
Abstract
Aberrant levels of the G2/M cyclin cyclin B1 (gene CCNB1) have been associated with multiple cancers; however, the literature lacks a focused and comprehensive analysis of the regulation of this important regulator of cell proliferation in cancer. Through this work, we performed a pancancer analysis of the levels of CCNB1 and dissected aspects of regulation and how this correlates with cancer prognosis. We comprehensively evaluated the expression and promoter methylation of CCNB1 across 38 cancers based on RNA sequencing data obtained from the Cancer Genome Atlas (TCGA). The correlation of CCNB1 with prognosis and the tumor microenvironment was explored. Using lung adenocarcinoma data, we studied the potential upstream noncoding RNAs involved in the regulation of CCNB1 and validated the protein levels and prognostic value of CCNB1 for this disease site. CCNB1 was highly expressed, and promoter methylation was reduced in most cancers. Gene expression of CCNB1 correlated positively with poor prognosis of tumor patients, and these results were confirmed at the protein level using lung adenocarcinoma. CCNB1 expression was associated with the infiltration of T helper cells, and this further correlated with poor prognosis for certain cancers, including renal clear cell carcinoma and lung adenocarcinoma. Subsequently, we identified a specific upstream noncoding RNA contributing to CCNB1 overexpression in lung adenocarcinoma through correlation analysis, expression analysis and survival analysis. This study provides a comprehensive analysis of the expression and methylation status of CCNB1 across several forms of cancer and provides further insight into the mechanistic pathways regulating Cyclin B1 in the tumorigenesis process.
Similar content being viewed by others


Introduction
The mechanism of tumorigenesis is very complicated, and genetic changes play an important role in it. In recent years, whole-genome sequencing analysis has furthered the understanding of tumor molecular mechanisms and has played an important role in promoting genome-driven oncology care1,2. There may be some common mechanisms for the occurrence and development of different tumors, and it is meaningful to perform a pancancer expression analysis of different genes and explore their correlation with clinical characteristics and potential molecular mechanisms. The Cancer Genome Atlas (TCGA) database contains DNA, RNA, protein, and epigenetic datasets, thus allowing us to perform pancancer analysis3.
The cell cycle-related protein cyclin B1 (CCNB1) is an important member of the cyclin family, which refers to proteins that differ in their levels to activate specific cyclin-dependent kinases (CDKs) required for progression in the cell cycle4. Activated CCNB1 can promote cells to enter the M phase from the G2 phase and initiate mitotic progression5. An increasing number of studies have shown that CCNB1 is closely related to the abnormal proliferation of cells and the occurrence of tumors, such as CCNB1 overexpression in liver cancer, breast cancer, esophageal cancer, and cervical cancer6,7. However, there is still a lack of pancancer analysis on the relationship between CCNB1 and various tumors based on clinical databases. In this study, we first constructed a pancancer analysis of CCNB1 based on the TCGA database. Gene expression, tumor stage, survival status, immune infiltration and other factors were included in the analysis to explore the potential molecular mechanism of CCNB1 in different cancers.
Materials and methods
Gene expression analysis
The TIMER2 website (http://timer.cistrome.org/)8 was used to analyze the expression difference of CCNB1 in different tumors and adjacent normal tissues of the TCGA database. For some tumors without normal adjacent tissues in TIMER2, such as adrenocortical carcinoma (ACC), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), and testicular germ cell tumors (TGCT), the GEPIA2 (http://gepia2.cancer-pku.cn/#analysis)9 website was used to compare the expression of CCNB1 in tumor tissues and adjacent normal tissues and obtain box plots. In addition, violin plots of CCNB1 expression in different pathological stages of all TCGA tumors were obtained in the “Stage Plot” module of GEPIA2.
Survival prognosis analysis
The “survival analysis” module of GEPIA2 was used to obtain the overall survival (OS) and disease-free survival (DFS) significance map data of CCNB1 in all TCGA tumors, with 50% as the cutoff to split the high-expression and low-expression cohorts.
DNA methylation and immune infiltration analysis
The UALCAN portal (http://ualcan.path.uab.edu/analysis.html) was used to explore the DNA methylation level of CCNB1 between TCGA tumors and corresponding normal tissues. The “Immune-Gene” module of TIMER2 was used to explore the association between CCNB1 expression and immune infiltrates across all TCGA tumors. The P values and partial correlation values were obtained via the purity-adjusted Spearman’s rank correlation test.
