differential expressed lncRNAs were blasted with miRbase to explore the miRNA precursors, predicting 2224 pre-miRNAs. The regulatory relationship was based on lncRNA, miRNA, and mRNA, and two ceRNA interaction networks were built with ceRNA_score. One ceRNA network consisted of 31 upregulated lncRNAs, 11 downregulated miRNAs, and 34 upregulated mRNAs (Fig. 6A), while another ceRNA network contained 128 downregulated lncRNAs, 40 upregulated miRNAs, and 113 downregulated mRNAs (Fig. 6B).

Construction of ceRNA networks. Two ceRNA interaction networks were built with ceRNA_score. (A) upregulated lncRNAs mediated ceRNA. (B) downregulated lncRNAs mediated ceRNA.
Discussions
Pre-eclampsia (PE) is a severe obstetric syndrome that has a strong association with maternal mortality3. Nevertheless, the underlying causes of PE remain unclear. Various studies have reported on the diverse factors that contribute to the development and progression of PE. Non-coding RNAs, specifically long non-coding RNAs (lncRNAs), have been identified to play significant roles in the development of PE. In addition, increasing data showed lncRNAs play critical roles in multiple cancers and other diseases. Nonetheless, the regulatory mechanisms of lncRNAs in PE, particularly via lncRNA-mediated competing endogenous RNA (ceRNA) networks, remain poorly understood. In this study, we comprehensively investigated the expression of lncRNAs in PE and identified that hundreds of lncRNAs were significantly differentially expressed, which were predominantly related to lipid metabolism and immunity pathways based on bioinformatics analysis.
Notably, PE can occur at any point after 20 weeks of gestation and can be classified into two subtypes: early onset PE (before 34 weeks) and late-onset PE (after 34 weeks)36. Previous studies indicated that poor placental development is closely related to PE. Consequently, many studies have focused on identifying the regulatory mechanisms of lncRNAs in placental tissue in PE and have predicted their potential as biomarkers of PE or for their involvement in the pathogenesis of PE through lncRNA-mediated ceRNA networks19,20,21,22,23,24,25.
However, analyzing the expression of lncRNAs in whole blood samples from PE patients, while excluding red blood cells, may be more relevant and useful for the development of biomarkers. Furthermore, studies have suggested that metabolic abnormalities of steroid hormones in placental tissue may have a significant role in the etiology of PE12. Therefore, we investigated the distinct expression profiling of lncRNAs in the whole blood removing the red blood cell may be more meaningful. Our analysis identified 896 significantly differentially expressed lncRNAs between PE and healthy pregnant women, comprising 586 upregulated and 310 downregulated lncRNAs. We also conducted gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses to gain a deeper understanding of the regulatory roles of these lncRNAs and their associated genes. Our analysis indicated that the upregulated lncRNAs primarily influenced pathways related to the metabolism of fats, such as fat digestion and absorption, ether lipid metabolism, glycerolipid metabolism, and sphingolipid metabolism. They were also associated with lysine degradation, the NF-kappa B signaling pathway, glycerophospholipid metabolism, phospholipase D signaling pathway, choline metabolism in cancer, and Fc gamma R-mediated phagocytosis. The downregulated lncRNAs, on the other hand, were primarily associated with ubiquitin-mediated proteolysis, thyroid cancer, Huntington's disease, microRNAs in cancer, FoxO signaling pathway, RNA transport, Fc epsilon RI signaling pathway, pyrimidine metabolism, long-term potentiation, and lysine degradation.
