
Abstract
The long noncoding RNA (lncRNA) LUCAT1 was recently reported to be upregulated and to play an essential role in multiple cancer types, especially colorectal cancer (CRC), but the molecular mechanisms of LUCAT1 in CRC are mostly unreported. Here, a systematic analysis of LUACT1 expression is performed with data from TCGA database and clinic CRC samples. LUCAT1 is identified as a putative oncogene, which is significantly upregulated in CRC and is associated with poor prognosis. Loss of LUCAT1 restricts CRC proliferative capacities in vitro and in vivo. Mechanically, NCL is identified as the protein binding partner of LUCAT1 by using chromatin isolation by RNA purification coupled with mass spectrometry (ChIRP-MS) and RNA immunoprecipitation assays. We also show that NCL directly binds to LUCAT1 via its putative G-quadruplex-forming regions from nucleotides 717 to 746. The interaction between LUCAT1 and NCL interferes NCL-mediated inhibition of MYC and promote the expression of MYC. Cells lacking LUCAT1 show a decreased MYC expression, and NCL knockdown rescue LUCAT1 depletion-induced inhibition of CRC cell proliferation and MYC expression. Our results suggest that LUCAT1 plays a critical role in CRC cell proliferation by inhibiting the function of NCL via its G-quadruplex structure and may serve as a new prognostic biomarker and effective therapeutic target for CRC.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most common malignancy and the third leading cause of cancer-related death worldwide1,2. The CRC morbidity and mortality rates in China have steadily increased3. Therefore, it is a priority to increase our understanding of the mechanisms that drive CRC progression and to identify diagnostic and therapeutic targets to treat CRC.
Long noncoding RNA (lncRNA) is defined as noncoding RNA (ncRNA) sequences that are longer than 200 nucleotides4. Accumulating evidence indicates that lncRNAs play critical roles in multiple cellular and biological processes, including cell development, differentiation, growth, tumorigenesis, and metastasis5,6. LncRNAs execute their functions in the cytoplasm or nucleus through diverse mechanisms, such as protein binding, chromatin modification, cytoplasmic scaffolding, and RNA decay, acting as regulators of gene expression7,8,9,10. To date, several important lncRNAs, such as CRNDE11, SNHG712, and lncRNA-FEZF1-AS113, have been shown to participate in CRC development and can be regarded as potential, new diagnostic, and therapeutic targets. Lung cancer-associated transcript 1 (LUCAT1), also known as SCAL1, was first identified in smoking-induced lung cancer cells14. LUCAT1 dysregulation was observed in various cancers, including non-small-cell lung cancer, glioma, renal cell carcinoma, esophageal squamous cell carcinoma, prostate cancer, cutaneous squamous cell carcinoma, and CRC15,16,17,18,19,20,21. However, the specific function of LUCAT1 and its regulatory mechanism in CRC proliferation remain largely unknown.
Nucleolin (NCL) is one of the most multifunctional RNA-binding proteins (RBPs). It is most abundant in the nucleolus and has been demonstrated to contribute to G-quadruplex (G4) formation in the promoter regions of some oncogenes. Studies have demonstrated that G4 is a negative regulator of transcription22. Small RNAs have been shown to be involved in changes in the G4 structure or to act as molecular decoys for G4-binding proteins, altering gene expression, or inhibiting protein activity23,24. However, studies touching upon whether lncRNAs can regulate the G4 formation of oncogenes are rarely reported, and it is unclear whether LUCAT1 could function along with NCL to regulate G4-associated gene expression and CRC progression.
In this study, we found that LUCAT1 upregulation in CRC leads to cancer cell proliferation. The colocalization and binding of LUCAT1 and NCL suggest that LUCAT1 plays a regulatory role by antagonizing the function of NCL, leading to an increase in MYC expression. Overall, our results provide new evidence that LUCAT1 exerts an oncogenic function, and the LUCAT1/NCL/MYC axis might be a potential prognostic marker and therapeutic target for CRC.
Materials and methods
Cell lines and cell culture
SW620 (RRID: CVCL_0547) and SW480 (RRID: CVCL_0546) cells were cultured in L15 (KeyGEN BioTECH, Jiangsu, China) medium supplemented with 10% fetal bovine serum (FBS, Biological Industries, Israel) and 1% antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin; Life Technologies, Inc., Grand Island, NY, USA) and cultured at 37 °C in 5% CO2. SW480 (primary colon adenocarcinoma) and SW620 (subsequent lymph node metastasis) cells were derived from the same patient. HCT116 (RRID: CVCL_0291), HT29 (RRID: CVCL_0320), and NCM460 (RRID: CVCL_0460) cells were cultured in McCoy’s 5 A (KeyGEN BioTECH) medium supplemented with 10% FBS and 1% antibiotics and cultured at 37 °C in 5% CO2. All cell lines were obtained from KeyGEN BioTECH (Jiangsu, China). All cell lines have been authenticated using STR (or SNP) profiling within the last 3 years. All experiments were performed with mycoplasma-free cells.
Patients and tissue sampling
CRC tissues and adjacent normal tissues used were obtained from the 36 patients who underwent surgical resection for CRC at the Third XiangYa Hospital of Central South University (Changsha, China) after informed consent was obtained. The study was approved by the ethics committee of the Third XiangYa Hospital of Central South University (No. 2016-S086). Before the surgical resections, no preoperative treatment was administered. Tissue specimens were collected during surgery and immediately stored in liquid nitrogen. All tissue samples were diagnosed and confirmed by a pathologist.
Lentivirus infection and transfection
Lentiviruses, including Cas9, sgRNA, and scramble (negative control) for LUCAT1, were obtained from GeneChem (Shanghai, China) for in vivo experiments. Cells were seeded at 60–70% confluence and infected with lentivirus culture (at an MOI of 10). The medium was replaced with fresh medium containing puromycin after 24 h, and noninfected cells were eliminated with puromycin. To generate stable lentivirus-transduced lines, cells were infected with virus and polybrene following the manufacturer’s recommendations, and stable cell lines were selected with 4 µg/ml puromycin treatment after 72 h of transfection. The efficiency in different cells was determined by qRT-PCR and Western blot. The shRNAs for NCL were obtained from RiboBio Co., LTD. (Guangzhou, China). The shRNAs were transfected with HiPerFect Transfection Reagent (Qiagen, Hilden, Germany). For each well of a 6-well plate (7 × 105 cells/well), 10 µL of shRNA (20 µM) and an RNAi negative control were diluted in 100 µL of serum-free culture medium, and then 12 µL of HiPerFect Transfection Reagent was added. The mixture was incubated for 10 min at room temperature; then, the mixture was added dropwise onto cells, and the plate was swirled gently. The cells were incubated for an additional 72 h for further analysis. All sequences are provided in Supplementary Table 1.
