
Abstract
Lung adenocarcinoma (LUAD) is the most common malignant subtype of lung cancer (LC). miR-200 family is one of the prime miR regulators of epithelial-mesenchymal transition (EMT) and worst overall survival (OS) in LC patients. The study aimed to identify and validate the key differentially expressed immune-related genes (DEIRGs) regulated by miR-200 family which may serve for therapeutic aspects in LUAD tumor microenvironment (TME) by affecting cancer progression, invasion, and metastasis. The study identified differentially expressed miRNAs (DEMs) in LUAD, consisting of hsa-miR-200a-3p and hsa-miR-141-5p, respectively. Two highest-degree subnetwork motifs identified from 3-node miRNA FFL were: (i) miR-200a-3p-CX3CR1-SPIB and (ii) miR-141-5p-CXCR1-TBX21. TIMER analysis showed that the expression levels of CX3CR1 and CXCR1 were significantly positively correlated with infiltrating levels of M0-M2 macrophages and natural killer T (NKT) cells. The OS of LUAD patients was significantly affected by lower expression levels of hsa-miR-200a-3p, CX3CR1 and SPIB. These DEIRGs were validated using the human protein atlas (HPA) web server. Further, we validated the regulatory role of hsa-miR-200a-3p in an in-vitro indirect co-culture model using conditioned media from M0, M1 and M2 polarized macrophages (THP-1) and LUAD cell lines (A549 and H1299 cells). The results pointed out the essential role of hsa-miR-200a-3p regulated CX3CL1 and CX3CR1 expression in progression of LC TME. Thus, the study augments a comprehensive understanding and new strategies for LUAD treatment where miR-200 family regulated immune-related genes, especially chemokine receptors, which regulate the metastasis and invasion of LUAD, leading to the worst associated OS.
Similar content being viewed by others


Introduction
LUAD is a common malignant subtype of NSCLC, accounting for more than \(40{\%}\) of LC cases worldwide1. It is a multifactor and multistage process associated with multiple genes where the absolute risk of distant metastasis is very high at every stage of disease2. This primarily enhances the disease’s systemic threat, leading to poor prognosis, higher recurrence rates, and lower OS of patients3.
miRNAs are a class of small (\(19-25\) nucleotides), highly conserved, ncRNA molecules that can modulate various molecular mechanisms by inhibiting translational or mRNA degradation. miR-200 family (i.e., miR-200a, miR-200b, miR-200c, miR-141, and miR-429) are among the crucial miRNAs which primarily regulate EMT in LC. It can be divided into two groups based on single nucleotide seed sequence difference—(i) miR-200b, miR-200c, and miR-429 (AAUACUG) and (ii) miR-200a and miR-141 (AACACUG). Even after being recognized as a crucial tumor-associated miRNA, the precise molecular mechanism involved in miR-200 family-mediated LC progression and metastasis is unknown mainly due to the controversial results as tumor suppressor or oncogenic levels in serum and/or tissue as well as at various stages of cancer. The miR-200 family has been associated with poor prognosis and worst OS in NSCLC patients, especially LUAD4,5. Therefore, further insight into the miR-200 regulated mechanisms for developing and progressing LUAD is required.
TME is a key player in tumor progression and metastasis, which may lead to identifying novel targets and developing novel therapies. Immune cells, a primary part of TME, play a critical role in tumor growth and development6. Cellular cross-talk between tumor cells and their TME, which consists of CAFs, leukocytes, various infiltrating immune cells, and non-cell components of the ECM, contributes to cancer progression. This is facilitated by various soluble factors like growth factors and chemokines7. Chemokine receptors are extensively expressed on tumor and TME cells, facilitating various processes such as tumor cell survival, angiogenesis, vascular permeability, leukocyte recruitment, immune suppression, tumor cell adhesion, proliferation, EMT, and metastasis8.
The present study identified the DEGs and DEMs between tumor and normal tissues based on TCGA-LUAD cohort followed by DEIRGs identification. Since our study focused on the miR-200 family, hence a closed 3-node FFL was created, which showed regulation of these DEIRGs (chemokine receptors) by miR-200 family and corresponding TFs. The OS of LUAD patients affected by miR-200 family-associated FFL hub elements was depicted using KM plots. Furthermore, the TIMER database revealed the correlation between the expression of miR-200 family-associated DEIRGs and tumor-infiltrating immune cells in LUAD patients. The expression results of miR-200 targeted DEIRGs obtained in our study were validated with HPA database. To further validate the regulatory role of miR-200a-3p in TME, the expression of CX3CR1 and CX3CL1 were studied in miR-200a-3p transfected LUAD cell lines (A549 and H1299 cells) which were cultured in conditioned media from M0, M1 and M2 polarized macrophages (THP-1 cell line).
Therefore, our study emphasizes on the critical role of miR-200 family-targeted DEIRGs which have an essential role in TME during metastasis. Targeting these miR-chemokine receptor axes might have therapeutic potential in treating LUAD.
Materials and methods
TCGA RNA-seq data extraction and DEA
mRNA HTseq and miRNA-seq count data of TCGA-LUAD were retrieved from the UCSC Xena browser (https://xenabrowser.net/)9. Back-log-transformation and cross-checking of the mRNA-seq and miRNA-seq LUAD cohort samples with TCGA-GDC10 data portal was performed as discussed previously11,12. Pre-processing (i.e., normalization and \({\mathrm{log}}_{2}\) transformation) of mRNA-seq and miRNA-seq cohorts was performed as discussed previously11,12. Batch correction of pre-processed values was performed as discussed previously12. Ensembl IDs to their corresponding HGNC symbol(s) mapping in the mRNA-seq cohort was performed as discussed previously12. Duplicate genes were handled as discussed previously13,14,15,16,17,18. Limma package19 was used for the identification of DEGs and DEMs corresponding to a threshold of \(\left|{\mathrm{log}}_{2}\left(\mathrm{fold change}\right)\right|>2\) and \(\mathrm{BH}-\mathrm{ p}-\mathrm{value}<0.0001.\)
Enrichment and PPIN analyses of LUAD-associated immune genes
The ImmPort (https://immport.niaid.nih.gov)20 is a critical repository for immunology-associated clinical and molecular data. The overlapping genes between human IRGs acquired from ImmPort and our filtered DEGs were categorized as the DEIRGs. Pathway and GO term enrichment analyses of our DEIRGs were performed using the ReactomePA package21. A \(\mathrm{q}-\mathrm{value }<0.0001\) was used as the preferred cutoff for selecting significantly enriched pathway and GO terms. The sequential steps of PPIN formation and cluster selection were performed as discussed previously22. The genes present in the topmost-scoring PPIN cluster were regarded as the hub DEIRGs, respectively.
miRNA-mRNA-TF regulatory network construction and ROC curve analysis
Significant human TFs interacting with our hub DEIRGs were acquired from ChEA v3.0 database (https://maayanlab.cloud/chea3/)23 corresponding to a \(p-\mathrm{value}\le 0.001\). miRNAs which were interacting with our hub DEIRGs and TFs were extracted as discussed previously15. The miRNAs overlapping with DEMs were retained and regarded as validated miRNAs. Sequential steps of 3-node miRNA FFL formation and visualization were performed as discussed previously11,15. ROC curve analysis was performed to assess the diagnostic sensitivity of miR-200 family-associated hub mRNAs in LUAD patients. The AUC assessed the diagnostic values.
Tumor immune infiltration analysis
TIMER (http://timer.cistrome.org/)24 is a web resource for systematically analyzing the immune infiltration across various cancer types. Using TIMER, we assessed the correlation between the expression of miR-200 family-associated hub mRNAs and tumor-infiltrating immune cells in LUAD patients. Spearman correlation was utilized to evaluate the statistical significance. The gene expression levels were \({\mathrm{log}}_{2}\) RSEM expressed.
