
Abstract
A flexible wearable electrode consisting of nickel–cobalt sulfide (NCS) nanowires was fabricated in this study. Self-supporting NCS was grown in situ on porous carbon nanofibers without a binder as a novel material for supercapacitor electrodes. The NCS nanowires were grown using cyclic voltammetry electrodeposition, which proved to be a fast and environmentally friendly method with good controllability of the material structure. One-dimensional carbon nanofibers (C) have high surface-area-to-volume ratios, short ion transmission distances, excellent mechanical strengths, and remarkable flexibilities. Moreover, the NCS@C flexible electrode exhibited a synergetic effect with the active compounds, and the dense active sites were uniformly distributed across the entire surface of the carbon fibers, enabling rapid electron transport and enhancing the electrochemical properties of the NCS@C nanowires. The NCS@C achieved specific capacitances of 334.7 and 242.0 mAh g−1 at a current density of 2 A g−1 and high current densities (up to 40 A g−1), respectively, corresponding to a 72.3% retention rate. An NCS@C-nanofilm-based cathode and an activated-carbon-based anode were used to fabricate a flexible asymmetric supercapacitor. The device exhibited high energy and power densities of 12.91 Wh kg−1 and 358 W kg−1, respectively.
Similar content being viewed by others


Introduction
Flexible and wearable electronic devices can be used as foldable capacitive touch screens and curved smartphones in numerous applications, such as motion detection and microrobotics. Intelligent, flexible energy storage, and conversion devices with low weight, high safety, small volume, excellent electrochemical performance, and good mechanical durability are in great demand1. Flexible supercapacitors are promising candidates because of their strong mechanical flexibility and high safety even under mechanical distortion. However, they must also have a long cycle life, high energy density, and excellent electrical conductivity2. A flexible supercapacitor generally comprises a compatible electrolyte and a separator in a flexible assembly and a flexible electrode with excellent electrochemical characteristics3. Carbon materials are widely used as flexible electrode materials, conductive additives, and supporting substrates in electrochemical energy storage devices because of their large surface areas, good electrical conductivities, and outstanding chemical stabilities4. Carbon fibers containing > 90% carbon are expected to improve the mechanical properties of flexible supercapacitors owing to their high elastic moduli and appropriate mechanical strength5. Carbon fiber textiles have outstanding flexibility but a low surface electrochemical reactivity and small surface area6. Nonetheless, the synthesis of hierarchical pores and various pseudocapacitive oxygenic groups transforms carbon fiber textiles into highly active electrodes with adequate cycling durability and high capacitance7. Transition metal sulfides with nanoscale structures have attracted broad interest because of their unique advantages in energy storage applications8. The electrochemical performance of most cobalt sulfide nanosheet arrays is unsatisfactory owing to the limited number of redox-active sites because their electronic structure is not properly regulated. Partially replacing the electrochemically inactive Co2+ ions that control the cobalt sulfide bond with inexpensive, environmentally benign, first-row transition metal cations would be beneficial for achieving a high charge storage capability, ionic conductivity, and extended cycle durability. In particular, nickel, which neighbors cobalt (atomic number 27; ionic radius = 0.79 Å) in the periodic table and has a similar atomic radius (0.83 Å) to Co, would be a superior choice for controlling cobalt sulfide bonds. Adding Ni2+ ions to the parent cobalt sulfide bonds results in the Ni2+ ions replacing inactive Co2+ ions close to the active sites, thus inducing more electrochemically active sites, higher redox activity, and high charge storage capability9. Nickel–cobalt sulfide (NCS) exhibits appealing properties for energy storage devices, including low toxicity, low cost, and high performance, making the energy storage devices competitive electrochemical devices10. Owing to their narrower bandgaps and higher electrical conductivities, binary metal sulfides are expected to outperform metal oxides in electrochemical devices. Therefore, NCS-based electrode materials with adjustable nanomorphologies, such as nanosheets, nanoparticles, and nanotubes, have been extensively used to mitigate the issues mentioned above. NiCoS exhibits a high specific capacitance (SC) because of its multiple valences11. For example, Soram et al. synthesized a flexible and transparent core–shell