Quantitative real-time PCR assays
Total RNA from cells and tissues was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was synthesized using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). Quantitative real-time PCR (qRT-PCR) was performed on a LightCycler 480 Real-Time PCR instrument (Roche, Basel, Switzerland) using SYBR Green Real-time PCR Master Mix (TOYOBO). Each sample was quantified in triplicate, and the experiment was repeated three times. Analysis was performed using the 2−∆∆Ct method, with GAPDH as the endogenous control. All primer pairs were purchased from Sangon Biotech (Shanghai, China), and sequences are provided in Supplementary Table 2.
Genomic PCR assays
Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen). PCR was performed on an Applied Biosystems instrument using Taq PCR StarMix (GenStar, Beijing, China). The amplification products were detected using 1% agarose gel electrophoresis and visualized on a Syngene Bio Imaging system. Primer sequences are provided in Supplementary Table 2.
Cell proliferation and colony formation assays
A cell counting kit 8 (CCK8) assay (Dojindo, Kumamoto, Japan) was used to measure cell proliferation in 96-well plates. Cells were seeded at 4 × 103 or 8 × 103 cells per well, with five replicates for each condition. CCK8 was added at 0, 24, 48, and 72 h and incubated at 37 °C for 2 h. Cell numbers were determined by measuring the absorbance at 450 nm using a 96-well format plate reader (Perkin Elmer, MA, USA). For colony formation assays, 500 cells were seeded in each well of a 6-well plate in triplicate for each condition and incubated for 10 or 14 days. The colonies were fixed with methanol, stained with crystal violet, and counted. The average colony counts were calculated, and a paired t-test was used to test statistical significance. Each experiment was repeated three times.
RNA immunoprecipitation
RIP experiments were carried out with the Magna RIP Kit (Millipore, #17-700, CA, USA) according to the manufacturer’s instructions using 5 µg of a rabbit anti-NCL antibody (Cell Signaling Technology, MA, USA, #14574) or rabbit IgG. Proteins isolated from beads were analyzed by Western blot. The coprecipitated RNAs were detected by real-time PCR.
Chromatin isolation by RNA purification (ChIRP) assays
ChIRP experiments were performed using the Magna ChIRP RNA Interactome Kit (Millipore, #17-10494) according to the manufacturer’s protocol. The cells were crosslinked using 1% glutaraldehyde and then sonicated at high intensity 40 times for 15-s pulses and 1-min intervals at 4 °C. The lengths of the sheared DNA were confirmed by 1% agarose gel electrophoresis analysis. The probes (100 pmol) for LUCAT1 were used for hybridization with sonicated cell lysate and streptavidin-coated magnetic beads. For RNA isolation, TRIzol Reagent and a Qiagen miRNeasy Mini Kit (Qiagen) were used. DNA isolation was performed according to a standard protocol. For protein collection, 3% formaldehyde was used for crosslinking, and the isolated proteins were washed and heated several times. The proteins were analyzed by Western blot. Probe sequences are provided in Supplementary Table 3.
RNA fluorescence in situ hybridization (FISH) and immunofluorescence
The cell smears were prepared for FISH using the standard methods. After fixation with 4% formaldehyde at room temperature for 10 min, the slides were permeabilized with 0.5% Triton X-100 at 4 °C for 10 min. The slides were incubated overnight at 37 °C in hybridization solution with the FISH probes. After hybridization, the slides were washed with wash buffer I (4 × SCC, 0.1% Tween-20), wash buffer II (2 × SCC), wash buffer III (1 × SCC) and PBS for 5 min at 42 °C. The slides were stained with 1 mg/ml 4′,6-diamidino-2-phenylindole (DAPI) for 10 min and then washed three times for 3 min. For colocalization studies, after RNA FISH, immunofluorescence was performed according to a standard protocol. A rabbit anti-NCL antibody (Cell Signaling Technology, #14574, 1:100) was used to detect the colocalization of LUCAT1 with NCL in CRC cells. Images were taken with a microscope (Olympus). All experiments were repeated three times. All FISH probes were commercially synthesized by RiboBio Co., LTD.
Western blot assays
Whole-cell lysates were prepared in 1 × RIPA buffer (KeyGEN) containing 1% PMSF (KeyGEN). The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE), transferred to polyvinylidene fluoride membranes (Millipore), blocked with 5% skim milk for 1 h at room temperature, immunoblotted with primary antibodies overnight and secondary antibodies for 1 h, and visualized on an Odyssey CLx Infrared Imaging System (LI-COR Biosciences, NE, USA). Antibodies used for Western blot analysis are provided in Supplementary Table 4.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were carried out using the EZ-ChIP Kit (Millipore, #17-371) according to the manufacturer’s protocol. Briefly, the cells were grown to 90% confluence in a 150 cm2 culture dish and crosslinked with 1% formaldehyde at room temperature for 10 min. Glycine (10×) was used to quench excess formaldehyde, and the cells were scraped and pelleted. Chromatin in lysis buffer was sonicated on ice to 200-1000 bp with a Vibra-Cell sonicator (Sonics & Materials, Inc., CT, USA) at 75% intensity two times (65% once and 55% once for 10-s pulses and 50-s intervals). The sheared DNA lengths were confirmed by agarose gel electrophoresis, and the chromatin was precleared with protein G-agarose beads (50% slurry). Precleared chromatin was incubated with an anti-NCL antibody (Novus Biologicals, CO, USA, NB600-241) or IgG and rotated at 4 °C for 12 h. Elution and reverse crosslinking of the protein-DNA complexes were performed according to the manufacturer’s protocol. Spin columns were used for the ChIP DNA extraction. Standard PCRs were performed, and products were detected using 1% agarose gel electrophoresis and visualized on a Syngene Bio Imaging system. The PCR primers used to amplify the promoter region are provided in Supplementary Table 5.