Validation of miR-200 family-associated hub DEIRGs using HPA and GEO
The protein expression levels of hub DEIRGs targeted by miR-200 family members were determined using the HPA database (https://www.proteinatlas.org/)25,26,27,28,29 in normal and LUAD tissues. NCBI-GEO (https://www.ncbi.nlm.nih.gov/geo/)30 was inquired utilizing “LUAD” and “lung adenocarcinoma” as keywords for extracting mRNA expression profiles. The search results were filtered as per the inclusion and exclusion criteria mentioned previously31. Sequential steps of batch correction, gene mapping, duplicate genes removal, DEGs identification were performed as discussed previously31. The presence of hub DEIRGs targeted by miR-200 family members was cross-checked in the DEGs list.
Survival analysis of miR-200 family-associated FFL hub items
KM plots showing the OS of 3-node miRNA FFL-associated subnetwork motif items were displayed as per parameters discussed previously32 via KM plotter database (https://kmplot.com/analysis/)33 corresponding to TCGA-LUAD cohort patient samples.
Cell culture, macrophage differentiation and transient transfection
Human LUAD cell lines-A549 and H1299 along with THP-1 (Human monocytes) were procured from NCCS, Pune, India. These cell lines were cultured in RPMI1640 (Cat # 61,870,036, Gibco, Waltham, MA, USA), \(1{\%}\) antibiotic–antimycotic (Cat # 15,240,096, Gibco, Waltham, MA, USA) at \(37\, ^\circ \mathrm{C}\) and \(5{\%}\) \({\mathrm{CO}}_{2}\). Differentiation of THP-1 cells were done by using \(5\,\mathrm{ ng}/\mathrm{mL}\) PMA (P8139, Sigma, Saint Louis, Missouri 63113, USA). They were stimulated using LPS (\(100\,\mathrm{ ng}/\mathrm{ml}\)) and IL-4 (\(10\,\mathrm{ ng}/\mathrm{mL}\), Cat # I4269, Sigma Aldrich, Bangalore, India) for \(24\,\mathrm{ h}\) for obtaining M1 and M2 macrophage cell population, respectively. A differentiated but non-stimulated macrophage population was termed as M0. Adherent cells were washed and cultured with serum-free RPMI medium for \(24\,\mathrm{ h}\), then the resulting macrophage-conditioned medium from M0, M1 and M2 macrophage population was collected and clarified by centrifugation at \(13,000\) rpm at \(4\, ^\circ \mathrm{C }\) for \(5\,\mathrm{ min}\). A549 and H1299 cells were transiently transfected with \(30\,\mathrm{ pmol}/\mathrm{mL}\) scrambled miRNA mimic (Qiagen, Hilden, Germany) and \(30\,\mathrm{ pmol}/\mathrm{mL}\) miR-200a-3p mimic (Qiagen, Hilden, Germany) using lipofectamine 3000 (Invitrogen, Waltham, MA, USA) for \(24\,\mathrm{ h}\). These scrambled and miR-200a-3p transfected cells from both A549 and H1299 cell lines were culture with \(1:5\) concentration of conditioned media from M0, M1 and M2 macrophages for \(18\,\mathrm{ h}\). The treated cells were further subjected to RNA isolation.
qRT-PCR analysis
Total RNA was isolated from M0, M1 and M2 THP-1 macrophages and scrambled/miR-200a-3p transfected A549 and H1299 cells which were treated with M0/M1/M2 macrophage-conditioned media using TRIzol reagent (Ambion, CA, USA) following the manufacturer’s protocol. The iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) was used to reverse-transcribe \(1\,\upmu \mathrm{g}\) of this isolated RNA. qRT-PCR analyses for quantifying mRNA of CX3CR1, and its ligand gene CX3CL1 and actin were performed by using iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). PCR using actin as an endogenous control was performed using 7900HT Fast Real-time PCR System (Applied Biosystems, Waltham, MA, USA). Relative quantification from real-time data is presented based on the calculation of \({2}^{-\Delta \Delta \mathrm{Ct}}\). The primer sequences used are CX3CR1-forward primer 5′-AAATACCCCATCATTCATGC-3′, reverse primer 5′-TTGTTCCAAACGTTTCTAGG-3′, CX3CL1-forward primer 5′-AGATACCTGTAGCTTTGCTC-3′, reverse primer 5′-TCTCGTCTCCAAGATGATTG-3′, Actin- forward primer 5′-AGCACAGAGCCTCGCCTT-3, reverse primer 5′- CATCATCCATGGTGAGCTGG-3′.
Cell proliferation assay
A549 and H1299 cells were harvested, seeded in \(96\)-well plates (\(100\ \upmu \mathrm{ L}\)/well), and cultured for \(24\,\mathrm{ h}\). They were both transiently transfected with \(30\,\mathrm{ pmol}/\mathrm{mL}\) scrambled miRNA mimic (Qiagen, Hilden, Germany) and \(30\,\mathrm{ pmol}/\mathrm{mL}\) miR-200a-3p mimic (Qiagen, Hilden, Germany) by using lipofectamine 3000 (Invitrogen, Waltham, MA, USA) for \(24\,\mathrm{ h}\). A549 and H1299 cancer cells were then cultured in a \(20{\%}\) conditioned medium from M0/M1/M2 macrophages in RPMI complete medium. MTT assay was performed to evaluate cell proliferation on completion of \(24\,\mathrm{ h}\) (\(\mathrm{N}=3\) for each group).\(5\mathrm{ \mu L}\) of MTT solution (Sigma-Aldrich, St Louis, MO, USA) (\(5\,\mathrm{ mg}/\mathrm{mL}\)) were added to each well, and incubated for \(3-4\mathrm{ h}\) at \(37\, ^\circ \mathrm{C}\) and \(5{\%}\) \({\mathrm{CO}}_{2}\). The supernatant was discarded, then \(100\,\mathrm{ \mu L}\) of DMSO was added to each well to dissolve the formazan crystal violet. The absorbance was measured at \(570\,\mathrm{ nm}\). Percent proliferation was calculated for all the groups.
Statistical analysis
All the experiments were repeated thrice independently, and the values obtained were expressed as the means \(\pm\) SEM. Data obtained from the study were analyzed by using GraphPad Prism version 7.0. Significance levels between two corresponding groups under comparison were checked using students t-test. \(p-\mathrm{value}<0.05\) was considered to be statistically significant.
Results
RNA-seq data extraction and DEA
mRNA and miRNA LUAD cohorts comprised \(444\) samples (\(433\) tumor and \(11\) healthy normal samples). After batch correction, gene mapping, and duplicacy removal, we were left with \(19193\) genes. Similarly, after removing low-count miRBase IDs, we were left with \(1355\) miRBase IDs out of \(1881\). Using limma, we identified a total of \(1053\) DEGs and \(46\) DEMs corresponding to the abovementioned threshold i.e., \(\left|{\mathrm{log}}_{2}\left(\mathrm{fold change}\right)\right|>2\) and \(\mathrm{BH}-p-\mathrm{value}<0.0001\). Figure S1A shows an annotation heatmap plot of top \(10\) down and top 10 upregulated DEGs. The sample annotation bar at top of the heatmap clearly illustrates more female samples (\(54.72{\%}\)) than male samples (\(45.72{\%}\)). The heatmap shows healthy normal samples clustered distinctly from tumor samples. Chromosome number \(10\) was populated with the highest number of DEGs (SFTPA1, SFTPA2, GDF10, and SFTPD) and all of these were downregulated.