Cu@Ni@NiCoS nanofiber electrode that exhibited an outstanding high area capacity of 6.94 Ah cm−2 and a transmittance of 76.83%12. Because NiCoS electrodes are highly dependent on the Faraday redox reaction and ion diffusion during rapid charge–discharge processes, they cannot be used for large-scale practical applications because of their poor rate performance and electrochemical stability. Qiu et al. synthesised NCS using single- and two-step hydrothermal methods13, and the NCS exhibited a remarkable SC of 242.0 mAh g−1 as a pseudosupercapacitor electrode. However, the actual percentage of electroactive materials can decrease significantly when severe aggregation of NCS occurs, resulting in hindered electron transfer and ion diffusion14. In this study, a facile two-step process was developed to fabricate novel nanoarray-structured NCS@C electrodes. The conversion of polymer fibers into carbon fibers requires both stabilization and carbonization. In this process, the removal of heteroatoms and volatilization of residual solvents affect the molecular structure, and the fiber diameter decreases owing to thermal shrinkage, forming a larger specific surface area, as compared to that of the original polymer fibers15. Optimization of the electrodeposition time using the appropriate NCS precursors can create a nanoarray morphology that increases the ion content in the electrode construct and facilitates electron transfer16. The NCS precursor was prepared using a special pulse electrodeposition method on porous carbon fibers, making the nanoarray structure controllable, fast, simple, and suitable for commercial batch production. Optimization of the electrodeposition time revealed that 40 s yielded the best-performing materials with the highest SC (334.7 mAh g−1 at a 2 Ag−1 current density) and cycling stability after 4,000 cycles (80.8% preserved capacitance), as compared to the 20 and 80 s electrodeposition time. Moreover, a device with an NCS@C cathode and an activated carbon-based anode yielded an energy density of 12.91 Wh kg−1 and a power density of 358 W kg−1, demonstrating the application of the new materials in a real-world device. These results suggest the strong potential of the electrochemically deposited NCS@C nanowires as competitive candidates for supercapacitor electrodes.
Experimental
Synthesis of carbon nanofibers
To prepare the electrospinning precursor solution, polyacrylonitrile (PAN; 3 g) was mixed with N,N-dimethylformamide (22 g, AR, ≥ 95), and the mixture was vigorously stirred at 60 °C until a homogeneous solution was formed (12 w% PAN)17. The PAN nanofiber precursor was electrospun from the as-prepared solution onto an aluminum foil collector. A voltage of 18 kV was applied between the aluminum collector and the 20# needle tip, which were 18 cm apart18. A constant flow rate of 0.8 ml/h was adopted. The collected PAN nanofiber membrane precursor was baked in a furnace at 260 °C for 4 h in air using a 1 °C min−1 heating rate for the pre-oxidization process19. The pre-oxidized fiber membrane was baked at 800 °C for 2 h in a tube furnace for carbonization (using a 1 °C min−1 heating rate) in the presence of argon gas20,21,22.
Synthesis of NiCo2S4 nanoarrays
All chemicals used for the synthesis of NCS were of analytical grade and used without additional purification. An electrodeposition hydrothermal process was used to synthesize NiCo2S4 nanoarrays with different morphologies23. Prior to NCS synthesis, the carbon nanofibers (substrates) were cleaned successively with nitric acid, ethanol, and deionized (DI) water24. In the first electrodeposition process, NixCo1-x(OH)2 with a layered morphology was deposited on the carbon nanofibers25. The material was electrodeposited using a standard three-electrode setup (working electrode: Ni2xCox(OH)2-coated carbon nanofibers, reference electrode: saturated Ag/AgCl, and counter electrode: platinum foil) at room temperature26. A metal hydroxide solution (0.1 M, 70 ml) with a Ni2+/Co2+ mole ratio of 1:2 was used as the electrodeposition electrolyte. Under a − 1.2 to 0.5 V potential, the Co2xNix(OH)2 acicular crystals were deposited on the carbon fiber cloth within 40 s. Dynamic potential cyclic scanning was used for the electrodeposition. The potential sweeping cycle was 0 to − 1.2 to 0.5 to 0 V. Samples with different deposition times, namely, 20, 40, and 80 s, were prepared. Cyclic voltammetry (CV) revealed that the sample with the 40 s deposition time exhibited the best electrochemical performance27. Next, the precursors were mixed with a Na2S solution (35 mL, 0.2 M) in an 80 mL Teflon-lined stainless-steel autoclave. The solution was incubated at 120 °C for 14 h28. The NCS@C electrode was removed from the liquid phase and rinsed with DI water and ethanol. The mixture was then maintained at 70 °C for 12 h29.