CCNB1-related gene enrichment analysis
The available experimentally determined CCNB1-binding proteins were obtained from the STRING website (https://string-db.org/) with the following main parameters: minimum required interaction score “Low confidence (0.150)”, meaning of network edges “evidence”, active interaction sources (“experiments”) and max number of interactors to show “no more than 50 interactors”.
The “Similar Gene Detection” module of GEPIA2 was used to obtain the top 100 CCNB1-correlated targeting genes based on the TCGA database. Intersecting genes between CCNB1-binding proteins and the top 100 CCNB1-related genes were obtained through a Venn diagram. Kyoto Encyclopedia of Genes and Genomes (KEGG)10,11 pathway (Permission ID: 231307) and Gene Ontology (GO) analyses of the intersecting genes were performed and visualized with a bubble diagram. Finally, we showed the partial correlation and P value of intersecting genes in the purity-adjusted Spearman’s rank correlation test through a heatmap obtained from TIMER2 and performed survival analysis on all intersecting genes in Kaplan‒Meier Plotter (http://kmplot.com/analysis/index.php?p=service&cancer=pancancer_rnaseq).
Candidate microRNA prediction
The StarBase database (http://starbase.sysu.edu.cn/) was employed to predict the upstream binding miRNAs of CCNB1, and only the predicted microRNAs that commonly appeared in more than two programs (PITA, RNA22, miRmap, microT, miRanda, PicTar, and TargetScan) were included for subsequent analyses. Subsequently, the prognostic values of selected microRNAs were analyzed by the Kaplan‒Meier Plotter database, and candidate microRNAs were finally determined.
Identification of upstream long noncoding RNAs (lncRNAs) of candidate microRNAs
The predicted upstream lncRNAs of candidate microRNAs were obtained from the StarBase database. According to the competing endogenous RNA (ceRNA) hypothesis, lncRNAs were selected based on a negative relationship with candidate microRNAs (P value < 0.05, correlation coefficient < − 0.1) and a positive relationship with CCNB1 (P value < 0.05, correlation coefficient > 0.1) and were chosen based on the prognostic values determined by Kaplan‒Meier Plotter.
Patient tissue specimens
Tissue specimens from 60 LUAD patients and 11 benign pulmonary lesion (BPL) patients who underwent surgery from April 2014 to July 2017 were collected for tissue chips. Written informed consent was obtained from each patient (Supplementary File). None of the patients had a history of preoperative chemotherapy or radiotherapy. The project was approved by the Ethics Committee of Zhongnan Hospital of Wuhan University (approval number: 2021100K), and the informed consent requirement was waived. All methods were performed in accordance with the relevant guidelines and regulations.
Statistical analysis
Most of the statistical analyses in this study were calculated by the online database or tools mentioned above. A P value < 0.05 or log rank P value < 0.05 was considered statistically significant.
Results
Pancancer analysis of CCNB1 expression
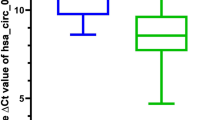
The expression differences of CCNB1 between tumor and adjacent normal tissues for the different tumors of the TCGA database are shown in Fig. 1A. For certain tumors without corresponding normal tissues in TCGA, the expression differences in the GTEx (Genotype-tissue expression) database were analyzed, and box plots were obtained via the GEPIA2 website (Fig. 1B). According to the results, CCNB1 was significantly upregulated in 23 cancer types compared with corresponding normal tissues, including adrenocortical carcinoma (ACC), bladder urothelial carcinoma (BLCA), bladder urothelial carcinoma (BRCA), cervical squamous cell carcinoma (CESC), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), ovarian serous cystadenocarcinoma (OV), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), sarcoma (SARC), stomach adenocarcinoma (STAD), testicular germ cell tumors (TGCT), uterine corpus endometrial carcinoma (UCEC), and uterine carcinoma (UCS).