Enrichment and PPIN modular analyses of DEIRGs
We retrieved \(1793\) IRGs from the ImmPort database and \(129\) DEIRGs overlapped between our DEGs and IRGs. Venn plot representing the number of DEGs, IRGs, and DEIRGs is shown in Fig. S1B. A total of \(110\) DEIRGs out of \(129\) were actively involved in \(52\) significantly enriched GO terms (i.e., \(14\) BP terms \(+\) \(32\) MF terms \(+\) \(6\) CC terms). Treemap illustrating the significant BP terms with respect to the number of DEIRGs is shown in Fig. 1A. The number of DEIRGs involved in BP terms ranged from \(13\) to \(27\), respectively. A lollipop plot showing the significant MF terms as lollipops and their corresponding DEIRG count is shown in Fig. 1B. The number of genes in these MF terms ranged from \(4\) to \(41\). Circos plot showing the association of DEIRGs and significant CC terms is shown in Fig. 1C. The number of DEIRGs in these CC terms ranged from \(7\) to \(14\). Moreover, \(52\) DEIRGs actively participated in \(8\) significantly enriched pathways. Circular barplot showing the significant pathways with respect to their gene count is shown in Fig. 1D. A total of \(9\) DEIRGs overlapped among all the significantly enriched pathways and GO terms and is shown by the Venn plot in Fig. S2. Amongst these DEIRGs, only \(7\) took part in the PPIN corresponding to the aforementioned threshold (i.e., \(\mathrm{confidence score }>0.9\)). Figure 2A shows the PPIN comprising \(7\) DEIRGs linked by \(11\) edges. The top-scoring PPIN cluster identified using MCODE comprised \(4\) hub DEIRGs linked by \(6\) edges as shown in Fig. 2B, respectively. A split violin plot showing the distribution of expression of \(4\) hub DEIRGs is shown in Fig. 2C. S1PR1 had a considerably higher expression level than the other three hub DEIRGs, as evidenced from the violin plot.

(A) Treemap chart illustrating significant GO-BP terms associated with DEIRGs. Each rectangular shape represents individual terms and their sizes vary according to the number of genes present in them. All the rectangles have a unique color signifying the distinct \(14\) BP terms. The term “cell chemotaxis” has the highest number of genes present in it, i.e., \(27\). While the terms “neutrophil chemotaxis” and “positive regulation of endothelial cell proliferation” has the lowest number of genes present in it, i.e., \(13\). (B) Lollipop plot showing the distribution of significant GO-MF terms associated with DEIRGs. The \(\mathrm{x}\) and \(\mathrm{y}\) axes represents the MF terms and gene count. The term “signaling receptor activator activity” has highest number of genes present in it, i.e., \(41\). The term “protein tyrosine kinase activator activity” has lowest number of genes present in it, i.e., \(4\). (C) Circos plot representing significant GO-CC terms associated with DEIRGs. Outer boundary of the circle consists of \(6\) CC terms (on the left) linked with the DEIRGs present in them (on the right). Each gene and pathway are denoted by unique color strips with an undirected edge showing the corresponding associations. (D) Circular barplot showing the distribution of \(8\) significant pathways associated with DEIRGs. The color and sizes of bars depend on the \(\mathrm{q}-\mathrm{value}\) and gene count, respectively.

(A) Unweighted and undirected PPIN of significantly enriched DEIRGs comprising \(7\) nodes and \(30\) edges. (B) Top-scoring PPIN cluster comprising a total of \(4\) nodes and \(30\) edges. Cyan-colored nodes signify downregulated expression status of DEIRGs. (C) Split violin plot displaying the expression intensity distribution of \(4\) hub DEIRGs. The normal and tumor samples are represented by magenta and sea green colors. The top and bottom of the boxes inside the splitted violin depicts the \(75\)th and \(25\)th percentile of the distribution, respectively. The horizontal lines within the boxes signifies the median values. Axis endpoints are labelled by the minimum and maximum values, respectively.
miRNA-mRNA-TF regulatory network analysis and ROC curve validation
Our \(3\)-node miRNA FFL regulatory network included a total of \(16\) nodes and \(37\) edges as shown in Fig. 3A. Amongst all the edges, \(12\), \(10\), and \(15\) edges belonged to TF-mRNA, miRNA-mRNA, and miRNA-TF pairs, respectively. Amongst all the nodes, \(7\), \(4\), and \(5\) nodes belonged to miRNAs, mRNAs, and TFs, respectively. The degree values of miRNAs, mRNAs, and TFs ranged from \(1\) to \(7\), \(1\) to \(6\), and \(1\) to \(7\), respectively. The average degrees of miRNAs, mRNAs, and TFs were \(3.125\), \(2.75\), and \(3.375\), respectively. We observed two highest-order subnetwork motifs with respect to the miR-200 family. The first subnetwork motif included one miRNA (miR-200a-3p), one mRNA (CX3CR1), and one TF (SPIB) as shown in Fig. 3B. Whereas the second one included one miRNA (miR-141-5p), one mRNA (CXCR1), and one TF (TBX21) as shown in Fig. 3C. The ROC curve analysis of CX3CR1 and CXCR1 is shown in Figs. 4A-B, respectively. The \(95\%\mathrm{CI}\) and AUC of CX3CR1 were \(0.906-1\) and \(0.953\), and those of CXCR1 were \(0.846-1\) and \(0.938\), respectively. Pairwise scatterplot matrices exhibiting association between CX3CR1, CXCR1, miR-200a-3p, and miR-141-5p is shown in Fig. 4C. Both the miRNAs (i.e., miR-141-5p and miR-200a-3p) had the highest correlation value. i.e., \(0.560\) as evidenced from the plot.

(A) Unweighted LUAD-specific \(3\)-node miRNA FFL comprising \(30\) nodes and \(30\) edges. (B) miR-200 family-associated highest-order subnetwork motif comprising one miRNA (hsa-miR-200a-3p), one TF (SPIB), and one hub DEIRG (CX3CR1). (C) miR-200 family-associated second highest-order subnetwork motif comprising one miRNA (hsa-miR-141-5p), one TF (TBX21), and one hub DEIRG (CXCR1). The green, red, and magenta-colored nodes represents the TFs, hub DEIRGs, and miRNAs, respectively.

ROC curve analyses of (A) CX3CR1 and (B) CXCR1. (C) Pairwise scatter plot of miR-200 associated hub items, i.e., CX3CR1, CXCR1, hsa-miR-200a-3p, and hsa-miR-141-5p. The upper triangular section represents the Spearman’s correlation coefficients between these hub items. While the lower triangular section represents the scatterplot and histogram distribution between these hub items. The diagonal consists of kernel densities for each hub item. Significant levels at \(0.05\), \(0.01\), and \(0.001\) are represented by *, **, and ***, respectively.
Tumor immune infiltration analysis
CXCR1 and CX3CR1 expression levels were significantly correlated with tumor purity and the infiltrating levels of macrophages and NKT in LUAD. M0 macrophages were significantly correlated with CX3CR1 (\(\mathrm{r}=-0.257\), \(p=7.51\times {10}^{-9}\)) whereas nonsignificantly correlated with CXCR1 (\(\mathrm{r}=0.015\), \(p=7.34\times {10}^{-1}\)) as shown in Fig. 5. Infiltrating levels of M1 macrophages had significant correlation with the expression levels of both CX3CR1 (\(\mathrm{r}=-0.105\), \(p=1.96\times {10}^{-2}\)) and CXCR1 (\(\mathrm{r}=-0.121\), \(p=7.16\times {10}^{-3}\)) (Fig. 5). But, infiltration levels of M2 polarized macrophages were significantly associated with the expression levels of both CX3CR1 (\(\mathrm{r}=0.333\), \(p=3.15\times {10}^{-14}\)) and CXCR1 (\(\mathrm{r}=0.125\), \(p=5.61\times {10}^{-3}\)) (Fig. 5). Similarly, CXCR1 (\(\mathrm{r}=-0.156\), \(p=5.07\times {10}^{-4}\)) had significant negative while CX3CR1 (\(\mathrm{r}=0.357\), \(p=2.78\times {10}^{-16}\)) had significant positive correlation with NKT infiltrating levels as shown in Fig. 5. In addition, both these genes were significantly negatively correlated with tumor purity in LUAD. These results indicate the clear role of both CX3CR1 and CXCR1 in immune responses in LUAD TME as suggested by their significant positive correlation with M2 macrophage infiltration and significant correlations with NKT infiltrations.