Assembly of asymmetric supercapacitor (ASC) devices
ASC devices with a two-electrode structure were assembled with the as-prepared NCS nanowires@C (cathode), AC (anode), and a polymer electrolyte separator (polyvinyl alcohol (PVA)-KOH) between them30. To prepare the AC electrode, polytetrafluoroethylene, acetylene black, and the active materials were mixed in a 1:1:8 mass ratio. The preparation of the PVA-KOH polymer electrolyte was based on a standard process31. First, PVA (6 g) was mixed with distilled water (60 mL) and the mixture was stirred for 2 h at 85 °C. Subsequently, a KOH solution was prepared by dissolving KOH (4 g) in DI water (20 mL). Finally, the PVA and KOH solutions were mixed and continuously stirred at 80 °C until a clear mixture was obtained. The prepared materials were submerged in the electrolyte solution for 8 min. The materials were dried under ambient conditions before the two electrodes were assembled. After the PVA-KOH solidified, the final ASC device was obtained.
Characterization
X-ray diffractometry (XRD) with Cu-Kα (l = 1.5418 Å) radiation at 40 kV and 40 mA was used to characterize the crystal structure and phase purity of the NCS. All samples were measured at 5–90° in an ambient environment32. X-ray photoelectron spectroscopy (XPS) analyses were performed using an ESCA2000 with an Al Kα excitation laser to determine the chemical state of the elements33. Field emission scanning electron microscopy (FE-SEM) and high-resolution transmission electron microscopy (HRTEM; JEM-2100F) were used to characterize the morphology of the NCS nanostructures. The Brunauer–Emmett–Teller (BET) surface area and porous properties of NCS@C were examined using nitrogen adsorption/desorption isotherms obtained using a surface area and porosity analyzer (TriStar II 3020, version 3.02, Micromeritics Instrument Corporation).
Electrochemical measurements
All electrochemical experiments were conducted on an RST 5100F platform using a conventional three-electrode setup34. A 3 M KOH solution was used as the electrolyte. The as-fabricated NCS@C electrodes were used as the working electrode, a platinum plate was used as the counter electrode, and a Hg/HgO electrode was used as the reference electrode. In the CV measurements, 0–0.6 V was used as the window for the potential scan35. The scan rate was set at 5, 10, 30, 50, 70, and 100 mV s−1. The galvanostatic charge–discharge (GCD) process was conducted in the potential range of 0–0.55 V using current densities of 1, 2, 6, and 10 A g−1. An ASC device was fabricated using NCS@C and AC@C as the cathode and anode, respectively, and a PVA-KOH gel was used as the electrolyte32. The weight of the anode material was calculated by charge-balancing between the two electrodes.
Results and discussion
Synthesis and characterization
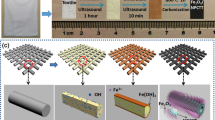
Figure 1e illustrates the NCS@C nanowire array synthesis process. Figure 1a–f shows a SEM image of the carbon nanofibers, a photographic image of the flexible NCS@C electrode, a SEM image of NCS@C, a TEM image of the carbon nanofiber, an illustration of the NCS@C electrode preparation process, and the EDX spectrum of the NiCoS electrode, respectively. NCS nanowires are generally grown on carbon fiber substrates via hydrothermal electrodeposition, followed by hydrothermal processes. First, NCOH was directly electrodeposited onto a carbon fiber substrate. The subsequent hydrothermal process yielded an NCS nanowire array. During electrodeposition, the NCOH precursors are typically Ni(II) and Co(III) nitrates, and a cathodic potential is applied to reduce these nitrates and H2O. OH− ions (or consumed H+) can be produced close to the electrode surface, leading to an increase in the pH and precipitation of NCOH. The mechanisms are shown in Eqs. (1–3)36:

(a) SEM image of the carbon nanofibers, (b) photographic image of the flexible NCS@C electrode, (c) SEM image of NCS@C, (d) TEM image of a carbon nanofiber, (e) illustration of the NCS@C electrode preparation process, and (f) the EDX spectrum of the NiCoS electrode.
Consequently, Na2S·9H2O was added to the composite electrode during hydrothermal processing. Na2S released reactive sulfide ions (S2−) during hydrolysis, which induced the nucleation of NiCo2S4 crystals owing to anion exchange in the sulfidation step. Nickel and Co ions in NCOH can receive two electrons from the S2− anion and cluster with it after dissociation with OH− (Eq. 4)37.