Expression level of the CCNB11 gene in different cancers and pathological stages. (A) The expression levels of CCNB1 in different cancers or specific cancer subtypes were analyzed through TIMER2. *P < 0.05; **P < 0.01; ***P < 0.001. Red indicates tumor tissue, and blue indicates normal tissue. (B) The expression of CCNB1 in certain cancers without corresponding normal tissues in the TCGA project. *P < 0.05; **P < 0.01; ***P < 0.001. Red indicates tumor tissue, and blue indicates normal tissue. (C) Based on the TCGA data, the expression levels of CCNB1 were analyzed by the main pathological stages of ACC, BRCA, COAD, ESCA, KICH, KIRC, KIRP, LIHC, LUAD, and LUSC. Log2 (TPM + 1) was applied for log-scale. The ordinate is the expression amount of CCNB1.
Additionally, violin plots of CCNB1 expression in different pathological stages of all TCGA tumors were obtained via the “Pathological Stage Plot” module of HEPIA2. As shown in Fig. 1C, CCNB1 was significantly related to pathological stages in 10 types of tumors, including ACC, BRCA, COAD, ESCA, KICH, KIRC, KIRP, LIHC, LUAD, and LUSC.
The prognostic values of CCNB1 in pancancer
Survival analysis for CCNB1 in pancancer was conducted. Overall survival and disease-free survival were included. As shown in Fig. 2, highly expressed CCNB1 was linked to poor prognosis of OS for tumors of ACC (P < 0.001), KICH (P = 0.0018), KIRP (P = 0.0096), LGG (P < 0.001), LIHC (P < 0.001), LUAD (P < 0.001), MESO (P < 0.001), PAAD (P = 0.011), and SKCM (P = 0.025) within the TCGA project, while COAD and THYM patients with higher expression of CCNB1 indicated better prognosis (P = 0.041, 0.034). The DFS analysis data showed that high CCNB1 expression indicated poor prognosis among all cancer types (ACC (P = 0.011), HNSC (P = 0.042), KIRC (P = 0.0029), KIRP (P < 0.001), LGG (P = 0.0051), LIHC (P < 0.001), LUAD (P = 0.0079), MESO (P = 0.034), PAAD (P = 0.0091), PRAD (P = 0.0032), SARC (P = 0.05), and UVM (P = 0.0013)).

The prognostic value of CCNB1 for cancers in TCGA. The GEPIA2 tool was used to perform overall survival (A) and disease-free survival (B) analyses of different cancers in TCGA by CCNB1 expression. The Kaplan‒Meier curves with positive results are given.
Enrichment analysis of CCNB1-related genes
To further explore the molecular mechanism of CCNB1 in tumorigenesis, we attempted to identify CCNB1-binding proteins and SND112 expression-correlated genes for KEGG pathway and GO enrichment analyses. A total of 51 CCNB1-binding proteins supported by experimental evidence were obtained on the STRING website, and the protein‒protein interaction (PPI) network is shown in Fig. 3A. The top 100 CCNB1-related genes were obtained via the GEPIA2 tool. As shown in Fig. 3B, 10 intersecting genes (CCNA2, CCNB2, CDC20, CDC25C, PLK1, CDK1, CKS1B, ESPL1, CKS2, PCNA) between CCNB1-binding proteins and the top 100 CCNB1-related genes were obtained through a Venn diagram. The KEGG analysis of intersecting genes indicated that CCNB1 was mainly involved in the “cell cycle” in tumor pathogenesis (Fig. 3C). The results of GO analysis further indicated that most of the intersecting genes were involved in cell proliferation, cell cycle, cell division, and others (Fig. 3D–F). As shown in Fig. S3A, the expression level of CCNB1 was positively correlated with all 10 intersecting genes. Further survival analysis showed that high expression of all intersection genes was associated with poor prognosis of lung adenocarcinoma (Fig. S3B,C), and similar results were observed in KIRP and LIHC (Fig. S4).

CCNB1-related gene enrichment analysis. (A) Fifty-one experimentally determined CCNB1-binding proteins were identified via the STRING tool. (B) An intersection analysis of CCNB1-binding and the top 100 CCNB1-correlated genes was conducted, and 10 intersecting genes were obtained. (C) Based on the 10 intersecting genes, KEGG pathway analysis was performed. (D–F) The top 10 items of GO analysis: biological processes (F), cellular components (D), and molecular functions (E) of the 10 intersecting genes.
Prediction and analysis of upstream miRNAs of CCNB1
According to the StarBase database, 11 upstream microRNAs were identified.