Luciferase reporter assays
Cells were seeded at a density of 8 × 104 cells per well in a 24-well plate. Cells were transfected with the reporter plasmid sh-NCL or the corresponding shRNA negative control and the LUCAT1 overexpression plasmid or the corresponding negative control plasmid by using Lipofectamine 2000 according to the manufacturer’s instructions (Invitrogen). The Renilla luciferase sequence in the pRL-TK vector (Promega, WI, USA) was used as an internal control. The luciferase activity was measured 24 h after transfection by using a Luciferase Assay System (Promega) with a luminometer (Perkin Elmer). The firefly luciferase activity was normalized to the Renilla luciferase activity. The data are expressed as the percent of luciferase activity in control cells (100%). All plasmids were commercially synthesized by RiboBio Co., LTD., and their information is provided in Supplementary Table 9.
Nude mouse xenograft tumor growth model and immunohistochemistry assays
Experiments with animals were performed in accordance with the guidelines for experimental animal management established by Kagawa University and guidelines for the welfare and use of animals in cancer research25. Experiments were carried out under pathogen-free conditions with randomly chosen littermates of the same sex, matched by age and body weight. The health status of mouse lines was routinely checked by veterinary staff. Five-week-old male BALB/c nude mice (nu+/nu+) were obtained from the Department of Laboratory Animals of Central South University. To investigate tumor growth in vivo, 2 × 106 cells were harvested and injected subcutaneously into the left or right flank of nude mice (n = 5 per group). After 30 days of the injection, the mice were killed by an overdose of pentobarbital (250 mg/kg, intraperitoneal injection), and the tumors were measured with calipers and an electronic scale to estimate the tumor volume and weight, respectively. The tumor volumes were calculated based on the formula: volume (mm3) = length (mm) × width (mm) × width (mm)/2. Tumors were further embedded in paraffin for H&E and IHC.
Bioinformatics analysis
Gene expression analysis of LUCAT1 was carried out using the starBase database v3.0 project (http://starbase.sysu.edu.cn)26. The survival analysis of LUCAT1 was carried out using the Gene Expression Profiling Interaction Analysis database (http://gepia.cancer-pku.cn)27. The relationship between LUCAT1 and NCL was predicted with the RNA-Protein Interaction Prediction software (http://pridb.gdcb.iastate.edu/RPISeq/)28. The potential binding sites between LUCAT1 and NCL were predicted with the QGRS Mapper software (http://bioinformatics.ramapo.edu/QGRS/index.php)29.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 to evaluate differences among experimental groups. Data were presented as mean ± s.d. of three independent experiments except where otherwise indicated. All data met the assumptions of the tests (e.g., normal distribution). Student’s t-test was used to analyze the assays. One-way analysis of variance (ANOVA) was used for comparison between the different groups. The relationship between gene expression and clinical pathological indicators is examined through chi-square test. The results with p < 0.05 were considered statistically significant. One asterisk, two asterisks, three asterisks and four asterisks indicate p < 0.05, p < 0.01, p < 0.001 and p < 0.0001, respectively. No samples or animals were excluded. No statistical methods were used to predetermine sample sizes. Sample sizes were similar to those generally employed in the field.
Results
LUCAT1 is upregulated in CRC and correlates with a poor prognosis
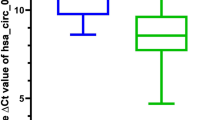
XLOC_004924, also named LUCAT1, was identified as one of the most significantly upregulated lncRNAs in CRC based on the gene profiling data that we have previously reported11. According to the ENSEMBL database, there are 61 transcriptomes of LUCAT1, and all isoforms are noncoding RNAs. LUCAT1-201 (ENST00000511918.6) was reported to be one of the functional transcripts, and its sequence was consistent with XLOC_004924. For the remainder of the manuscript, all instances where LUCAT1 is mentioned were LUCAT1-201. Next, we found that LUCAT1 was also highly expressed in CRC according to the data from starBase v3.0 (Fig. 1A). In addition, LUCAT1 upregulation was also reported in other cancer types (Fig. 1B). To validate this result, we performed qRT-PCR to evaluate the expression level of LUCAT1 in 36 pairs of CRC and adjacent normal tissues. The results showed that LUCAT1 expression in cancer tissues was higher than that in adjacent normal tissues in CRC patients (p = 0.024, Fig. 1C). Next, we summarized the clinicopathological characteristics of these patients, and the data demonstrated that patients with higher LUCAT1 expression showed a poorer histopathological grade and more lymph node metastasis than the patients with lower LUCAT1 expression (Table 1). Additionally, we examined the correlation between the LUCAT1 expression level and the prognosis of CRC patients through the GEPIA database by using the same cutoff point. Kaplan–Meier survival analysis showed that patients with higher levels of LUCAT1 had shorter overall survival and disease-free survival times than those who had lower levels of LUCAT1 (Fig. 1D, E). Moreover, the expression level of LUCAT1 was detected in 4 human CRC cell lines (HCT116, HT-29, SW620, and SW480) and normal human colonic epithelial NCM460 cells, and the results showed that LUCAT1 was significantly higher in cancer cell lines than in NCM460 cells (Fig. 1F). HCT116 and SW620 cells with relatively higher LUCAT1 expression were selected for subsequent functional assays.