The phases and structures of the NCS nanowire arrays were characterized using XRD. As depicted in Fig. 2, the peaks of the NCS NWAs were consistent with those of cubic-phase NiCo2S4 (JCPDS No. 00-020-0782)28. The typical peaks for the (220), (311), (511), and (440) planes of the NCS phase indicate that highly crystalline NCS nanosheets were formed on the carbon fiber. The XRD patterns of the carbon nanofibers are shown in Fig. 2.

XRD patterns of NCS@C.
XPS was used to further examine the chemical compositions and valence states of the active materials. The full XPS spectrum in Fig. 3a shows that the material contained Ni, C, and S. Based on Gaussian fitting, two doublets due to spin–orbit coupling and two shakeup satellite peaks were observed in the original Ni 2p spectrum (Fig. 3). The coexisting Ni(II) and Ni(III) were indicated by two strong peaks at 874.3 and 856.0 eV for Ni 2p3/2 and Ni 2p1/2, respectively34. Figure 3 shows the S 2p region of S. The peaks at 160.9 (2p3/2) and 162.85 eV (2p1/2) clearly demonstrate the presence of NiCo2S4 in the product, and no oxide impurities, such as NiCo2O4, were detected. The XPS analysis showed that the sulfide nanosheets were composed of multivalent transition metal elements that can provide rich redox reaction sites and excellent conductivity.

Wide survey of the XPS spectrum of NCS@C. (a) High-resolution XPS spectra of (b) Ni 2p, (c) C 2p, and (d) S 2p.
The capacitance and cycle stability of ASCs are highly dependent on the morphology of the electrode material. The SEM images (Fig. 1a and c) show that the NCS nanosheets uniformly covered the carbon fibers, developing a hierarchical structure, and that the nanosheets were homogeneously wrapped around the nanowires. As shown in Fig. 1a, the obtained carbon nanofiber samples exhibited a smooth surface, relatively uniform diameter distribution, and no bead string structure. Uniform NiCo2O4 nanowires were successfully grown on the carbon fiber surfaces (Fig. 1c). To comprehensively understand and analyze the morphology- and structure-dependent properties of the NCS@C nanowires, the electrodeposition time was varied (20, 40, and 80 s) using well-controlled solution concentrations and temperatures. A deposition time of 40 s yielded the optimal morphology, and the sample outperformed the others in terms of electrochemical properties. The separated nanowires were supportive; they offered more surface area to the network structure of the carbon fibers and provided channels for ion transport. These orderly nanowire network structures produced numerous open channels and surfaces with dense electroactive sites, which offer more active sites for redox reactions and expose more active material to the electrolyte, leading to an enhanced SC and cycle stability. Different deposition times resulted in different material structures, which can affect conductivity and cause a shift in the redox peaks. The microstructure of NCS@C was examined using HRTEM. Figure 4a shows that nanoflakes were distributed on the surfaces of the carbon fibers. The HRTEM image of a single nanoflake shows clear lattice fringes with a spacing of 0.28 nm (Fig. 4b), which were indexed to the (220) planes of NCS. Figure 4c shows the diffraction rings in the SAED pattern, which matched well with the (220), (311), and (400) planes of the NCS, indicating the presence of NCS@C. Figure 4d shows the EDS elemental mapping used to determine the elemental distribution in the NCS@C sample. Overall, Co, Ni, and S were uniformly distributed on NCS@C.

(a) TEM, (b) HRTEM, and (c) SAED images of an individual NCS@C nanosheet and (d) the corresponding EDS mapping results.
Nitrogen adsorption/desorption measurements were performed to examine the porosity and BET surface area of the NCS@C. All N2 adsorption/desorption isotherms in Fig. 5 were typical type IV isotherms with a hysteresis loop in the P/P0 range of 0.3–0.8, suggesting that the materials had a mesoporous structure. These curves are based on the IUPAC classification of type IV isotherms with loop hysteresis. The single-point surface area at P/Po = 0.2915360 was 9.3382 m2/g. The BET and Langmuir surface areas of P/Po were 9.7151 and 16.6330 m2/g, respectively. The t-plot micropore area and external surface area were 7.1754 and 2.5397 m2/g, respectively. The BJH adsorption–desorption cumulative surface area of pores with a diameter of 1.000–3,000.000 Å were 13.460 and 16.2958 m2/g, respectively.

BET analysis results of the nitrogen adsorption and desorption isotherms for the NCS@C.