Furthermore, we investigated the regulatory mechanisms of these lncRNAs via ceRNA analysis. ceRNA regulatory mechanism suggests that some RNAs including lncRNAs contain miRNA response element, which sites competitively bind miRNAs and subsequently impact their target genes’ expression. The role of ceRNA regulatory networks has been extensively studied in various diseases. Besides, lncRNA PGK1P2 was confirmed to regulate the expression level of Phosphoglycerate kinase 1 via miR-330-5p in PE25. An identified ceRNA network in early onset PE consisted of 21 lncRNAs, 3 mRNAs, and 69 miRNAs and could potentially be involved in the regulatory process of PE23. According to one study, Lnc-CTD-2383M3.1 may regulate ADAMTS6 expression via miR-210-3p sponging and may potentially have a regulatory role in the development of PE22. However, the etiopathogenesis of PE is still vague. To gain further insight into the etiopathogenesis of PE, we constructed two ceRNA regulatory networks using the ceRNA_score algorithm, with one network consisting of 31 lncRNAs, 11 miRNAs, and 34 mRNAs, and the other one containing 128 lncRNAs, 40 miRNAs, and 113 mRNAs.
Additionally, the ceRNA networks involved several hub genes, including ephrin B1, pannexin 1 (Panx 1), a member of the solute carrier family, IL-6, and TNF receptor superfamily members37. Panx 1 and TLR4 might induce ferroptosis in PE via solute carrier family 7 member 11-mediated signaling pathways38. The IL-6/STAT3 signaling pathway could be repressed by pravastatin to alleviate oxidative stress, improve preeclampsia, and decrease the apoptosis of placental trophoblastic cells in rats with PE39. In pregnancy, Tumor necrosis factor-alpha (TNF-α) was closely associated with hormone synthesis, placental architecture, and embryonic development, and the increasing levels of TNF-α were related to pregnancy loss and PE40. Epidermal Growth Factor-like Domain 7 mediated trophoblast migration and invasion and plays a role in placental development41. Matrix metalloproteinases-2 and -9 might be developed as biomarkers for predicting the development of PE in the first trimester42. circRNA zinc finger DHHC-type palmitoyltransferase 20 mediated the expression of GRHL2 via miR-144 sponges, resulting in the inhibition of proliferation, migration, and invasion in trophoblast cells43. These findings indicate that these target genes may be critical in the etiopathogenesis of PE, but further validation is necessary. In this study, we demonstrated that the differentially expressed lncRNAs between PE and healthy pregnant women were primarily related to pathways involving lipid metabolism, innate immunity, and ubiquitin-mediated proteolysis. In addition, a large subset of target genes was mediated by the lncRNA-mediated ceRNA networks and the rols of these dysregulated lncRNAs were still need further validated in the etiopathogenesis of PE. Taken together, these findings provide new insights into the underlying mechanisms of PE.
Data availability
The RNA sequencing data were deposited into the published data with the accession number of PRJNA665923 and are available at the following URL: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA665923. mRNA Database: ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.38_GRCh38.p12/GCF_000001405.38_GRCh38.p12_rna.fna.gz
References
Al-Jameil, N., Aziz Khan, F., Fareed Khan, M. & Tabassum, H. A brief overview of preeclampsia. J. Clin. Med. Res. 6(1), 1–7 (2014).
Eiland, E., Nzerue, C. & Faulkner, M. Preeclampsia 2012. J. Pregnancy 2012, 586578 (2012).
Poston, L. et al. Preconceptional and maternal obesity: Epidemiology and health consequences. Lancet Diabetes Endocrinol. 4(12), 1025–1036 (2016).
Griffith, M. I., Mann, J. R. & McDermott, S. The risk of intellectual disability in children born to mothers with preeclampsia or eclampsia with partial mediation by low birth weight. Hypertens. Pregnancy 30(1), 108–115 (2011).
Peng, Y., Hong, H., Gao, N., Wan, A. & Ma, Y. Bioinformatics methods in biomarkers of preeclampsia and associated potential drug applications. BMC Genomics 23(1), 711 (2022).
Lynch, A., McDuffie Jr, R., Murphy, J., Faber, K. & Orleans, M. Preeclampsia in multiple gestation: The role of assisted reproductive technologies. Obstet. Gynecol. 99(3), 445–451 (2002).
Wimalasundera, R. C. et al. Pre-eclampsia, antiretroviral therapy, and immune reconstitution. Lancet 360(9340), 1152–1154 (2002).