(A) LUCAT1 had higher expression in CRC samples than in matched normal tissues from TCGA database (colon adenocarcinoma (COAD), Normal=41, tumor=471; rectum adenocarcinoma (READ), Normal=10, tumor=167). (B) The relative expression level of LUCAT1 in kidney renal clear cell carcinoma (KIRC), liver hepatocellular carcinoma (LIHC), COAD, lung adenocarcinoma (LUAD), esophageal carcinoma (ESCA), stomach adenocarcinoma (STAD), head and neck squamous cell carcinoma (HNSC), lung squamous cell carcinoma (LUSC), and uterine corpus endometrial carcinoma (UCEC). (C) LUCAT1 in CRC tissues was upregulated compared with that in adjacent normal tissues, n = 36 (measured by qRT-PCR; GAPDH was used as an internal control). (D) Kaplan–Meier estimated overall survival in patients with high or low LUCAT1 expression, higher LUCAT1 expression with poorer overall survival, p = 0.031. Group cutoff-point: 25% (high) and 75% (low). (E) Kaplan–Meier estimated disease-free survival in patients with high or low LUCAT1 expression, higher LUCAT1 expression with poorer disease-free survival, p = 0.017. Group cutoff points were 25% (high) and 75% (low). (F) LUCAT1 expression in colorectal cancer cell lines (HT29, SW480, HCT116, and SW620) was increased compared to that in NCM460 cells, a normal colon cell line (measured by qRT-PCR; GAPDH was used as an internal control). The results are presented as the mean ± s.d. and are representative of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
LUCAT1 is required for the proliferation of CRC cells in vitro and in vivo
To clarify the functional significance of LUCAT1 in CRC cells, we generated LUCAT1-knockout (LUCAT1-KO) cell lines for the first time by using the CRISPR/Cas9 system. The LUCAT1-specific dual sgRNAs were designed to knock out the full length of the LUCAT1 coding sequence, excluding the possible biological activity of different LUCAT1 transcripts as well. Cas9 and sgRNA were delivered by lentiviruses into HCT116 and SW620 cells. The deletion of LUCAT1 was verified by genomic PCR using primers outside of the sgRNA-targeted region (positive control primers) and primers across the sgRNA-targeted region (negative control primers) (Supplementary Fig. 1a). The band detected in LUCAT1-KO and scramble cell lines (termed NC cells in the following text) confirmed the successful knockout of LUCAT1, and their sequences also correctly matched as expected (Supplementary Fig. 1b). Consistently, the mRNA levels of LUCAT1 also dramatically decreased (Fig. 2A). Therefore, we investigated the role of LUCAT1 in the malignant behavior of HCT116 and SW620 cells. Growth curves generated from CCK8 proliferation assays showed that LUCAT1 knockout (LUCAT1-KO) significantly inhibited HCT116 and SW620 cell proliferation (Fig. 2B). Similarly, colony formation assays revealed that the clonogenic survival of HCT116 and SW620 cells was significantly impaired in LUCAT1-KO cell lines (Fig. 2C). However, our results showed that LUCAT1 knockout did not induce cell cycle arrest (Fig. 2D). These findings suggested that LUCAT1 played an important role in CRC proliferation in vitro.

(A) The expression of LUCAT1 was detected by qRT-PCR in HCT116 cells, and cells transfectedwith the CRISPR/Cas9 system differed significantly between the LUCAT1 knockout (LUCAT1-KO) and control (NC) groups in HCT116 and SW620 cells. GAPDH was used as an internal control. (B) Reduction in the proliferation ability of LUCAT1-KO HCT116 and SW620 cells compared with the control (NC) cells by a CCK8 assay. (C) Reduction in colony formation ability of LUCAT1-KO HCT116 and SW620 cells compared with that of the control (NC) cells by a colony formation assay. The bar graph indicates the number of colonies, *p < 0.05. (D) Cell cycle of LUCAT1-KO HCT116 and SW620 cells compared with the control (NC) was analyzed by flow cytometry. The distribution of the cell cycle is shown in the graphs. The results are presented as the mean±s.d. and are representative of at least three independent experiments. (E) Images of xenograft-transplantednude mousemodels (n = 6) and dissected tumors 30 days after injection with LUCAT1-KO SW620 cells, LUCAT1-KO HCT116 cells and their corresponding NC cell lines. (F) Tumor growth curves of LUCAT1-KO HCT116 and SW620 cell and control (NC) cell groups in the xenograft model. (G) Xenograft tumor weight for LUCAT1-KO HCT116 and SW620 cell and control (NC) cell groups in the xenograft model. The results are presented as the mean±s.d. and are representative of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns p > 0.05.
To further evaluate the role of LUCAT1 in vivo, we injected HCT116 LUCAT1-KO cells, SW620 LUCAT1-KO cells, and their corresponding NC cells into nude mice. All mice developed tumors at the injection site (Fig. 2E). However, the average size and weight of the tumors generated by LUCAT1-KO cells were significantly smaller than those generated by control cells (Fig. 2F, G). Consistently, immunohistochemical (IHC) staining showed a lower Ki67 expression in LUCAT1-KO tumor tissue compared to NC group (Supplementary Fig. 2a), suggesting a hindered proliferative ability of LUCAT1-KO cancer cells. All tumor tissues were verified by H&E staining (data not shown). Collectively, these results confirmed the oncogenic activity of LUCAT1 in CRC in vivo, which was consistent with what we observed in vitro.
LUCAT1 directly binds to NCL in the nucleus
It has been shown that the function of lncRNAs is consistent with their subcellular localization and exerted by interacting with various proteins30. We first performed assays in HCT116 cells to separate the cytoplasmic and nuclear RNA components and measured the expression ratio of LUCAT1 by qRT-PCR to confirm the subcellular localization of LUCAT1. We found that LUCAT1 was predominantly enriched in the nuclei of CRC cells (Fig. 3A). One of the most important roles of lncRNAs in the nucleus is their interaction with specific proteins to regulate the expression of target genes5,31,32,33; thus, we speculated that LUCAT1 may also function in this manner. To verify our hypothesis, we used biotinylated LUCAT1 probes to investigate proteins that potentially interact with LUCAT1 by performing ChIRP assays coupled with mass spectrometry (LC–MS/MS) in HCT116 cells and SW620 cells. In total, 52 specific LUCAT1-associated proteins were isolated from HCT116 cells, 17 specific LUCAT1-associated proteins were isolated from SW620 cells, and only NCL, RPL18A, and C7orf24 were detected in both cells (Supplementary Table 6). Among these proteins, NCL was selected for further verification due to its better mass spectrometric data and a higher binding score (Supplementary Table 7). Then, the parallel reaction monitoring (PRM) results further verified and quantified NCL among the products precipitated by the LUCAT1 probes, showing that compared with the negative control, the product was enriched with NCL, indicating that NCL might be the LUCAT1 interaction protein (Fig. 3B). To validate the potential interaction between LUCAT1 and NCL, we performed RIP assays in HCT116 and SW620 cells. The results revealed that LUCAT1 was significantly more enriched with the anti-NCL antibody than with the IgG (control antibody) (Fig. 3E). Furthermore, we simultaneously carried out FISH with the LUCAT1 probe and IF with the anti-NCL antibody. The results showed that NCL was significantly abundant in the nucleus (Fig. 3C), and the colocalization of LUCAT1 and NCL in the nucleus was clearly observed in HCT116 cells (Fig. 3D). Taken together, these findings demonstrated that LUCAT1 is an NCL-binding lncRNA.