Electrochemical characterization of electrodes
Three electrochemical methods (electrochemical impedance spectroscopy (EIS), GCD, and CV) were used to characterize the NCS samples38. The correlation between the deposition time and the electrochemical properties was explored. The samples with the electrochemical deposition times of 20, 40, and 80 s were labeled as NCS@C-20 (0.6 mg/cm2), NCS@C-40 (1.4 mg/cm2), and NCS@C-80 (0.78 mg/cm2), respectively. CV scans of the NCS@C nanowires at various electrodeposition times and a scanning rate of 10 mV s−1 are shown in Fig. 6a. Representative CV scans of the electrodeposited NCS@C electrodes with scanning rates ranging from 5 to 50 mV s−1 are shown in Fig. 6b–d. The currents of the four samples increased with an increasing scanning rate. The oxidation and reduction peaks shifted to more negative and positive potentials, respectively. This phenomenon is due to the relatively slow shift of the alkali ions with increasing scanning rates. Because the alkali ion rate shift became relatively slow with an increasing scanning rate, only the outer layer of the active electrode surface was used to store charge in the redox reaction process. Mathematical analysis revealed that the CV curve of the NCS@C-40 nanowire electrode material was higher than those of NCS@C-20 and NCS@C-80, suggesting a higher SC for the NCS@C electrode obtained with an electrodeposition time of 40 s.

(a) CV curves at a scanning rate of 10 mV s−1 and (b), (c), and (d) CV scans of the three samples at the scanning rates of 5–50 mV s−1.
GCD measurements were performed to understand the influence of the deposition time on the electrochemical characteristics of the NCS@C electrode. The SC was computed from the GCD curve using Eq. (5)3:
where, ∆V, t, and I refer to the discharge potential range, discharge time (s), and discharge current (A), respectively, and m is the weight of the electroactive material on the working electrode (g). Figure 6a–d show the GCD measurement results of the NCS@C electrode materials prepared in the range of 0–0.55 V at various current densities (2–40 A g−1) in a 2 M KOH aqueous solution. Nonlinear charge–discharge curves, which were close to the level of the discharge platform, were observed. This is because the quasi-reversible redox reaction at the electrode/electrolyte interface induces pseudocapacitance behavior in the material. The symmetric GCD curves demonstrate that the material possessed excellent charge/discharge characteristics and electrochemical reversibility.
Figure 7a shows that, at a current density of 2 A g−1, the electrochemical properties of the NCS@C electrode were tuned by adjusting the electrodeposition time. The NCS@C-20 electrode with an electrodeposition time of 20 s exhibited a specific capacity of 183 mAh g−1 (Fig. 7b). The NCS@C electrode material with an electrodeposition time of 40 s exhibited the highest SC (334.7 mAh g−1) (Fig. 7c). When the deposition time was extended to 80 s, the SC of NCS@C (310.3 mAh g−1) exhibited a decreasing trend (Fig. 7d). Therefore, the GCD curves of NCS@C exhibited a higher SC when the electrodeposition time was 40 s. Based on these findings, the advantages of the NCS nanosheet with an electrodeposition time of 40 s for enhancing the capacitance are clear.

(a) Charge/discharge curves at a current density of 2 A g−1. Charge/discharge curves at different current densities of 2, 4, 10, 20, and 40 A g−1 for (b) NCS@C-20, (c) NCS@C-40, and (d) NCS@C-80.
According to Fig. 8a, NCS@C-40 exhibited the highest electrochemical property at the current densities of 2–40 A g−1. NCS@C-40 outperformed NCS@C-20 by approximately three-fold based on the SC at the same current density. A 72.3% SC was retained at a high current density of up to 2 A g−1, as compared to that with 40 A g−1, demonstrating the excellent rate capability of the material. The improvement in the NCS@C-40 sample rate capability and SC mainly benefited from its architecture with hierarchical pores, which enabled adequate exposure of the pseudocapacitance-active components. Moreover, the chemical binding between the NCS and carbon fiber promoted charge transportation between the current collector and the active components, which was conducive to a superior rate capability.

(a) SC of each sample at the current densities of 2, 4, 10, 20, 30, and 40 A g−1 and (b) Nyquist impedance plots of NCS@C.
The fundamental behavior of the electrodes was examined using EIS analysis (Fig. 8b), which was performed in the frequency range of 100 kHz–0.01 Hz. The spectra of the two samples exhibited one arc in the high-frequency region and one inclined line in the low-frequency region39,40. The intercept of the plot at the solid line (Z') was consistent with the internal resistance in the high-frequency region (Rs), representing the contact resistance at the electrolyte-active material and active material-collector interfaces. By fitting the low-frequency region, the slope of the curve was used to calculate the Warburg impedance (W), which indicates how the electrolyte diffuses through the pores of the electrode and protons in the material41. The EIS results showed that NCS possessed a lower Rs (0.23 Ω) and larger slope (W), demonstrating that NCS is an excellent base material. The NCS@C nanowire electrodes exhibited approximately the same impedance as the other samples (0.24 Ω) but the largest slope, indicating that the ion diffusion rate and conductivity were improved at the electrode/electrolyte interface.