Scatterplots showing significant correlations of (A) CXCR1 and (B) CX3CR1 mRNA expression with M0, M1, M2, NKT infiltrating levels across the TCGA-LUAD cohort. mRNA expression levels against tumor purity are demonstrated on the left panel. Spearman’s correlation and estimated statistical significance are displayed for each scatter plot.
Validation of miR-200 family-associated hub DEIRGs using HPA and GEO
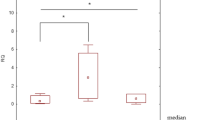
In HPA database, the protein expression levels of both CX3CR1 and CXCR1 in the LUAD sample tissues were distinct from the corresponding normal lung tissue samples (Fig. 6). GSE116959 (\(11\) healthy control + \(57\) tumor tissues) and GSE43458 (\(30\) healthy control + \(80\) tumor tissues) mRNA expression profiles were chosen in accordance with the abovementioned exclusion/inclusion criteria. Both CXCR1 and CX3CR1 were present in the DEGs list, thus affirming their validation in external GEO datasets. Figure S3 depicts box-and-whisker plots of the relative expression distributions of CXCR1 and CX3CR1 across LUAD patient samples compared to healthy normals. As observed, both genes' mRNA expression levels were significantly downregulated in tumor samples compared to healthy normals.

Representative IHC images of (A) CX3CR1 and (B) CXCR1 across normal and LUAD tissues via HPA database.
Prognostic assessment of miR-200 family-associated hub items
The corresponding KM plots shown in Fig. 7A–C depicted that lower expression levels of CX3CR1 (\(\mathrm{HR}=1.75\); \(95\%\mathrm{CI}=1.2-2.5\); \(p<0.05\)), miR-200a-3p (\(\mathrm{HR}=1.67\); \(95\%\mathrm{CI}=1.2-2.2\); \(p<0.05\)), and SPIB (\(\mathrm{HR}=1.67\); \(95\%\mathrm{CI}=1.2-2.3\); \(p<0.05\)) worsened the OS in \(513\) LUAD patients. The low and high expression cohort median survival times of each item is detailed in table S1, respectively.

KM plots showing OS of (A) CX3CR1, (B) miR-200a-3p, and (C) SPIB. The black and red colored lines corresponds to LUAD samples with lower and higher expression levels, respectively. The lower expression of all these subnetwork motif items correlates with lower OS in LUAD patients. All these motif items were highly significant (\(\mathrm{log rank } p < 0.05\)).
Expression of fractalkine receptor CX3CR1 and its ligand CX3CL1 in overexpression model of miR-200a-3p cultured in macrophage-conditioned media
To study the critical role of miR-200a-3p regulated identified hub gene CX3CR1 (fractalkine receptor) in TME, scrambled or miR-200a-3p transfected LUAD cell lines A549 and H1299 were grown in M0/M1/M2 macrophage-conditioned media. The expression of CX3CR1 and its only known ligand, the chemokine CX3CL1 (a.k.a Fractalkine), was quantified using qRT-PCR in M0/M1/M2 macrophages as well as in scrambled/miR-200a-3p transfected A549 and H1299 cells treated with macrophage-conditioned media.
The expression of CX3CL1 was very low in M0 subset macrophages and even further lower in M1 macrophages while it was undetectable in case of M2 polarized macrophages (data not shown). The CX3CR1 expression in M1 polarized macrophages was significantly decreased as compared to M0 and M2 subsets. While it was significantly higher in M2 polarized macrophages than both M0 and M1 macrophages subsets (Fig. 8A).

(A) Relative fold change in expression of CX3CR1 in M0 (unstimulated), M1 (LPS stimulated) and M2 (IL4 stimulated) macrophages. Relative fold change in the expression of (B) CX3CL1 and (C) CX3CR1 upon transfection of scrambled/ miR-200a-3p in A549 cells indirectly co-cultured in M0/M1/M2 macrophage conditioned medium as determined by qRT-PCR. (D) Percentage proliferation of scrambled/ miR-200a-3p transfected A549 cells when indirectly co-cultured in M0/M1/M2 macrophage conditioned medium, as determined by MTT assay. Relative fold change in the expression of (E) CX3CL1 and (F) CX3CR1 upon transfection of scrambled/ miR-200a-3p in H1299 cells indirectly co-cultured in M0/M1/M2 macrophage conditioned medium as determined by qRT-PCR. (G) Percentage proliferation of scrambled/ miR-200a-3p transfected H1299 cells when indirectly co-cultured in M0/M1/M2 macrophage conditioned medium, as determined by MTT assay. \(\upbeta\)-actin was used as an endogenous control. ns- Nonsignificant, *p < 0.05, **p < 0.01, ***p < 0.001.
M0 macrophages are the undifferentiated subset of macrophages that can differentiate into a specific polarized state of macrophages like M1 which are pro-inflammatory or M2 which are anti-inflammatory. In the presence of M0 macrophage-conditioned media, miR-200a-3p transfected A549 and H1299 cells showed a highly significant increase in the expression of CX3CL1 (Fig. 8B,E) in comparison to scrambled transfected cells. Similarly, a significant increase in the relative expression of fractalkine receptor CX3CR1 was also quantified in both miR-200a-3p transfected A549 and H1299 cells cultured in M0-conditioned media (Fig. 8C,F). This indicated a clear role of miR-200-3p in increasing CX3CL1 and CX3CR1 expression in cancer cells. The expression of CX3CL1 was significantly lower in both miR-200a-3p transfected A549 and H1299 cells when grown in the presence of M1 and M2 conditioned media as compared to scrambled transfected corresponding control cells. However, the expression level of CX3CL1 was much higher when cells were grown in M1 macrophage-conditioned media than when the cells were grown in M2 macrophage-conditioned media (Fig. 8B,E). This indicates that CX3CL1 (Fractalkine) expression is much higher in pro-inflammatory TME, suggesting the cancer-eradicating role of CX3CL1-CX3CR1 axis, possibly by inducing apoptosis in cancer or immune targeting of cancer cells leading to anti-cancer processes. However, the significant decrease in CX3CL1 after miR-200a-3p transfection in both pro-inflammatory and anti-inflammatory TMEs suggests the association of miR-200a with decreased immune effector cell infiltration and decreased tumor-suppressive activity, which may also result in higher metastasis. Although, CX3CL1 additionally has tumor-suppressive activity as CX3CL1 overexpression has been shown to increase the chemotactic efficiency and infiltration of immune effector cell, resulting in an improved prognosis.
The expression of the fractalkine receptor CX3CR1 also increased in both miR-200a-3p transfected A549 and H1299 cell lines when cultured in M0-conditioned media (Fig. 8C,F). The expression of CX3CR1 decreased significantly in miR-200a transfected A549 cell line, and a highly significant decrease was observed in miR-200a transfected H1299 cell line when cultured in M1 macrophage-conditioned medium indicating a chemotactic role of CX3CR1 here in pro-inflammatory TME. Surprisingly in M2 conditioned medium, miR-200a-3p transfected A549 cell line showed a highly significant increase in CX3CR1 receptor as compared to scrambled transfected cells. However, in H1299, there was a significant reduction in CX3CR1 expression in miR-200a-3p transfected cells when grown in M2-conditioned media. Increased CX3CR1 expression is associated with increased M2 macrophage infiltration and increased CX3CR1 expression has also been associated with metastasis-initiating cells.