Ramma, W. et al. The elevation in circulating anti-angiogenic factors is independent of markers of neutrophil activation in preeclampsia. Angiogenesis 15(3), 333–340 (2012).
Saito, S., Shiozaki, A., Nakashima, A., Sakai, M. & Sasaki, Y. The role of the immune system in preeclampsia. Mol. Aspects Med. 28(2), 192–209 (2007).
Cantonwine, D. E. et al. Estrogen metabolism pathways in preeclampsia and normal pregnancy. Steroids 144, 8–14 (2019).
Shin, Y. Y. et al. Regulation of steroid hormones in the placenta and serum of women with preeclampsia. Mol. Med. Rep. 17(2), 2681–2688 (2018).
Feng, Y., Lian, X., Guo, K., Zhang, G. & Huang, X. A comprehensive analysis of metabolomics and transcriptomics to reveal major metabolic pathways and potential biomarkers of human preeclampsia placenta. Front. Genet. 13, 1010657 (2022).
Guo, Y., Gao, Y. & Liu, S. lncRNA XIST is associated with preeclampsia and mediates trophoblast cell invasion via miR-340-5p/KCNJ16 signaling pathway. Transpl. Immunol. 74, 101666 (2022).
Li, K., Lv, C., Zhang, W. & Fang, J. CircFN1 upregulation initiated oxidative stress-induced apoptosis and inhibition of proliferation and migration in trophoblasts via circFN1-miR-19a/b-3p-ATF2 ceRNA network. Reprod. Biol. 22(2), 100631 (2022).
Shu, C., Xu, P., Han, J., Han, S. & He, J. Upregulation of circRNA hsa_circ_0008726 in pre-eclampsia inhibits trophoblast migration, invasion, and EMT by regulating miR-345–3p/RYBP axis. Reprod. Sci. (Thousand Oaks, Calif.) 29(10), 2829–2841 (2022).
Hu, Z., Dong, C. & Dong, Q. Circ_0015382 is associated with preeclampsia and regulates biological behaviors of trophoblast cells through miR-149-5p/TFPI2 axis. Placenta 108, 73–80 (2021).
Tang, X., Cao, Y., Wu, D., Sun, L. & Xu, Y. Downregulated DUXAP8 lncRNA impedes trophoblast cell proliferation and migration by epigenetically upregulating TFPI2 expression. Reprod. Biol. Endocrinol. 21(1), 58 (2023).
Peng, C. Y. et al. Long non-coding RNA TLR8-AS1 induces preeclampsia through increasing TLR8/STAT1 axis. J. Hypertens. 41(8), 1245–1257 (2023).
Long, W. et al. Distinct expression profiles of lncRNAs between early-onset preeclampsia and preterm controls. Clin. Chim. Acta Int. J. Clin. Chem. 463, 193–199 (2016).
Luo, S. et al. Identification of key molecules and lncRNA–miRNA–mRNA ceRNA network in preeclampsia. Int. J. Gen. Med. 14, 7579–7590 (2021).
Chen, D. et al. Identification of mRNA-, circRNA- and lncRNA-associated ceRNA networks and potential biomarkers for preeclampsia from umbilical vein endothelial cells. Front. Mol. Biosci. 8, 652250 (2021).
Jiang, R. et al. Bioinformatics-based identification of miRNA-, lncRNA-, and mRNA-associated ceRNA networks and potential biomarkers for preeclampsia. Medicine 99(45), e22985 (2022).
Zhang, Z. et al. Identification of key genes and long noncoding RNA-associated competing endogenous RNA (ceRNA) networks in early-onset preeclampsia. BioMed Res. Int. 2020, 1673486 (2020).
Liu, S., Xie, X., Lei, H., Zou, B. & Xie, L. Identification of key circRNAs/lncRNAs/miRNAs/mRNAs and pathways in preeclampsia using bioinformatics analysis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 25, 1679–1693 (2019).
Tong, J. et al. Dysfunction of pseudogene PGK1P2 is involved in preeclampsia by acting as a competing endogenous RNA of PGK1. Pregnancy Hypertens. 13, 37–45 (2018).