Cycle performance tests were conducted as a key measurement index to evaluate the capability of the supercapacitors in real applications. Figure 8 shows the cycling durability of the specimens measured using a consecutive charge–discharge process at 40 A g−1. Based on these results, the NCS@C electrodes were expected to be in a state of inactivation, as confirmed by the cycling test. The specific capacity value decreased from 242 to 160 mAhg−1 at 500 cycles, then increased slowly and remained at approximately 195.53 mAhg−1 at the 4000th cycle. The initial increase in the specific capacity of the device was due to the activation of the electrodes through the exposure of more active sites in the hierarchical porous NCS and carbon nanofiber materials during cycling. As shown in Fig. 9, at a low current density, some side reactions occur during the electrochemical redox reaction, leading to an incomplete discharge. The charge and discharge times decreased with an increasing current density. The electrochemical process was primarily affected by the electric double layer; thus, the Coulombic efficiency increased. The NCS@C-40 electrode demonstrated exceptional cycling durability, maintaining 80.8% of the original capacity after 4000 cycles, suggesting that the complex and porous nanostructures and carbon layer can increase the cycling stability.

Cycling performance of the NCS@C nanotube arrays (4000 cycles) at a large current density of 40 A g−1.
To further evaluate the potential applications of the NCS@C nanowires, an ASC test was performed with the NCS@C nanowires as the cathode and AC on the NF (denoted as NCS@C//AC) as the anode in a 6 M KOH aqueous electrolyte solution (Fig. 10).

(a) CV curves of the device at different scan rates, (b) CV curves of the AC and NCS@C electrodes in the three-electrode system at a scan rate of 5 mV s−1, (c) schematic diagram of the ASC device, and (d) Ragone plot of the energy and power densities of the ASC device.
The CV scans of the NCS@C//AC ASC at different scanning rates over a voltage range of 0–1.6 V are shown in Fig. 10b. The shapes of the CV curves of the NCS@C//AC ASC and NCS@C nanowire electrodes were similar in the three-electrode system, which is in agreement with the charge storage mechanism of the NCS@C nanowire electrode. The ASC device exhibited identical CV curves at different scanning rates from 5 to 50 mV s−1, showing that this material has appropriate rapid charging and discharging characteristics and displays Faraday pseudocapacitance characteristics as an electric double-layer capacitor. Moreover, a GCD test was used to characterize the electrochemical performance of the ASC devices at various current densities. When the current densities of the ASC were 0.5, 1, 2, and 5 A g−1, the corresponding SCs were 15.69, 12.22, 9.44, and 5.55 mAh g−1, respectively. Both the charging and discharging curves were highly symmetric, indicating that the ASC exhibited good electrochemical reversibility.
The power and energy densities are two critical parameters for evaluating the performance of the electrode material of the ASC in practical applications, and can be obtained using Eqs. (6) and (7):
where E and P refer to the energy density (Wh kg−1) and power density (W kg−1), respectively, C is the SC, V is the potential range, and t is the discharging time. A high energy density of 12.91 Wh kg−1, corresponding to a power density of 358 W kg−1, was achieved using the ASC at a current density of 0.5 A g−1 (Fig. 9d). These results suggest that the NCS@C nanowires can be used as positive electrodes in ASC devices to obtain high energy density, which is required for the practical application of flexible supercapacitors.
Conclusions
NCS nanowires were grown on carbon fibers as binder-free electrodes for supercapacitors by combining a hydrothermal treatment and electrochemical deposition. NCS nanowires with a reactive surface area were evenly distributed on the surface of the carbon fiber to achieve rapid transport of electrons and improve the electrochemical performance. At a current density of 2 A g−1, the NCS@C-40 exhibited a high SC of up to 334.7mAh g−1. At a high current density (40 A g−1), the SC of NCS@C was 242.0 mAh g−1, corresponding to a 72.3% retention rate. The SC of NCS@C remained at 80.8% after 4,000 cycles. A high energy density of 12.91 Wh kg−1 and a power density of 358 W kg−1 were achieved by the ASC at a current density of 0.5 A g−1. These findings strongly suggest that the NCS@C nanowire is a competitive candidate as a positive electrode for ASC devices to obtain high energy density, which is required for the practical application of flexible supercapacitors (Supplementary File 1).