According to the mechanism by which miRNAs regulate target gene expression, upstream microRNAs should be negatively correlated with CCNB1. As shown in Fig. 4A, CCNB1 was significantly negatively correlated with hsa-miR-181a-5p, hsa-miR-181b-5p, hsa-miR-181c-5p and hsa-miR-181d-5p and positively correlated with hsa-miR-183-5p and hsa-miR-93-5p. To further determine the upstream microRNAs, the correlation between microRNAs and the prognosis of lung adenocarcinoma was explored. The results showed that low expression of hsa-miR-181c-5p and hsa-miR-181d-5p was associated with poor prognosis of lung adenocarcinoma (P = 0.0014, 0.017, Fig. 4A,C). The above results suggested that hsa-miR-181c-5p and hsa-miR-181d-5p might be the most potential upstream microRNAs of CCNB1 in LUAD.

Identification of hsa-miR-181c-5p and hsa-miR-181d-5p as potential upstream microRNAs of CCNB1 in LUAD. (A) The expression correlation between predicted microRNAs and CCNB1 in LUAD analyzed by the starBase database. (B,C) The expression of hsa-miR-181c-5p and hsa-miR-181d-5p in lung adenocarcinoma was negatively correlated with CCNB1 (all P < 0.001), and high expression was associated with a better prognosis (P = 0.0014, 0.017).
Prediction and analysis of upstream lncRNAs of hsa-miR-181c-5p and hsa-miR-181d-5p
The upstream lncRNAs of hsa-miR-181c-5p and hsa-miR-181d-5p were predicted via the StarBase database. A total of 58 possible lncRNAs were forecasted. Based on the competing endogenous RNA (ceRNA) hypothesis, lncRNAs can increase the expression of target mRNAs by competitively binding to microRNAs. Therefore, LINC01468, which was not only negatively related to microRNA but also positively related to CCNB1, was chosen. As shown in Fig. 5B, LINC01468 was negatively related to hsa-miR-181c-5p (R = − 0.145, P < 0.001, Fig. 5A) and hsa-miR-181d-5p (R = − 0.126, P = 0.0043, Fig. 5B) and positively related to CCNB1 (R = 0.258, P < 0.001, Fig. 5C). Similarly, LINC01468 was highly expressed in lung adenocarcinoma (P < 0.001, Fig. 5D), and high expression was associated with poor prognosis (P = 0.042, Fig. 5E). As shown in Fig. 5E, we constructed a protein network based on the ceRNA hypothesis. In this network, the overexpression of LINC01468 caused a decrease in hsa-miR-181c-5p and hsa-miR-181d-5p. The deletion of hsa-miR-181c-5p and hsa-miR-181d-5p promoted the translation of CCNB1, which promoted the progression of LUAD by regulating the cell cycle.

Identification of LINC01468 as the potential upstream lncRNA of hsa-miR-181c-5p and hsa-miR-181d-5p in LUAD. Correlation analysis showed that the expression of LINC01468 in LUAD was negatively correlated with hsa-miR-181c-5p (A) and hsa-miR-181d-5p (B) but positively correlated with CCNB1 (C). (D) The expression of LINC01468 in LUAD was significantly higher than that in control normal samples. (E) The results of overall survival analysis showed that high expression of LINC01468 was related to poor prognosis. (F) CCNB1-related ceRNA regulation network.
DNA methylation and immune infiltration analysis
The DNA methylation level of CCNB1 between TCGA tumors and corresponding normal tissues was explored in the UALCAN portal. Contrary to the expression level of CCNB1, a reduced methylation level of CCNB1 was observed in most tumor tissues (BLCA, CESC, COAD, ESCA, HNSC, KIRC, KIRP, LIHC, LUAD, LUSC, PRAD, SARC, TGCT, THCA, UCEC) compared with corresponding normal tissues (Fig. S1). CCNB1 expression was positively related to Th2 cell infiltration in all tumors (Fig. S2A,B), and high Th2 cell infiltration in ACC, KIRC, LGG, LUAD, PAAD, and UVM suggested poor prognosis (Fig. S2B).
Validation in LUAD tissue
To verify the results of the above bioinformatics analysis, immunohistochemical (IHC) staining was performed on tissue microarray slides containing 60 LUAD tissues and 11 normal tissues. The results showed that CCNB1 was highly expressed in LUAD compared to normal tissue (P < 0.001, Fig. 6A,D). Furthermore, the expression of CCNB1 was related to TNM and T stage (P = 0.0246, 0.0033, Fig. 6B,C). Survival analysis in patients with LUAD showed that patients with high CCNB1 expression had a poorer prognosis (P = 0.048, Fig. 6E). The above results were all consistent with the results of the bioinformatics analysis.