Tumor cell proliferation upon miR-200a-3p overexpression on culturing with macrophage-conditioned medium
To further assess if the expression of CX3CL1-CX3CR1 and miR-200a-3p was correlated with the proliferation ability of A549 and H1299 cell line when grown in the presence of M0, M1 and M2 macrophage-conditioned media, MTT assay was performed. There was a nonsignificant difference in the proliferation ability of scrambled or miR-200a-3p transfected A549 cells when cultured in M0 and M1 macrophage-conditioned media. However, there was a significant decrease in proliferation of miR-200a transfected A549 cells compared to scrambled transfected when cultured in M2 macrophage-conditioned media indicating that miR-200a overexpression leads to decreased proliferation of cancer cells in TME. However, this difference was insignificant when cells were grown in either M0, M1 or M2 macrophage-conditioned media (Fig. 8D).
In H1299 cell line there was no difference in the proliferation ability of miR-200a transfected cells when cultured in M1 or M2-conditioned medium (Fig. 8G). The proliferation ability of H1299 cell line decreased when cultured in M0 macrophage condition medium. The proliferation assay in both A549 and H1299 cells indicated that the LUAD cell proliferation ability was not correlated with the expression of CX3CL1 or CX3CR1. This is indicative of the dual role of CX3CL1-CX3CR1 axis in TME.
Discussion
Carcinogenesis of LUAD is among one of the most lethal malignancies, which is a complex and multistage process regulated by many genes and miRNAs34. Genetic regulations involved in TME including tumor inflammation and immunity play a critical decisive role at various stages of tumor development6. Our study assumes TME as a key player in tumor growth and metastasis which may pave the way for identifying therapeutic and prognostic targets for early detection and treatment of LUAD for increased OS of LC patients. The present study identified DEMs and DEGs in the TCGA-LUAD cohort followed by \(129\) DEIRGs detection. Our study specifically targeted regulatory network motifs associated with miR-200a-3p and miR-141-5p which were our DEMs of interest in LUAD. The pathway and GO term analysis results showed that the BP were enriched in cell chemotaxis, second messenger mediated signaling, etc., affirming the critical role of IRGs in immune response in TME. In this study, we further validated the role of miR-200a-3p in TME in vivo using miR-200a-3p transfected A549 (wild type p53 LUAD) and H1299 (null p53 LUAD) cells cultured in M0, M1 (pro-inflammatory) and M2 (anti-inflammatory) macrophage-conditioned medium.
The first highest order miR-200 family-associated subnetwork motif comprised miR-200a-3p, CX3CR1 and SPIB. miR-200 family has been reported to function as an oncogene or tumor-suppressor in several carcinogenesis, but its crucial biological importance and functions in NSCLC are subtle. The miR-200 family is known to target ZEB1 and ZEB2, which maintains the epithelial phenotype of cells mediated by transcriptional repression of E-cadherin. ZEB1 and ZEB2 can induce EMT leading to cancer cell migration, invasion, and metastasis. \({\varvec{\upbeta}}\)-catenin mRNA is a direct target of miR-200a, and it can suppress the β-catenin/Wnt signaling pathway which is commonly involved in cancer35. Moreover, miR-200a expression was negatively associated with cyclin D1 and \(\upbeta\)-catenin in human meningioma tumor tissues36. It was suggested that miR-200a-3p was able to induce EMT in NSCLC and serve as a tumor promoter.
CX3CL1 is the only known member of chemokine CX3C family and CX3CR1 is the only receptor of CX3CL1 (Fractalkine). CXCL1 (membrane-bound/secreted) can potentially regulate tumor-related inflammatory response. CX3CR1 (the fractalkine receptor) is expressed in NKT cells, \({\mathrm{CD}8}^{+}\) T cells, DCs, and monocytes. The CX3CL1–CX3CR1 axis is reported to be upregulated in LC, colon cancer, breast cancer, gastric cancer, prostate cancer, and other malignancies. Our study emphasizes the different pattern of expression and the role of CX3CL1-CX3CR1 axis in pro-inflammatory (M1 macrophages) and anti-inflammatory (M2 macrophages) TME. Indeed, the high expression of CX3CR1 in M2 macrophages obtained in our study corroborated with other studies considering CX3CR1-expressing \({\mathrm{CD}68}^{+}{\mathrm{CD}206}^{+}\) cells in the lungs M2-macrophages37. While M1 macrophages are shown to increase the expression of CX3CL1-CX3CR1 axis supporting the theory that CX3CL1 can attract immune effector cells to the tumor location site and exert an anti-tumor immune effect. However, the downregulation of both CX3CL1 and CX3CR1 when cultured in M2 macrophage-conditioned medium supports the dual functional role of CX3CL1. This discrepancy may be attributed to the dual role of CX3CL1 acting both as a chemoattractant for leukocytes as well as an adhesion molecule for the tumor cells.
The clinical role of CX3CL1-CX3CR1 signaling has been reported to be contradictory and it may exert both pro-tumor and anti-tumor effects based on tumor tissue and histological grade of tumor38,39. CX3CL1 may also promote the adhesion of CX3CR1-positive tumor cells to target organs, thus causing the migration of tumor cells and promoting tumorigenesis40. miR-200a expression is downregulated in NSCLC, thus acting as tumor suppressor. Our study observed that miR-200a-3p upregulates the expression of CX3CR1, indicating the critical role of miR-200a-3p as a regulator of metastasis to distant organs and EMT. And hence the results can be corroborated with the study suggesting a correlation of miR-200a with advanced stages of NSCLC and low survival. Thus the upregulation of CX3CR1 in our study may be correlated with the poor prognosis and role of miR-200a in EMT. Our study supports the tumorigenic and metastatic role of miR-200a via modulating the expression of CX3CR1 in TME.
In contrast to this, the study shows the reduction in CX3CR1 expression on transfection of miR-200a-3p transfection in H1299. This indicates the role of p53-mediated effect of miR-200a in regulating CX3CR1 expression. Several studies pointed out the essential role of Src/FAK signaling pathway in CX3CL1-CX3CR1 axis-mediated migration and invasion of LC and breast cancer cells41,42. Further study will be needed to elucidate the most probable involvement of Src/FAK pathway regulation via miR-200a in LC. Interestingly, these cells were also reported to have a higher CX3CR1 mRNA expression. Emerging studies also suggest CX3CR1 as a marker of stem-like tumor cells and cells with relatively higher CX3CR1 expression show transcriptomic profiles enriched in pathways regulating pluripotency. In murine models, these cells resist chemotherapy and metastasis-initiating behavior43.
Expression of CX3CR1 is heterogenous even between cancer subtypes is associated with histological grade and stage-dependent progression of various malignancies38,44. Tumor cells which express CX3CL1 can induce the invasion and metastasis of CX3CR1-positive tumor cells45,46. It was shown earlier that the mRNA and protein expression of CX3CL1 and CX3CR1 was significantly high in primary LC and secondary bone metastasis. Serum levels of both were positively correlated to the LC progression. Thus, it was suggested that the CX3CL1–CX3CR1 axis is associated with LC growth and metastasis47. Similarly, increased CX3CR1 expression was correlated with bone metastasis in prostate cancer, similarly in breast cancer, expression of CX3CR1 predicted the occurrence of BM40,48. Fractalkine and CX3CR1 are recognizably capable of facilitating the adhesion and extravasation of CX3CR1-expressing circulating tumor cells into skeleton and soft-tissue organs and facilitating colonization and progression of tumor into secondary organs49. CX3CL1 induced cell migration in human osteosarcoma cells via upregulating ICAM-1 expression mediated by CX3CR1/PI3K/Akt/NF-κBpathway44. FKN/CX3CR1 activated JAK/STAT signaling in PDAC, which could further regulate cell growth and EMT. Inhibition of CX3CR1 was reported to inhibit the cancer cell survival and increase sensitivity towards chemotherapy in NSCLC50. CX3CR1 activates pro-survival signaling pathways in normal and cancer cells thus promoting cell viability51. It is known to act via Wnt and notch signaling pathways52,53. It may be predicted that miR-200a targets Wnt and notch signaling possibly via CX3CR1 axis to target metastasis in LUAD.