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Muralee Gopi, C. V. V., Vinodh, R., Sambasivam, S., Obaidat, I. M. & Kim, H. Recent progress of advanced energy storage materials for flexible and wearable supercapacitor: From design and development to applications. J. Energy Storage 27, 101035 (2020).
Choi, H.-J. et al. Graphene for energy conversion and storage in fuel cells and supercapacitors. Nano Energy 1, 534–551 (2012).
Gao, Z., Yang, W., Wang, J., Song, N. & Li, X. Flexible all-solid-state hierarchical NiCo2O4/porous graphene paper asymmetric supercapacitors with an exceptional combination of electrochemical properties. Nano Energy 13, 306–317 (2015).
Xiong, G. et al. Bioinspired leaves-on-branchlet hybrid carbon nanostructure for supercapacitors. Nat. Commun. 9, 790 (2018).
Trung, T. Q. & Lee, N. E. Flexible and stretchable physical sensor integrated platforms for wearable human-activity monitoring and personal healthcare. Adv. Mater. 28, 4338–4372 (2016).
Liu, B. T. et al. Extraordinary pseudocapacitive energy storage triggered by phase transformation in hierarchical vanadium oxides. Nat. Commun. 9, 1375 (2018).
Ma, L. et al. Synthesis of a three-dimensional interconnected oxygen-, boron-, nitrogen-, and phosphorus tetratomic-doped porous carbon network as electrode material for the construction of a superior flexible supercapacitor. ACS Appl. Mater. Interfaces 12, 46170–46180 (2020).
Wang, D. et al. Kelp-like structured NiCo2S4-C-MoS2 composite electrodes for high performance supercapacitor. J. Alloys Compd. 735, 1505–1513 (2018).
Ojha, G. P., Pant, B., Acharya, J., Lohani, P. C. & Park, M. Solvothermal-localized Selynazation transformation of cobalt nickel MOFs templated heterointerfaces enriched monoclinic Co3Se4/CoNi2Se4@Activated knitted carbon cloth for flexible and bi-axial stretchable supercapacitors. Chem. Eng. J. 464, 142621 (2023).
Jin, L., Liu, B., Wu, Y., Thanneeru, S. & He, J. Synthesis of mesoporous CoS2 and NixCo1–xS2 with superior supercapacitive performance using a facile solid-phase sulfurization. ACS Appl. Mater. Interfaces 9, 36837–36848 (2017).
Zhang, S. et al. Porous NiCoS nanosheets decorated activated carbon cloth for flexible asymmetric supercapacitors. Diam. Relat. Mater. 127, 109154 (2022).
Soram, B. S. et al. Flexible transparent supercapacitor with core-shell Cu@ Ni@NiCoS nanofibers network electrode. Chem. Eng. J. 395, 125019 (2020).
Yang, J. et al. Electroactive edge site-enriched nickel–cobalt sulfide into graphene frameworks for high-performance asymmetric supercapacitors. Energy Environ. Sci. 9, 1299–1307 (2016).
Hou, L. et al. Hollow mesoporous hetero-NiCo2S4/Co9S8 submicro-spindles: Unusual formation and excellent pseudocapacitance towards hybrid supercapacitors. J. Mater. Chem. A 5, 133–144 (2017).
Wang, Y., Wu, X., Han, Y. & Li, T. Flexible supercapacitor: Overview and outlooks. J. Energy Storage 42, 103053 (2021).
Chen, X. et al. Electrodeposited nickel aluminum-layered double hydroxide on Co3O4 as binder-free electrode for supercapacitor. J. Mater. Sci. Mater. Electron. 30, 2419–2430 (2019).
Li, H., Chen, R., Ali, M., Lee, H. & Ko, M. J. In situ grown MWCNTs/MXenes nanocomposites on carbon cloth for high-performance flexible supercapacitors. Adv. Funct. Mater. 30, 2002739 (2020).
Zhang, L., Hui, K. N., Hui, K. S. & Lee, H. Facile synthesis of porous CoAl-layered double hydroxide/graphene composite with enhanced capacitive performance for supercapacitors. Electrochim. Acta 186, 522–529 (2015).
Zhou, C., Zhang, Y., Li, Y. & Liu, J. Construction of high-capacitance 3D CoO@polypyrrole nanowire array electrode for aqueous asymmetric supercapacitor. Nano Lett. 13, 2078–2085 (2013).