Validation of CCNB1 in LUAD. (A) Immunohistochemical results of the tissue chip showed that the expression level of CCNB1 in LUAD was significantly higher than that in normal tissues, and the expression of CCNB1 was correlated with TNM (B) and T stages (C). (D) Representative immunohistochemical staining showing CCNB1 expression in different LUAD stages. (E) The Kaplan‒Meier results showed that high CCNB1 expression was related to poor prognosis.
Discussion
Increasing evidence shows that CCNB1 is overexpressed in most human tumors4, which is consistent with the results of this study (Fig. 1A,B). Our study also showed that CCNB1 was significantly related to OS and DFS (Fig. 2). Recent studies have shown that in addition to directly regulating the cell cycle, CCNB1 can also coordinate the cell cycle by enhancing mitochondrial activity, which is closely related to the pathological characteristics of tumors13. A correlation between CCNB1 and tumor stage was observed in this study (Fig. 1C). We explored the potential mechanism of CCNB1 in tumors through the tumor microenvironment (TME) and DNA methylation.
TP53 gene mutations are observed in most human malignancies and play an important role in tumorigenesis14. Among the 10 CCNB1-related genes, CCNB2 and CDK1 participated in the P53 pathway according to the results of enrichment analysis (Fig. 3). A study in pancreatic cancer showed that CCNB1 silencing inhibited cell proliferation and induced cell apoptosis via the p53 signaling pathway4,15. A recent study indicated that P53 can inhibit the transcription of CCNB2, CDK, and other cyclin genes through the p53-p21-DREAM-CDE/CHR pathway, leading to cell cycle arrest and apoptosis16. Cyclin-dependent kinase 1 encoded by CDK1 is the only kinase essential for cell cycle regulation17. Cyclin A2 (CCNA2) is also an important molecule in the cyclin family. Studies in BRCA showed that the dysregulated expression of CDK1/CCNA2/CCNB1 was related to prognosis18. Other CCNB1-related genes have shown different diagnostic significance in different studies19,20,21,22,23.
Recent studies have fully confirmed the regulatory role of noncoding RNAs (microRNAs, lncRNAs, circular RNAs, etc.) in cancers24. Competing endogenous RNAs link the function of protein-coding mRNAs with noncoding RNAs and can regulate the expression level of each other by competing for shared microRNAs25. Based on the good performance of CCNB1 in multiple analyses of lung adenocarcinoma, we mainly explored the ceRNA regulatory mechanism of CCNB1 in lung adenocarcinoma. Hsa-miR-181c-5p and hsa-miR-181d-5p were recognized as CCNB1 upstream regulatory microRNAs in lung adenocarcinoma. Research has shown that hsa-miR-181c-5p is involved in the target pathway of cell proliferation and the cell cycle in pediatric cancer stem cells26. In breast cancer, head and neck squamous cell carcinoma, and other tumor studies, it has been shown that hsa-miR-181c-5p affects the prognosis of tumors by regulating the expression of multiple genes27,28,29. Research has shown that hsa-miR-181d-5p can repress the activation/phosphorylation of the transcription factors STAT3/5A and further inhibit the progression of glioblastoma30. Another in vitro study showed that overexpression of hsa-miR-181d significantly inhibits cell proliferation, migration, and cell cycle progression in esophageal squamous cell carcinoma31. Hsa-miR-181c-5p and hsa-miR-181d-5p both showed strong predictive effects on the prognosis of lung adenocarcinoma in this study (Fig. 4B,C). This study shows that LINC01468 is significantly related to the prognosis of lung adenocarcinoma. Similar results were observed in bladder urothelial carcinoma, and the research showed that the combined detection of LINC01468 and other lncRNAs had important predictive significance for bladder urothelial carcinoma32. According to the results of this study, hsa-miR-181c-5p and hsa-miR-181d-5p inhibited the expression of CCNB1 in LUAD, while LINC01468 promoted the expression of CCNB1 through competitive sharing of hsa-miR-181c-5p and hsa-miR-181d-5p.