(A) The amounts of LUCAT1 in the nuclear and cytoplasmic fractions of HCT116 cells were quantified by qRT-PCR. GAPDH was used as a control for cytoplasmic transcripts. MALAT1 was used as a nuclear marker. A nuclear/cytoplasmic ratio >1 proved that LUCAT1 was a nuclear-enriched lncRNA. (B) ChIRP assay using LUCAT1 probes followed by PRM analysis, which detected and quantified the amounts of NCL. Production pattern and chromatograph of two labeled NCL peptides (GYAFIEFASFEDAK and GFGFVDFNSEEDAK). Different colors represent different fragment ions of the same polypeptide. Each peptide was quantified using six fragment ions. U1 probes were used as a positive control. (C) Immunofluorescence staining using anti-NCL antibodies (green) in HCT116 cells. Scale bars, 10 µm. DAPI, 4′,6-diamidino-2-phenylindole. (D) Colocalization analysis: RNA FISH assay of LUCAT1 (red) combined with the immunofluorescence detection of NCL (green) in HCT116 cells. Scale bars, 20 µm. (E) A RIP assay was performed in HCT116 and SW620 cells using anti-NCL and anti-IgG antibodies (control). The amount of LUCAT1 mRNA was quantified by qRT-PCR. The results are presented as the mean±s.d. and are representative of at least three independent experiments. (F–H) Expression changes in four G4-associated genes, MYC, HIF-1α, VEGF, and KRAS, were determined in the NC and sgRNA HCT116 and SW620 cells with qRT-PCR (G) and Western blot analyses (F, H). (I) ChIP analysis of the interaction between the NCL protein and the MYC promoter in HCT116 cells. ChIP-qPCR of NCL at the MYC promoter when LUCAT1 or the vector was overexpressed in HCT116 cells. (J) NCL binding to the MYC promoter was detected by a luciferase assay. The relative luciferase activity of the reporter containing the MYC NHE III1 promoter/mutant was cotransfected with the indicated constructs in HCT116 cells. LUCAT1 interfered with NCL binding to the MYC promoter via its potential G4-forming sequence. (K) Relative luciferase activity of the reporter containing the MYC NHE III1 promoter/mutant that was cotransfected with the indicated constructs into HCT116 cells. The results are presented as the mean±s.d. and are representative of at least three independent experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001, ns p > 0.05.
LUCAT1 regulates MYC expression by antagonizing the function of NCL
It has been demonstrated that NCL can regulate gene expression by binding to the G4 sequence and promoting G4 structure formation with its high affinity and selectivity18,34. Therefore, we hypothesized that LUCAT1 has a potential G4 sequence that can be recognized by NCL and then affected NCL-mediated regulation of gene expression. To verify this idea, we first screened several important oncogenes, including KRAS, MYC, VEGF, and HIF-1α, by qPCR and Western blot analyses; these oncogenes are reported to contain the G4 structure in their promoters and are closely associated with CRC34,35,36,37. We found that MYC mRNA and protein expression in LUCAT1-KO HCT116 and LUCAT1-KO SW620 cells was significantly decreased compared with that in NC cells (Fig. 3F–H). Down-regulation of MYC was also observed in LUCAT1-KO tumor tissue by IHC staining than NC group (Supplementary Fig. 2b). Further bioinformatic analysis revealed that LUCAT1 was correlated with MYC expression in CRC cancers (Supplementary Fig. 3). Together, these data suggest that MYC transcription is regulated by LUCAT1 in NCL-dependent way.
To determine whether NCL controls the transcription of MYC through the binding of G4-forming sequence in the promoter region, we next performed a ChIP assay in HCT116 cells. The ChIP results confirmed that NCL could directly bind to the G4 sequence of MYC (-141 bp to -114 bp) in the promoter region (named MYC NHE III138) (Supplementary Fig. 4b), while fragments of KRAS, VEGF and HIF-1α were not observed (Supplementary Fig. 4a).
To further confirm that NCL could directly bind to MYC NHE III1 and regulate gene expression, we assayed the expression of a luciferase reporter containing MYC NHE III1 sequences (-1000 bp to 0 bp). Because HCT116 cells expressed a high basal level of NCL, we first knocked down NCL in HCT116 cells by using NCL-specific small hairpin RNAs (sh-NCL) (Supplementary Fig. 5). We found that inhibition of NCL resulted in increased reporter luciferase activity, but this increase was abrogated when the G4 sequence of MYC NHE III1 was mutated (Fig. 3J).
Due to the high affinity of NCL for G4, we analyzed the potential G4 sequence of LUCAT1 using the QGRS Mapper program. The QGRS Mapper program predicted that the putative G4-forming region from nucleotides 717 to 746 of LUCAT1 (LUCAT1 717-746) had the highest score for G4-forming probability (Supplementary Table 8). We next investigated whether LUCAT1 717-746 was required for its interaction with NCL. We introduced mutations into the LUCAT1 region 717–746 to generate LUCAT1-Mut. We found that overexpression of LUCAT1 resulted in increased MYC reporter luciferase activity but not LUCAT1-Mut luciferase activity (Fig. 3K), indicating that the NCL-mediated inhibition of MYC transcription could be reduced by overexpressing LUCAT1. In addition, overexpression of both LUCAT1 and LUCAT1-Mut did not change the reporter luciferase activity when the G4 sequence of MYC NHE III1 was mutated (Fig. 3K). Consistently, overexpression of LUCAT1 also resulted in decreased binding affinity between NCL and the MYC promoter in the ChIP assay (Fig. 3I). These findings suggested that LUCAT1 may directly interact with NCL through the LUCAT1 region 717–746.
We further compared the sequence of LUCAT1 717-746 (Supplementary Fig. 4c, highlighted) with the sequence of PCR products produced by the RIP assay with an anti-NCL antibody (Supplementary Fig. 4c, underlined); interestingly, there was an overlap between them. In other words, these findings further verified the accuracy and correctness of the prediction of the binding site between LUCAT1 and NCL. Taken together, these results indicated that the G4-forming sequence of LUCAT1 could antagonize NCL binding to MYC NHE III1 in the promoter.