Walle, M., Getu, F., Gelaw, Y. & Getaneh, Z. The diagnostic value of hepatic and renal biochemical tests for the detection of preeclampsia among pregnant women attending the Antenatal Care Clinic at the University of Gondar Comprehensive Specialized Hospital, Gondar, Northwest Ethiopia. Int. J. Gen. Med. 15, 7761–7771 (2022).
Ping, Z. et al. Identification and comparison of circular RNAs in preeclampsia. PeerJ 9, e11299 (2021).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30(15), 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12(4), 357–360 (2015).
Roberts, A., Pimentel, H., Trapnell, C. & Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics (Oxford, England) 27(17), 2325–2329 (2011).
Kang, Y.-J. et al. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acid Res. 45(W1), W12–W16 (2017).
Sun, L. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41(17), e166 (2013).
Sonnhammer, E. L., Eddy, S. R., Birney, E., Bateman, A. & Durbin, R. Pfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 26(1), 320–322 (1998).
Li, A., Zhang, J. & Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 15(1), 311 (2014).
Anders, S. & Huber, W. J. H. Germany: European Molecular Biology Laboratory, Differential expression of RNA-Seq data at the gene level—The DESeq package. F1000Research 10, 4010 (2012).
Ching, T. et al. Genome-wide hypermethylation coupled with promoter hypomethylation in the chorioamniotic membranes of early onset pre-eclampsia. Mol. Hum. Reprod. 20(9), 885–904 (2014).
Adu-Gyamfi, E. A. et al. Ephrin and Eph receptor signaling in female reproductive physiology and pathology. Biol. Reprod. 104(1), 71–82 (2021).
El-Khalik, S. R. A., Ibrahim, R. R., Ghafar, M. T. A., Shatat, D. & El-Deeb, O. S. Novel insights into the SLC7A11-mediated ferroptosis signaling pathways in preeclampsia patients: Identifying pannexin 1 and toll-like receptor 4 as innovative prospective diagnostic biomarkers. J. Assist. Reprod. Genet. 39(5), 1115–1124 (2022).
Wang, G. J., Yang, Z., Huai, J. & Xiang, Q. Q. Pravastatin alleviates oxidative stress and decreases placental trophoblastic cell apoptosis through IL-6/STAT3 signaling pathway in preeclampsia rats. Eur. Rev. Med. Pharmacol. Sci. 24(24), 12955–12962 (2020).
Romanowska-Próchnicka, K. et al. The role of TNF-α and anti-TNF-α agents during preconception, pregnancy, and breastfeeding. Int. J. Mol. Sci. 22(6), 2922 (2021).
Massimiani, M. et al. Increased circulating levels of Epidermal Growth Factor-like Domain 7 in pregnant women affected by preeclampsia. Transl. Res. 207, 19–29 (2019).
Timokhina, E. et al. Matrix metalloproteinases MMP-2 and MMP-9 as markers for the prediction of preeclampsia in the first trimester. Ceska Gynekol. 86(4), 228–235 (2021).
Zhou, B. et al. CircZDHHC20 represses the proliferation, migration and invasion in trophoblast cells by miR-144/GRHL2 axis. Cancer Cell Int. 20, 19 (2020).
Acknowledgements
This study was supported by Suzhou Commission of Health and Family Planning (SZYJTD201709) and Suzhou science and Technology Development plan (SKYXD2022086).
Author information
Authors and Affiliations
Contributions
Funding acquisition, Y.S.; Investigation, L.Z.; Methodology, Y.X. and J.T.; Project administration, X.Z.; Resources, C.L.; Software, Y.Y. and C.Z.; Supervision, X.Z. and Y.S.; Visualization, X.Z.; Writing—original draft, X.Z.; Writing—review & editing, Y.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, L., Liu, C., Xu, Y. et al. Characterization of the lncRNA-mediated ceRNA regulatory networks in preeclampsia by integrated bioinformatics. Sci Rep 13, 17271 (2023). https://doi.org/10.1038/s41598-023-44059-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44059-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