In addition to genetics, epigenetic changes also play an important role in carcinogenesis, and DNA methylation is an important part of epigenetics33. DNA methylation is often described as a ‘silencing’ epigenetic mark and is involved in adding stability to the repression of transcription34. Changes in promoter methylation and histone acetylation levels are the result of imbalances in enzyme balance and ultimately lead to tumor progression35. According to the results of the study, CCNB1 was highly expressed in tumor tissues and was associated with a poor prognosis; therefore, we hypothesized that CCNB1 might be an oncogene. Compared with corresponding normal tissues, the promoter methylation level of CCNB1 in tumor tissues was significantly reduced (Fig. S1), which led to a decrease in the stability of the repression of transcription and ultimately led to an increase in the expression level of CCNB1 and promoted tumor progression.
The important role of the immune system in cancer control is beyond doubt36. Helper T cells (Th) are the key immune cells for regulating immunity and are divided into at least four subsets, T regulatory, Th1, Th2, and Th17 cells, which are jointly involved in regulating the immune response37. The results of this study showed that CCNB1 was positively correlated with Th2 cell infiltration in all TCGA tumors (Fig. S2A). Studies have shown that Th2 cells have both antitumor and protumor effects in the TME38. Th2 cell-mediated type 2 immune responses can contribute to antitumor immunity, while Th2 cells promote tumor progression through secreted cytokines38,39,40. In this study, high Th2 cell infiltration in ACC, KIRC, LGG, LUAD, PAAD, and UVM suggested a poor prognosis (Fig. S2B). The above results indicated that CCNB1 was related to Th2 cell infiltration in certain tumors, such as AAC and LUAD, and affected prognosis. In conclusion, our study indicated that the expression of CCNB1 is widely increased in many tumor types, and the expression is positively correlated with the poor prognosis of patients. At the same time, CCNB1 may be an oncogene, and it may affect the tumor microenvironment through Th2 cells. Furthermore, the LINC01468-hsa-miR-181c-5p/hsa-miR-181d-5p-CCNB1 network may play an important role in LUAD progression.
Data availability
GEPIA2 (http://gepia2.cancer-pku.cn/#analysis), UALCAN portal (http://ualcan.path.uab.edu/analysis.html), STRING website (https://string-db.org/), Kaplan–Meier Plotter (http://kmplot.com/analysis/index.php?p=service&cancer=pancancer_rnaseq), StarBase database (http://starbase.sysu.edu.cn/).
Abbreviations
- ACC:
-
Adrenocortical carcinoma
- BPL:
-
Benign pulmonary lesions
- BLCA:
-
Bladder urothelial carcinoma
- BRCA:
-
Bladder urothelial carcinoma
- CCNB1:
-
Cell cycle-related proteins cyclin B1
- CDKs:
-
Cyclin-dependent kinases
- CeRNA:
-
Competing endogenous RNA
- CESC:
-
Cervical squamous cell carcinoma
- CHOL:
-
Cholangio carcinoma
- COAD:
-
Colon adenocarcinoma
- DFS:
-
Disease-free survival
- DLBC:
-
Lymphoid neoplasm diffuse large B-cell lymphoma
- ESCA:
-
Esophageal carcinoma
- GBM:
-
Glioblastoma multiforme
- GO:
-
Gene Ontology
- HNSC:
-
Head and neck squamous cell carcinoma
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- KIRC:
-
Kidney renal clear cell carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LIHC:
-
Liver hepatocellular carcinoma
- LncRNAs:
-
Long noncoding RNAs
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- OV:
-
Ovarian serous cystadenocarcinoma
- OS:
-
Overall survival
- PRAD:
-
Prostate adenocarcinoma
- READ:
-
Rectum adenocarcinoma
- SARC:
-
Sarcoma
- STAD:
-
Stomach adenocarcinoma
- TCGA:
-
The Cancer Genome Atlas
- TGCT:
-
Testicular germ cell tumors
- UCEC:
-
Uterine corpus endometrial carcinoma
- UCS:
-
Uterine carcinosarcoma
- PPI:
-
Protein‒protein interaction
- IHC:
-
Immunohistochemical
- THYM:
-
Thymoma
- MESO:
-
Mesothelioma
References
International Cancer Genome Consortium. International network of cancer genome projects. Nature 464, 993. https://doi.org/10.1038/nature08987 (2010).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216. https://doi.org/10.1038/s41586-019-1689-y (2019).
Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120. https://doi.org/10.1038/ng.2764 (2013).
Zhang, H. et al. Effect of CCNB1 silencing on cell cycle, senescence, and apoptosis through the p53 signaling pathway in pancreatic cancer. J. Cell Physiol. 234, 619–631. https://doi.org/10.1002/jcp.26816 (2018).
Wang, Z. et al. Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev. Cell 29, 217–232. https://doi.org/10.1016/j.devcel.2014.03.012 (2014).
Egloff, A. M., Vella, L. A. & Finn, O. J. Cyclin B1 and other cyclins as tumor antigens in immunosurveillance and immunotherapy of cancer. Cancer Res. 66, 6–9. https://doi.org/10.1158/0008-5472.CAN-05-3389 (2006).
Jackman, M. et al. Cyclin B1-Cdk1 facilitates MAD1 release from the nuclear pore to ensure a robust spindle checkpoint. J. Cell Biol. 219, 7082. https://doi.org/10.1083/jcb.201907082 (2020).
Lin, S. et al. Cost-effectiveness of atezolizumab plus chemotherapy for advanced non-small-cell lung cancer. Int. J. Clin. Pharm. 42, 1175–1183. https://doi.org/10.1007/s11096-020-01076-3 (2020).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560. https://doi.org/10.1093/nar/gkz430 (2019).
Kanehisa, M. et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Cui, X. et al. A pan-cancer analysis of the oncogenic role of staphylococcal nuclease domain-containing protein 1 (SND1) in human tumors. Genomics 112, 3958–3967. https://doi.org/10.1016/j.ygeno (2020).
Xie, B., Wang, S., Jiang, N. & Li, J. J. Cyclin B1/CDK1-regulated mitochondrial bioenergetics in cell cycle progression and tumor resistance. Cancer Lett. 443, 56–66. https://doi.org/10.1016/j.canlet.2018.11.019 (2019).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harbor Persp. Biol. 2, a001008. https://doi.org/10.1101/cshperspect.a001008 (2010).
Innocente, S. A. et al. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. U.S.A. 96, 2147–2152. https://doi.org/10.1073/pnas.96.5.2147 (1999).
Fischer, M., Quaas, M., Steiner, L. & Engeland, K. The p53–p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 44, 164–174. https://doi.org/10.1093/nar/gkv927 (2016).
Santamaria, D. et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815. https://doi.org/10.1038/nature06046 (2007).
Xing, Z. et al. Expression and prognostic value of CDK1, CCNA2, and CCNB1 gene clusters in human breast cancer. J. Int. Med. Res. 49, 300060520980647. https://doi.org/10.1177/0300060520980647 (2021).
Hwang, J. S. et al. MicroRNA-1258 inhibits the proliferation and migration of human colorectal cancer cells through suppressing CKS1B expression. Genes 10, 0912. https://doi.org/10.3390/genes10110912 (2019).
Pirim, D. Integrative analyses of molecular pathways and key candidate biomarkers associated with colorectal cancer. Cancer Biomark. 27, 555–568. https://doi.org/10.3233/CBM-191263 (2020).
Xu, J. H., Wang, Y. & Xu, D. CKS2 promotes tumor progression and metastasis and is an independent predictor of poor prognosis in epithelial ovarian cancer. Eur. Rev. Med. Pharmacol. Sci. 23, 3225–3234. https://doi.org/10.26355/eurrev_201904_17681 (2019).
Yang, Q., Yu, B. & Sun, J. TTK, CDC25A, and ESPL1 as prognostic biomarkers for endometrial cancer. Biomed. Res. Int. 2020, 4625123. https://doi.org/10.1155/2020/4625123 (2020).
Zheng, C. & Yang, R. RCD24, B7–H4 and PCNA expression and clinical significance in ovarian cancer. J. BUON 24, 715–719 (2019).
Chan, J. J. & Tay, Y. Noncoding RNA:RNA regulatory networks in cancer. Int. J. Mol. Sci. 19, 1310. https://doi.org/10.3390/ijms19051310 (2018).
Qi, X. et al. ceRNA in cancer: Possible functions and clinical implications. J. Med. Genet. 52, 710–718. https://doi.org/10.1136/jmedgenet-2015-103334 (2015).