Silencing NCL restores LUCAT1-KO-mediated inhibition of MYC expression and CRC cell proliferation
To investigate the function of NCL in the LUCAT1-mediated modulation of MYC expression and proliferation, we examined the effect of NCL knockdown on MYC expression, as well as on the proliferation activity of LUCAT1-KO HCT116 and SW620 cells. We found that knockdown of NCL almost completely restored MYC expression in LUCAT1-KO HCT116 and SW620 cells (Fig. 4A, B). In addition, knockdown of NCL also restored the proliferative activities of LUCAT1-KO HCT116 and SW620 cells (Fig. 4C, D). These results further indicated that LUCAT1 promoted CRC cell proliferation by antagonizing the NCL-mediated inhibitory effects on MYC.

(A) LUCAT1-KO HCT116 and SW620 cells were transfected with the indicated shRNAs, and MYC expression was quantified by qRT-PCR. GAPDH was used as a control. (B) LUCAT1-KO HCT116 and SW620 cells were transfected with the indicated shRNAs, and MYC expression was quantified by a Western blot. GAPDH was used as a control. (C) LUCAT1-KO HCT116 and SW620 cells were transfected with the indicated shRNAs, and the proliferation ability was quantified by a CCK8 assay. (D) LUCAT1-KO HCT116 and SW620 cells were transfected with the indicated shRNAs, and the colony formation ability was quantified by a colony formation assay. The results are presented as the mean ± s.d. and are representative of at least three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Recent studies have shown that thousands of lncRNAs are dysregulated in many types of cancer and play important roles in carcinogenesis, but few studies have elucidated their specific mechanisms. In this study, we discovered that LUCAT1 promoted CRC cell proliferation by forming a G4 with NCL. This interaction blocked NCL from facilitating G4 formation in the MYC gene promoter region and led to MYC expression.
Our present study demonstrates the clinical significance of LUCAT1 in CRC. The clinical data revealed that LUCAT1 in CRC tissues was upregulated compared with that in adjacent normal tissues, indicating that LUCAT1 may be involved in the development and progression of CRC. We further investigated the relationship between LUCAT1 and clinical pathology of CRC and verified that higher LUCAT1 expression resulted in poorer histopathological grade and increased lymph node metastasis than lower LUCAT1 expression. Chen et al’s study shows that LUCAT1 up-regulation is significantly associated with CRC liver metastasis and poorer clinical prognosis18, supporting our findings that LUCAT1 is contributed to progression of CRC. In addition, a systematic analysis of online databases in our study discovered that LUCAT1 could be a risk factor for prognosis in CRC patients, suggesting it might be a potential therapeutic target for CRC. Similar condition is reported in cervical cancer39 and non-small-cell lung cancer15, showing that highly expressed LUCAT1 in peripheral blood or tissue of patients is related to poorer survival. To further investigate the oncogenic role of LUCAT1 in CRC, we transfected the CRISPR/Cas9 system with dual sgRNAs into HCT116 and SW620 cells to knock out the full-length LUCAT1 gene coding sequence. Compared with other studies, our study showed a causal role of LUCAT1 in cancer via the CRISPR/Cas9 system for the first time. Although shRNA offers the ability to create cells with stable suppression of individual genes, a genome-wide collection of individual gene-silenced cells is currently unavailable for any mammal. Conversely, CRISPR/Cas9-based gene-deleted cells are available for any mammal40. After knocking out LUCAT1, CRC cell proliferation was significantly inhibited. This is also consistent with the results of studies in cervical cancer, ovarian cancer, breast cancer, choroidal melanoma, clear cell renal cell carcinoma, non-small-cell lung cancer and esophageal squamous cell carcinoma, showing that loss of LUCAT1 hinders cell proliferation15,17,20,39,41,42,43. The results described above indicate the oncogenic role of LUCAT1 in CRC.
Like other lncRNAs, LUCAT1 has been proven to participate in carcinogenesis and cancer progression by regulating gene expression through a posttranscriptional mechanism19. For example, LUCAT1 can promote esophageal squamous cell carcinoma and clear cell renal cell carcinoma tumorigenesis by controlling the ubiquitination and stability of DNMT1 and the AKT/GSK-3β signaling pathway, respectively17,20. In addition, LUCAT1 has been reported to induce CRC cell cycle arrest and apoptosis by binding to UBA52 and activating the p53 pathway44. Some studies also explore the relationship between LUCAT1 and miRNAs, including miR-514a/b-3b in choroidal melanoma, miR-199a-5p in ovarian cancer, and miR-199b-5p in cervical cancer39,41,42. LUCAT1 are found to share conserved binding site with these miRNAs and then modulates cancer progression through their direct interaction. However, the mechanisms of LUCAT1 transcriptional regulation in CRC remain unclear. To further investigate the underlying mechanisms by which LUCAT1 regulated downstream effectors in CRC, ChIRP and RIP experiments were performed. Proteins interacting with LUCAT1 in CRC were pulled down by biotin-labeled LUCAT1 probes and investigated by LC/MS analysis, and NCL was identified as a LUCAT1-associated protein for the first time. LUCAT1 also could be precipitated by NCL antibody, confirming that LUCAT1 were directly interacted with NCL in CRC cells. We demonstrated that the depletion of NCL restored the cell proliferation ability of CRC cells with LUCAT1 knockout. These results indicate that LUCAT1 enhances CRC cell proliferation by negatively regulating NCL function.
It has been demonstrated that NCL localizes primarily in the nucleus and binds to the G4 to facilitate its formation38,45,46. The presence of G4s can be found in the promoter regions of many oncogenes. It has been reported that NCL binds to the G4 in the promoter, and this binding suppresses oncogene expression34. Some small RNAs have also been reported to stabilize or interfere with the G4 structure24,47. Therefore, we investigated four candidate genes, MYC, KRAS, VEGF, and HIF-1α, which were dysregulated in CRC and closely correlated with CRC progression48,49,50,51. They are also known to form G4 structures in their promoter regions and negatively regulate gene transcription22,34,36,52. We found that only MYC could be suppressed when LUCAT1 was knocked out, and this reduction could be reversed by knocking down NCL. Then, both ChIP and luciferase reporter assays confirmed that NCL could directly bind to MYC NHE III1, the promoter region that controlled up to 90% of total MYC transcription. Overexpression of LUCAT1 blocked NCL from binding to the MYC promoter and enhanced MYC transcription. Our results support the hypothesis that NCL can regulate MYC transcription in HeLa cells38. Thus, we disclosed that LUCAT1 enhanced MYC transcription and expression by interacting with NCL, which prevented NCL from being recruited to the MYC promoter and inducing G4 formation. Although other proteins, such as UBA52, were reported to interact with LUCAT1 in HCT116 cells53, they were not detected in our ChIRP-MS results. A possible reason is that LUCAT1 is multifunctional and can regulate CRC progression through a different pathway.