The cell proliferation ability of miR-200a-3p transfected A549 and H1299 cells did not correlate with the corresponding CX3CL1-CX3CR1 axis expression. This could be because the cytokines are mainly associated with EMT and metastasis processes and do not significantly affect the cancer cell apoptosis in TME. A recent report in human pancreatic cancer cells indicated that CX3CL1 protects against apoptosis. Pancreatic cancer cell lines upregulated the expression of anti-apoptotic molecules like BCL-2 and BCL-xl in response to exogenous CX3CL1; while CX3CL1 stimulation caused the decrease in the expression of the pro-apoptotic caspase 3. The mechanisms found depended on AKT phosphorylation54.
The lymphocyte-restricted protein, Spi-B, is ectopically expressed in LCs, and its increased expression level is correlated with poor prognosis in human LC. Spi-B was the hub TF involved in the regulatory network of miR-200a and CX3CR1. Earlier studies show that Spi-B was expressed in invasive cancer cells in human primary LC tissues. Vimentin was co-expressed with Spi-B whereas E-cadherin was repressed in LC. Spi-B-expression was also associated with lymphatic metastasis, short OS and tumor grade. Spi-B also downregulated claudin-2, disrupting intercellular junctions and enhancing invasiveness in LC cells55. SPIB activation significantly increased anoikis resistance from loss of attachment‐induced autophagy56,57.
In our study, another FFL subnetwork motif of miR-200 family-mediated regulation included miR-141, CXCR1 and TBX21. Role of miR-141-5p is largely unclear in NSCLC. Lower expression of miR-141-5p was associated with poor patient survival as an independent risk factor, lymph node metastasis, and advanced TNM stage58. In another study, higher expression of miR-141 in serum was associated with shorter OS in LUAD patients59. miR-141 expression also improved the secretion of VEGFA. VEGFA is associated with CAFs and tumor invasion in LUAD models in murine LUADs. This is mediated by downregulation of KLF65,60 and induced neoangiogenesis. miR-141 regulates the expression of PHLPP1 and PHLPP2, antagonists of PI3K/AKT signaling and promotes the proliferation of NSCLC cells61. This further indicates the critical role of miR-141-5p circuitry in regulating LUAD progression. Till now, no study has reported the role of miR-141 in the regulation of IRGs in TME of NSCLC which may enhance the invasion and metastasis. Expression of CXCL8 chemokine which has angiogenic and pro-inflammatory activity is also known to affect tumor cells, inducing the proliferation of LC cells through CXCR162.The CXCL8 (secreted by tumor cells)-CXCR1/2 (in TME) axis may also regulate CSC proliferation and self-renewal thus play a critical role in LC progression and metastasis in TME63.
CXCR1 can interact with both the ligands, CXCL6 and CXCL8. CXCL6 is known to be upregulated in LC64. miR-141 was found to be downregulated in the CXCL6 treated A549 cells65. CXCR1 is expressed on monocytes, some NK cells, granulocytes, and mast cells66. CXCR1 alone is associated with CXCL8-mediated chemotaxis67. TBX21, aTh1 cell-specific TF, was earlier identified as an independent predictive factor in LUAD. TBX21 was correlated with cancer stemness mediated by the TBX21-IL-4 pathway in LUAD patients68. TBX21 was associated with poor prognosis in LUAD.
There are controversial studies reported with respect to the high or low expression of miR-200 family in LUAD which significantly reduces the median OS in LUAD patients. Our study reported the survival plot of the LUAD patients with relation to miR-200a, CX3CR1 and Spi-B. All of them significantly (\({p }< 0.05\)) affected the OS in the LC patients with the median OS in lower expression cohort of miR-200a, CX3C1 and Spi-B being \(34.87\) months, \(42.17\) months and \(41.17\) months respectively (Fig. 7). The controversies reported in different reports may be due to the source of miR-200 family used in expression studies, i.e. either tissue or serum. The expression of CX3CR1 and CXCR1 was further correlated with the IHC images provided in the HPA database (Fig. 6).
The tumor immune infiltration analysis was done using TIMER. Our study reported that CXCR1 and CX3CR1 expressions were significantly negatively correlated with tumor purity whereas significantly positively correlated with infiltrating levels of macrophage in LUAD, especially M2 macrophages. CXCR1 had a significant negative correlation while CX3CR1 had a significant positive correlation with infiltrating levels of NKT in LUAD.
As cancer is designated as a chronic inflammatory disease, thus tumor progression is dependent on inflammatory microenvironment. CX3CR1 facilitates macrophage survival, leading to enhanced angiogenesis and metastasis of tumor cells. Deficiency of CX3CR1 diminished the extent of macrophages infiltrating the metastatic foci by inducing macrophage apoptosis69. Thus, our studies indicate that CX3CR1 regulated by miR-200a further aggravates the metastasis in LUAD mediated by inflammatory macrophages as indicated by positive correlation obtained in our studies. Few studies also indicated the possible role of CX3CR1-mediated angiogenic switch regulating malignant transition where macrophage may play a pivotal role in vascular remodelling in different tumor models69,70,71. However, this needs to be further correlated to TME studies. CX3CR1 is also expressed on T cell and NKT cell subsets. NKT, also called CD1d-restricted T cells, are a heterogeneous group of T cells that also share the properties of NK cells. TH1-like NKT cells may induce an antitumor response whereas, TH2- and Treg-like NKT cell subsets may mediate tumor progression and immune escape. Overstimulated NKT cells may skew toward TH2-/Treg-like subsets, thus mediating immune escape and tumor progression72. No studies had been done to reveal the association of CX3CR1 and NKT in TME.
Increased tumor angiogenesis and shorter median OS of LC are associated with the expression of IL8 mRNA, which is known to act through CXCR1/2, in the lung TME induced by infiltrating macrophages via the NFKB pathways73. One other study correlated CXCR1 expression intensity with tumor infiltration by macrophages and total number of tumor-infiltrating immune cell74, however the exact role of CXCR1 in macrophage or NKT cells mediated tumor progression has not been yet revealed. Our study could improve our comprehension in miR-200 family regulated role of IRGs in TME in LUAD.
Conclusion
TME is a complex player in the progression and metastasis of tumor. The study emphasizes the miR-200 family-regulated IRGs as key players in LC, leading to aggravated metastasis and reduced OS in LUAD patients. These IRGs could also affect tumor-infiltrating cell population in LUAD which was correlated with the possible role of these immune effector cells in tumor progression, invasion, and metastasis. The study provided insight into the role of miR-200a- 3p mediated regulation of CX3CL1-CX3CR1 axis, which may be associated with progression of LC. Thus, targeting miR-200a-CX3CR1-SPiB and miR-141-CXCR1-TBX21 axes may provide a potential prognostic as well as therapeutic target for efficient management of LUAD-related metastasis and poor survival.
Data availability
The data used in our study was downloaded from https://xenabrowser.net/datapages/?cohort=GDC%20TCGA%20Lung%20Adenocarcinoma%20(LUAD)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443.