Sanchez-Diaz, P. C. et al. De-regulated microRNAs in pediatric cancer stem cells target pathways involved in cell proliferation, cell cycle and development. PLoS ONE 8, e61622. https://doi.org/10.1371/journal.pone.0061622 (2013).
Liu, X. et al. Epithelial splicing regulatory protein 1 is overexpressed in breast cancer and predicts poor prognosis for breast cancer patients. Med. Sci. Monit. 27, e931102. https://doi.org/10.12659/MSM.931102 (2021).
Pereira, T. et al. MicroRNA profiling reveals dysregulated microRNAs and their target gene regulatory networks in cemento-ossifying fibroma. J. Oral Pathol. Med. 47, 78–85. https://doi.org/10.1111/jop.12650 (2018).
Sannigrahi, M. K. et al. DNA methylation regulated microRNAs in HPV-16-induced head and neck squamous cell carcinoma (HNSCC). Mol. Cell Biochem. 448, 321–333. https://doi.org/10.1007/s11010-018-3336-6 (2018).
Liu, H. W. et al. Enhanced Hsa-miR-181d/p-STAT3 and Hsa-miR-181d/p-STAT5A ratios mediate the anticancer effect of garcinol in STAT3/5A-addicted glioblastoma. Cancers 11, 888. https://doi.org/10.3390/cancers11121888 (2019).
Li, D. et al. MicroRNA-181d is a tumor suppressor in human esophageal squamous cell carcinoma inversely regulating Derlin-1. Oncol. Rep. 36, 2041–2048. https://doi.org/10.3892/or.2016.5028 (2016).
Gao, X. et al. Development of a novel six-long noncoding RNA signature predicting survival of patients with bladder urothelial carcinoma. J. Cell Biochem. 120, 19796–19809. https://doi.org/10.1002/jcb.29285 (2019).
Shen, H. & Laird, P. W. Interplay between the cancer genome and epigenome. Cell 153, 38–55. https://doi.org/10.1016/j.cell.2013.03.008 (2013).
Jones, P. A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. https://doi.org/10.1038/nrg3230 (2012).
Giacinti, L., Vici, P. & Lopez, M. Epigenome: A new target in cancer therapy. Clin. Terap. 159, 347–360 (2008).
Bruni, D., Angell, H. K. & Galon, J. The immune contexture and immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680. https://doi.org/10.1038/s41568-020-0285-7 (2020).
Li, Z., Zhang, Y. & Sun, B. Current understanding of Th2 cell differentiation and function. Protein Cell 2, 604–611. https://doi.org/10.1007/s13238-011-1083-5 (2011).
Schreiber, S. et al. Metabolic interdependency of Th2 cell-mediated type 2 immunity and the tumor microenvironment. Front. Immunol. 12, 632581. https://doi.org/10.3389/fimmu.2021.632581 (2021).
Aspord, C. et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J. Exp. Med. 204, 1037–1047. https://doi.org/10.1084/jem.20061120 (2007).
Prokopchuk, O., Liu, Y., Henne-Bruns, D. & Kornmann, M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: Evidence for autocrine and paracrine actions. Br. J. Cancer 92, 921–928. https://doi.org/10.1038/sj.bjc.6602416 (2005).
Acknowledgements
The authors thank all our colleagues who helped us during the current study. Thank you very much for the technical support provided by Mr. Pengju Gong.
Funding
This project was supported by the Program of Excellent Doctoral (Postdoctoral) of Zhongnan Hospital of Wuhan University (Grant No. ZNYB2019002), and Natural Science Foundation of Hubei Province (2020CFB703).
Author information
Authors and Affiliations
Contributions
X.L.C., W.Y.H. and T.H.X. conceived and designed the study. X.L.C. and W.Y.H. contributed to the literature search and data collection. X.L.C., D.P., W.Y.H., T.H.X., Z.X.F., and Z.J.P. contributed to data interpretation. X.L.C., W.Y.H., Z.J.P., and T.H.X. contributed to the figures and writing of the report. D.P., W.X.Y. contributed to revision of the manuscript. Z.J.P. directed the revision of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dai, P., Xiong, L., Wei, Y. et al. A pancancer analysis of the oncogenic role of cyclin B1 (CCNB1) in human tumors. Sci Rep 13, 16226 (2023). https://doi.org/10.1038/s41598-023-42801-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42801-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