Some studies have demonstrated that lncRNAs can act as molecular decoys to competitively bind to proteins by imitating their G4-forming structure sequence23. Since NCL can target G4 and facilitate its formation, we speculated that NCL might also be able to bind to LUCAT1 via putative G4-forming regions in LUCAT1. As we expected, the MYC reporter luciferase activity inhibited by NCL could be recovered by the overexpression of LUCAT1 but not by LUCAT1-Mut, which was mutated at the sequence of the predicted G4-forming region. Moreover, the sequence of the predicted G4-forming region in LUCAT1 was consistent with the sequences of the RNA fragments detected in the RIP experiment. These results indicate that nucleotides 716 bp to 746 bp of the LUCAT1 transcript contain a G4-forming sequence, which could be recognized by NCL to form G4 structures.
In summary, our present work revealed that the CRC-associated lncRNA LUCAT1 played an oncogenic role, contributing to CRC proliferation. We confirmed for the first time that LUCAT1 interfered with G4 formation in the MYC promoter by antagonizing the binding between NCL and MYC via its G4-forming region, promoting MYC transcription and expression. In conclusion, these results might indicate that the LUCAT1/NCL/MYC axis might be a potential prognostic marker and therapeutic target in CRC, potentially providing new insight into CRC progression and treatment.
Data availability
Gene expression analysis of LUCAT1 was carried out using the starBase database v3.0 project (http://starbase.sysu.edu.cn). The survival analysis of LUCAT1 was carried out using the Gene Expression Profiling Interaction Analysis database (http://gepia.cancer-pku.cn). The relationship between LUCAT1 and NCL was predicted via RNA-Protein Interaction Prediction (http://pridb.gdcb.iastate.edu/RPISeq/). The potential binding sites between LUCAT1 and NCL were predicted by QGRS Mapper (http://bioinformatics.ramapo.edu/QGRS/index.php). All the data will be made available upon reasonable request.
References
Siegel, R. L. et al. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 67, 177–193 (2017).
Chen, W. et al. Cancer statistics in China, 2015. CA Cancer J. Clin. 66, 115–132 (2016).
Chen, W. Q. et al. Report of cancer incidence and mortality in China, 2014. Zhonghua Zhong Liu Za Zhi 40, 5–13 (2018).
Wu, T. & Du, Y. LncRNAs: from basic research to medical application. Int J. Biol. Sci. 13, 295–307 (2017).
Ji, Q. et al. Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex. Br. J. Cancer 111, 736–748 (2014).
Fatica, A. & Bozzoni, I. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 15, 7–21 (2014).
Ulitsky, I. & Bartel, D. P. lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46 (2013).
Gong, C. & Maquat, L. E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470, 284–288 (2011).
Tsai, M. C. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693 (2010).
Xing, Z. et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 159, 1110–1125 (2014).
Wu, H. et al. Comprehensive analysis of differentially expressed profiles of lncRNAs and construction of miR-133b mediated ceRNA network in colorectal cancer. Oncotarget 8, 21095–21105 (2017).
Shan, Y. et al. LncRNA SNHG7 sponges miR-216b to promote proliferation and liver metastasis of colorectal cancer through upregulating GALNT1. Cell Death Dis. 9, 722 (2018).
Bian, Z. et al. LncRNA-FEZF1-AS1 promotes tumor proliferation and metastasis in colorectal cancer by regulating PKM2 signaling. Clin. Cancer Res. 24, 4808–4819 (2018).
Thai, P. et al. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am. J. Respir. Cell Mol. Biol. 49, 204–211 (2013).
Sun, Y. et al. Long non-coding RNA LUCAT1 is associated with poor prognosis in human non-small lung cancer and regulates cell proliferation via epigenetically repressing p21 and p57 expression. Oncotarget 8, 28297–28311 (2017).
Gao, Y. S., Liu, X. Z., Zhang, Y. G., Liu, X. J. & Li, L. Z. Knockdown of long noncoding RNA LUCAT1 inhibits cell viability and invasion by regulating miR-375 in glioma. Oncol. Res. 26, 307–313 (2018).
Zheng, Z. et al. Long non-coding RNA LUCAT1 promotes proliferation and invasion in clear cell renal cell carcinoma through AKT/GSK-3beta signaling pathway. Cell Physiol. Biochem. 48, 891–904 (2018).
Chen, Y., Yu, X., Xu, Y. & Shen, H. Identification of dysregulated lncRNAs profiling and metastasis-associated lncRNAs in colorectal cancer by genome-wide analysis. Cancer Med. 6, 2321–2330 (2017).
Liu, C. et al. Long noncoding RNA LUCAT1 promotes migration and invasion of prostate cancer cells by inhibiting KISS1 expression. Eur. Rev. Med. Pharm. Sci. 23, 3277–3283 (2019).
Yoon, J. H. et al. The long noncoding RNA LUCAT1 promotes tumorigenesis by controlling ubiquitination and stability of DNA methyltransferase 1 in esophageal squamous cell carcinoma. Cancer Lett. 417, 47–57 (2018).
Das Mahapatra, K. et al. A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci. Rep. 10, 3637 (2020).
Huppert, J. L. & Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic acids Res. 35, 406–413 (2007).
Matsumura, K. et al. The novel G-quadruplex-containing long non-coding RNA GSEC antagonizes DHX36 and modulates colon cancer cell migration. Oncogene 36, 1191–1199 (2017).
Chang, Y. C. et al. An XIST-related small RNA regulates KRAS G-quadruplex formation beyond X-inactivation. Oncotarget 7, 86713–86729 (2016).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412 (2010).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42(Database issue), D92–D97 (2014).
Tang, Z. et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids Res. 45(W1), W98–W102 (2017).
Muppirala, U. K., Honavar, V. G. & Dobbs, D. Predicting RNA-protein interactions using only sequence information. BMC Bioinforma. 12, 489 (2011).