Abbreviations
- NSCLC:
-
Non-small cell lung cancer
- LUAD:
-
Lung adenocarcinoma
- LC:
-
Lung cancer
- OS:
-
Overall survival
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- EMT:
-
Epithelial-mesenchymal transition
- TME:
-
Tumor microenvironment
- DEGs:
-
Differentially expressed genes
- DEMs:
-
Differentially expressed miRNAs
- TCGA:
-
The cancer genome atlas
- DEIRGs:
-
Differentially expressed immune-related genes
- FFL:
-
Feed-forward loop
- ECM:
-
Extracellular matrix
- CAFs:
-
Cancer-associated fibroblasts
- TFs:
-
Transcription factors
- KM:
-
Kaplan–Meier
- TIMER:
-
Tumor immune estimation resource
- HPA:
-
Human protein atlas
- HTseq:
-
High-throughput sequence
- HGNC:
-
HUGO gene nomenclature committee
- BH:
-
Benjamini–Hochberg
- PPIN:
-
Protein–protein interaction network
- DEA:
-
Differential expression analysis
- ImmPort:
-
Immunology database and analysis portal system
- IRGs:
-
Immune-related genes
- GO:
-
Gene ontology
- ROC:
-
Receiver operating characteristics
- AUC:
-
Area under curve
- LPS:
-
Lipopolysaccharide
- RPMI:
-
Roswell park memorial institute medium
- IL-4:
-
Interleukin 4
- rpm:
-
Revolutions per minute
- qRT-PCR:
-
Quantitative real-time PCR
- DMSO:
-
Dimethyl sulfoxide
- SEM:
-
Standard error of the mean
- BP:
-
Biological process
- MF:
-
Molecular function
- CC:
-
Cellular compartment
- CI:
-
Confidence interval
- NKT:
-
Natural killer T
- HR:
-
Hazard ratio
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- ZEB2:
-
Zinc finger E-box binding homeobox 2
- VEGFA:
-
Vascular endothelial growth factor A
- CSC:
-
Cancer stem cell
- SFTPA1:
-
Surfactant protein A1
- SFTPA2:
-
Surfactant protein A2
- GDF10:
-
Growth differentiation factor 10
- SFTPD:
-
Surfactant protein D
- S1PR1:
-
Sphingosine-1-phosphate receptor 1
- CX3CR1:
-
C-X3-C motif chemokine receptor 1
- SPIB:
-
Spi-B transcription factor
- CXCR1:
-
C-X-C motif chemokine receptor 1
- TBX21:
-
T-Box transcription factor 21
- CX3CL1:
-
C-X3-C motif chemokine ligand 1
- ICAM-1:
-
Intercellular adhesion molecule-1;
- UCSC:
-
University of California, Santa Cruz
- GDC:
-
Genomic data commons
- DC:
-
Dendritic cell
- ncRNA:
-
Non-coding RNA
- NCCS:
-
National centre for cell science
- cDNA:
-
Complementary DNA
- MTT:
-
[3′-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]
- MCODE:
-
Molecular complex detection
- IHC:
-
Immunohistochemistry
- BM:
-
Brain metastasis
- PDAC:
-
Pancreatic ductal adenocarcinoma
- NK:
-
Natural killer
References
Shi, J. et al. Somatic genomics and clinical features of lung adenocarcinoma: A retrospective study. PLoS Med. 13, e1002162 (2016).
Consonni, D. et al. Lung cancer prognosis before and after recurrence in a population-based setting. J. Natl. Cancer Inst. 107, djv059 (2015).
Liang, Y. & Wakelee, H. A. Adjuvant chemotherapy of completely resected early stage non-small cell lung cancer (NSCLC). Transl. Lung Cancer Res. 2, 403–410 (2013).
Pecot, C. V. et al. Tumour angiogenesis regulation by the miR-200 family. Nat. Commun. 4, 2427 (2013).
Tejero, R. et al. miR-141 and miR-200c as markers of overall survival in early stage non-small cell lung cancer adenocarcinoma. PLoS ONE 9, e101899 (2014).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, Inflammation, and Cancer. Cell 140, 883–899 (2010).
Rollins, B. J. Inflammatory chemokines in cancer growth and progression. Eur. J. Cancer Oxf. Engl. 1990(42), 760–767 (2006).
Borsig, L., Wolf, M. J., Roblek, M., Lorentzen, A. & Heikenwalder, M. Inflammatory chemokines and metastasis—Tracing the accessory. Oncogene 33, 3217–3224 (2014).
Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020).
Grossman, R. L. et al. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 375, 1109–1112 (2016).
Mohammad, T. et al. Differential gene expression and weighted correlation network dynamics in high-throughput datasets of prostate cancer. Front. Oncol. 12, 881246 (2022).
Solanki, R. et al. Combined transcriptomics and in-silico approach uncovers the role of prognostic biomarkers in hepatocellular carcinoma. Hum. Gene 35, 201154 (2023).
Ahmad, S. et al. Transcriptome meta-analysis deciphers a dysregulation in immune response-associated gene signatures during sepsis. Genes 10, 1005 (2019).
Arora, S. et al. Comprehensive integrative analysis reveals the association of KLF4 with macrophage infiltration and polarization in lung cancer microenvironment. Cells 10, 2091 (2021).
Mohsin, M. et al. Integrated transcriptomic and regulatory network analyses uncovers the role of let-7b-5p, SPIB, and HLA-DPB1 in sepsis. Sci. Rep. 12, 11963 (2022).
Shriwash, N. et al. Identification of differentially expressed genes in small and non-small cell lung cancer based on meta-analysis of mRNA. Heliyon 5, e01707 (2019).
Singh, P. et al. Survival-based biomarker module identification associated with oral squamous cell carcinoma (OSCC). Biology 10, 760 (2021).
Singh, P. et al. Network-based identification of signature genes KLF6 and SPOCK1 associated with oral submucous fibrosis. Mol. Clin. Oncol. https://doi.org/10.3892/mco.2020.1991 (2020).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015).
Bhattacharya, S. et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data 5, 180015 (2018).
Yu, G. & He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 12, 477–479 (2016).
Mushtaq, A. et al. Unravelling hub genes as potential therapeutic targets in lung cancer using integrated transcriptomic meta-analysis and in silico approach. J. Biomol. Struct. Dyn. 0, 1–14 (2022).
Keenan, A. B. et al. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224 (2019).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucl. Acids Res. 48, W509–W514 (2020).
Sjöstedt, E. et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947 (2020).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017).
Uhlen, M. et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 366, eaax9198 (2019).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017).
Uhlen, M. et al. Tissue-based map of the human proteome. Science 347, 1260419–1260419 (2015).
Clough, E. & Barrett, T. The gene expression omnibus database. Methods Mol. Biol. Clifton N.J. 1418, 93–110 (2016).
Tabassum, G. et al. Investigating the role of Kinesin family in lung adenocarcinoma via integrated bioinformatics approach. Sci. Rep. 13, 9859 (2023).
Singh, P., Gurung, R., Sultan, A. & Dohare, R. Understanding the role of adipokines and adipogenesis family in hepatocellular carcinoma. Egypt. J. Med. Hum. Genet. 24, 17 (2023).
Nagy, Á., Lánczky, A., Menyhárt, O. & Győrffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 8, 9227 (2018).
Nadal, E. et al. Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin. Cancer Res. 19, 6842–6852 (2013).
Zaravinos, A. The regulatory role of MicroRNAs in EMT and cancer. J. Oncol. 2015, 865816 (2015).
Saydam, O. et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol. Cell. Biol. 29, 5923–5940 (2009).
Movahedi, K. et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 70, 5728–5739 (2010).
Liu, J. et al. Increased CX3CL1 mRNA expression level is a positive prognostic factor in patients with lung adenocarcinoma. Oncol. Lett. 17, 4877–4890 (2019).
Liu, W. et al. Role of CX3CL1 in diseases. Arch. Immunol. Ther. Exp. (Warsz.) 64, 371–383 (2016).