Kikin, O., D’Antonio, L. & Bagga, P. S. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 34(Web Server issue), W676–W682 (2006).
Koch, L. Functional genomics: screening for lncRNA function. Nat. Rev. Genet. 18, 70 (2017).
Lu, Y. et al. The long non-coding RNA NEAT1 regulates epithelial to mesenchymal transition and radioresistance in through miR-204/ZEB1 axis in nasopharyngeal carcinoma. Tumour Biol. 37, 11733–11741 (2016).
Li, X. L. et al. Long noncoding RNA PURPL suppresses basal p53 levels and promotes tumorigenicity in colorectal cancer. Cell Rep. 20, 2408–2423 (2017).
Ma, Y. et al. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/beta-catenin signalling pathway via suppression of activator protein 2alpha. Gut 65, 1494–1504 (2016).
Sun, D., Guo, K. & Shin, Y. J. Evidence of the formation of G-quadruplex structures in the promoter region of the human vascular endothelial growth factor gene. Nucleic acids Res. 39, 1256–1265 (2011).
Siddiqui-Jain, A., Grand, C. L., Bearss, D. J. & Hurley, L. H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl Acad. Sci. USA 99, 11593–11598 (2002).
Cogoi, S. & Xodo, L. E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic acids Res. 34, 2536–2549 (2006).
Welsh, S. J. et al. Inhibition of the hypoxia-inducible factor pathway by a G-quadruplex binding small molecule. Sci. Rep. 3, 2799 (2013).
Gonzalez, V., Guo, K., Hurley, L. & Sun, D. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J. Biol. Chem. 284, 23622–23635 (2009).
Yang T., Xia S. Study of the biological function of LncRNA LUCAT1 on cervical cancer cells by targeting miR-199b-5p. Biosci Rep. 40, BSR20200422 (2020).
Unniyampurath, U., Pilankatta, R. & Krishnan, M. N. RNA Interference in the Age of CRISPR: will CRISPR Interfere with RNAi? Int. J. Mol. Sci. 17, 291 (2016).
Wang, L., Tang, D., Wu, T. & Sun, F. ELF1-mediated LUCAT1 promotes choroidal melanoma by modulating RBX1 expression. Cancer Med. 9, 2160–2170 (2020).
Liu, H. Z. et al. LncRNA LUCAT1 promotes proliferation of ovarian cancer cells by regulating miR-199a-5p expression. Eur. Rev. Med. Pharm. Sci. 24, 1682–1687 (2020).
Campbell, A., Mohl, J. E., Gutierrez, D. A., Varela-Ramirez, A. & Boland, T. Thermal bioprinting causes ample alterations of expression of LUCAT1, IL6, CCL26, and NRN1L genes and massive phosphorylation of critical oncogenic drug resistance pathways in breast cancer cells. Front Bioeng. Biotechnol. 8, 82 (2020).
Zhou, Q. et al. LUCAT1 promotes colorectal cancer tumorigenesis by targeting the ribosomal protein L40-MDM2-p53 pathway through binding with UBA52. Cancer Sci. 110, 1194–1207 (2019).
Bates, P. J., Kahlon, J. B., Thomas, S. D., Trent, J. O. & Miller, D. M. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 274, 26369–26377 (1999).
Dempsey, L. A., Sun, H., Hanakahi, L. A. & Maizels, N. G4 DNA binding by LR1 and its subunits, nucleolin and hnRNP D, A role for G-G pairing in immunoglobulin switch recombination. J. Biol. Chem. 274, 1066–1071 (1999).
Balasubramanian, S., Hurley, L. H. & Neidle, S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Disco. 10, 261–275 (2011).
Baba, Y. et al. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am. J. Pathol. 176, 2292–2301 (2010).
Boutin, A. T. et al. Oncogenic Kras drives invasion and maintains metastases in colorectal cancer. Genes Dev. 31, 370–382 (2017).
Rochlitz, C. F., Herrmann, R. & de Kant, E. Overexpression and amplification of c-myc during progression of human colorectal cancer. Oncology 53, 448–454 (1996).
Bendardaf, R. et al. VEGF-1 expression in colorectal cancer is associated with disease localization, stage, and long-term disease-specific survival. Anticancer Res. 28, 3865–3870 (2008).
De Armond, R., Wood, S., Sun, D., Hurley, L. H. & Ebbinghaus, S. W. Evidence for the presence of a guanine quadruplex forming region within a polypurine tract of the hypoxia inducible factor 1alpha promoter. Biochemistry 44, 16341–16350 (2005).
Wei, S. et al. Overexpression of Toll-like receptor 4 enhances LPS-induced inflammatory response and inhibits Salmonella Typhimurium growth in ovine macrophages. Eur. J. Cell Biol. 98, 36–50 (2019).
Acknowledgements
We thank all individuals who participated in this work.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81602568, 81773130, 81702336), China Postdoctoral Science Foundation (No.2018M643009), the National Natural Science Foundation of Hunan Province (No. 2019JJ50906).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics committee of the Third XiangYa Hospital of Central South University (No.2016-S086).
Consent for publication
Thirty-six pairs of CRC tissues and adjacent normal tissues used were obtained from the 36 patients who underwent surgical resection of CRC at the Third XiangYa Hospital of Central South University (Changsha, China) after informed consent was obtained.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Calin
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, R., Li, L., Bai, Y. et al. The long noncoding RNA LUCAT1 promotes colorectal cancer cell proliferation by antagonizing Nucleolin to regulate MYC expression. Cell Death Dis 11, 908 (2020). https://doi.org/10.1038/s41419-020-03095-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03095-4
This article is cited by
-
Targeting and engineering long non-coding RNAs for cancer therapy
Nature Reviews Genetics (2024)
-
LncRNA SNHG26 promotes gastric cancer progression and metastasis by inducing c-Myc protein translation and an energy metabolism positive feedback loop
Cell Death & Disease (2024)
-
G-quadruplexes from non-coding RNAs
Journal of Molecular Medicine (2023)
-
The N6-methyladenosine modification of circALG1 promotes the metastasis of colorectal cancer mediated by the miR-342-5p/PGF signalling pathway
Molecular Cancer (2022)
-
KRAS-related long noncoding RNAs in human cancers
Cancer Gene Therapy (2022)