Shulby, S. A., Dolloff, N. G., Stearns, M. E., Meucci, O. & Fatatis, A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 64, 4693–4698 (2004).
Liang, Y. et al. CX3CL1 involves in breast cancer metastasizing to the spine via the Src/FAK signaling pathway. J. Cancer 9, 3603–3612 (2018).
Liu, W., Liang, Y., Chan, Q., Jiang, L. & Dong, J. CX3CL1 promotes lung cancer cell migration and invasion via the Src/focal adhesion kinase signaling pathway. Oncol. Rep. 41, 1911–1917 (2019).
DiNatale, A. et al. Subsets of cancer cells expressing CX3CR1 are endowed with metastasis-initiating properties and resistance to chemotherapy. Oncogene 41, 1337–1351 (2022).
Liu, J.-F., Tsao, Y.-T. & Hou, C.-H. Fractalkine/CX3CL1 induced intercellular adhesion molecule-1-dependent tumor metastasis through the CX3CR1/PI3K/Akt/NF-κB pathway in human osteosarcoma. Oncotarget 8, 54136–54148 (2017).
Liu, P. et al. CX3CL1/fractalkine enhances prostate cancer spinal metastasis by activating the Src/FAK pathway. Int. J. Oncol. 53, 1544–1556 (2018).
Su, Y.-C. et al. Differential impact of CX3CL1 on lung cancer prognosis in smokers and non-smokers. Mol. Carcinog. 57, 629–639 (2018).
Liu, Y. et al. Clinical significance of expression level of CX3CL1–CX3CR1 axis in bone metastasis of lung cancer. Clin. Transl. Oncol. https://doi.org/10.1007/s12094-020-02431-6 (2020).
Andre, F. et al. Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 17, 945–951 (2006).
Shen, F. et al. Novel small-molecule CX3CR1 antagonist impairs metastatic seeding and colonization of breast cancer cells. Mol. Cancer Res. 14, 518–527 (2016).
Luo, W. et al. miRNA-296-3p modulates chemosensitivity of lung cancer cells by targeting CX3CR1. Am. J. Transl. Res. 8, 1848–1856 (2016).
Meucci, O., Fatatis, A., Simen, A. A. & Miller, R. J. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U.S.A. 97, 8075–8080 (2000).
Sansone, P. et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 7, 10442 (2016).
Zhu, N. et al. Challenging role of Wnt5a and its signaling pathway in cancer metastasis (review). Exp. Ther. Med. 8, 3–8 (2014).
Wang, H. et al. Fractalkine/CX3CR1 induces apoptosis resistance and proliferation through the activation of the AKT/NF-κB cascade in pancreatic cancer cells. Cell Biochem. Funct. 35, 315–326 (2017).
Du, W. et al. Spi-B–mediated silencing of claudin-2 promotes early dissemination of lung cancer cells from primary tumors. Cancer Res. 77, 4809–4822 (2017).
Zhang, H. et al. SPIB promotes anoikis resistance via elevated autolysosomal process in lung cancer cells. FEBS J. https://doi.org/10.1111/febs.15272 (2020).
Zhao, X. et al. SPIB acts as a tumor suppressor by activating the NFkB and JNK signaling pathways through MAP4K1 in colorectal cancer cells. Cell. Signal. 88, 110148 (2021).
Li, W., Cui, Y., Wang, D., Wang, Y. & Wang, L. MiR-141-3p functions as a tumor suppressor through directly targeting ZFR in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 509, 647–656 (2019).
Upton, M. P. et al. Expression of vimentin in surgically resected adenocarcinomas and large cell carcinomas of lung. Am. J. Surg. Pathol. 10, 560–567 (1986).
Roybal, J. D. et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol. Cancer Res. MCR 9, 25–35 (2011).
Mei, Z. et al. MicroRNA-141 promotes the proliferation of non-small cell lung cancer cells by regulating expression of PHLPP1 and PHLPP2. FEBS Lett. 588, 3055–3061 (2014).
Liu, Q. et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 31, 61–71 (2016).
Ha, H., Debnath, B. & Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 7, 1543–1588 (2017).
Khan, M. N. et al. CXCR1/2 antagonism with CXCL8/Interleukin-8 analogue CXCL8(3–72)K11R/G31P restricts lung cancer growth by inhibiting tumor cell proliferation and suppressing angiogenesis. Oncotarget 6, 21315–21327 (2015).
Li, J. et al. CXCL6 promotes non-small cell lung cancer cell survival and metastasis via down-regulation of miR-515-5p. Biomed. Pharmacother. 97, 1182–1188 (2018).
Chuntharapai, A., Lee, J., Hébert, C. A. & Kim, K. J. Monoclonal antibodies detect different distribution patterns of IL-8 receptor A and IL-8 receptor B on human peripheral blood leukocytes. J. Immunol. Baltim. Md 1950(153), 5682–5688 (1994).
Ramjeesingh, R., Leung, R. & Siu, C.-H. Interleukin-8 secreted by endothelial cells induces chemotaxis of melanoma cells through the chemokine receptor CXCR1. FASEB. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 17, 1292–1294 (2003).
Zhao, S., Shen, W., Yu, J. & Wang, L. TBX21 predicts prognosis of patients and drives cancer stem cell maintenance via the TBX21–IL-4 pathway in lung adenocarcinoma. Stem Cell Res. Ther. 9, 89 (2018).
Zheng, J. et al. Chemokine receptor CX3CR1 contributes to macrophage survival in tumor metastasis. Mol. Cancer 12, 141 (2013).
Lin, E. Y. et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66, 11238–11246 (2006).
Lin, E. Y. & Pollard, J. W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 67, 5064–5066 (2007).
Krijgsman, D., Hokland, M. & Kuppen, P. J. K. The role of natural killer T cells in cancer—A phenotypical and functional approach. Front. Immunol. 9, 367 (2018).
Chen, J. J. W. et al. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin. Cancer Res Off. J. Am. Assoc. Cancer Res. 9, 729–737 (2003).
Spaks, A. Role of CXC group chemokines in lung cancer development and progression. J. Thorac. Dis. 9, S164–S171 (2017).
Acknowledgements
Archana Sharma and Prithvi Singh would like to thank the Indian Council of Medical Research (ICMR), Government of India for awarding Research Associate with Grant Number: 5/3/8/2/ITR-F/2019-ITR and Senior Research Fellowship with Grant Number: BMI/11(89)/2020, respectively. Mansoor Ali Syed would like to thank ICMR for awarding him Extramural research grant (Grant Number: 5/30/59/2020/NCD3).
Author information
Authors and Affiliations
Contributions
A.S.: Conceptualization, Methodology, Software, Formal analysis, Data curation, Writing - Original Draft, Writing - Review & Editing, Validation, Visualization, Investigation. P.S.: Conceptualization, Methodology, Software, Formal analysis, Data curation, Writing - Original Draft, Writing - Review & Editing. R.J.: Software, Data curation, Writing - Review & Editing. S.A.A.: Writing - Review & Editing. F.A.: Writing - Review & Editing. A.H.R.: Writing - Review & Editing. H.O.A.: Writing - Review & Editing. R.D.: Writing - Review & Editing, Supervision, Project Administration. M.A.S.: Resources, Writing - Review & Editing, Supervision, Project Administration. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, A., Singh, P., Jha, R. et al. Exploring the role of miR-200 family in regulating CX3CR1 and CXCR1 in lung adenocarcinoma tumor microenvironment: implications for therapeutic intervention. Sci Rep 13, 16333 (2023). https://doi.org/10.1038/s41598-023-43484-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43484-1
This article is cited by
-
Recent advances on high-efficiency of microRNAs in different types of lung cancer: a comprehensive review
Cancer Cell International (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