Yan, J. et al. Advanced asymmetric supercapacitors based on Ni(OH)2/graphene and porous graphene electrodes with high energy density. Adv. Funct. Mater. 22, 2632–2641 (2012).
Ojha, G. P., Pant, B., Acharya, Jiwan & Park, M. Prussian red anions immobilized freestanding three-dimensional porous carbonaceous networks: A New Avenue to attain capacitor- and faradic-type electrodes in a single frame for 2.0 V hybrid supercapacitors. ACS Sustain. Chem. Eng. 10, 2994–3006 (2022).
Ojha, G. P., Pant, B., Acharya, Jiwan & Park, M. An electrochemically reduced ultra-high mass loading three-dimensional carbon nanofiber network: A high energy density symmetric supercapacitor with a reproducible and stable cell voltage of 2.0 V. Nanoscale 13, 19537–19548 (2021).
Zhao, J.-Y. et al. Metal–organic framework-derived Co9S8 embedded in N, O and S-tridoped carbon nanomaterials as an efficient oxygen bifunctional electrocatalyst. J. Mater. Chem. A 7, 7389–7395 (2019).
Li, H. et al. Electrodeposited NiCo layered double hydroxides on titanium carbide as a binder-free electrode for supercapacitors. Electrochim. Acta 261, 178–187 (2018).
Wu, H. B., Pang, H. & Lou, X. W. D. Facile synthesis of mesoporous Ni0.3Co2.7O4 hierarchical structures for high-performance supercapacitors. Energy Environ. Sci. 6, 3619–3626 (2013).
Wang, Q. et al. Flexible coaxial-type fiber supercapacitor based on NiCo2O4 nanosheets electrodes. Nano Energy 8, 44–51 (2014).
Umeshbabu, E., Rajeshkhanna, G. & Rao, G. R. Urchin and sheaf-like NiCo2O4 nanostructures: Synthesis and electrochemical energy storage application. Int. J. Hydrog. Energy 39, 15627–15638 (2014).
Wang, Y. et al. Hollow NiCo2S4 nanospheres hybridized with 3D hierarchical porous rGO/Fe2O3 composites toward high-performance energy storage device. Adv. Energy Mater. 8, 1703453 (2018).
Qiu, K. et al. Mesoporous, hierarchical core/shell structured ZnCo2O4/MnO2 nanocone forests for high-performance supercapacitors. Nano Energy 11, 687–696 (2015).
Ko, T. H. et al. Polypyrrole-decorated hierarchical NiCo2O4 nanoneedles/carbon fiber papers for flexible high-performance supercapacitor applications. Electrochim. Acta 247, 524–534 (2017).
Shen, L. et al. Formation of nickel cobalt sulfide ball-in-ball hollow spheres with enhanced electrochemical pseudocapacitive properties. Nat. Commun. 6, 6694 (2015).
Sun, S. et al. Metal–organic framework derived honeycomb Co9S8@C composites for high-performance supercapacitors. Adv. Energy Mater. 8, 1801080 (2018).
Filik, J., May, P. W., Pearce, S. R. J., Wild, R. K. & Hallam, K. R. XPS and laser Raman analysis of hydrogenated amorphous carbon films. Diam. Relat. Mater. 12, 974–978 (2003).
Chen, W., Xia, C. & Alshareef, H. N. One-step electrodeposited nickel cobalt sulfide nanosheet arrays for high-performance asymmetric supercapacitors. ACS Nano 8, 9531–9541 (2014).
He, G. et al. S, N-Co-doped graphene-nickel cobalt sulfide aerogel: improved energy storage and electrocatalytic performance. Adv. Sci. 4, 1600214 (2017).
Wang, C., Sun, P., Qu, G., Yin, J. & Xu, X. Nickel/cobalt based materials for supercapacitors. Chin. Chem. Lett. 29, 1731–1740 (2018).
Zhang, Z. et al. NiCo2S4 sub-micron spheres: An efficient non-precious metal bifunctional electrocatalyst. Nanoscale 6, 3540–3544 (2014).
Wei, J. S. et al. Carbon dots/NiCo2O4 nanocomposites with various morphologies for high performance supercapacitors. Small 12, 5927–5934 (2016).
Zhang, L. et al. Role of graphene on hierarchical flower-like NiAl layered double hydroxide-nickel foam-graphene as binder-free electrode for high-rate hybrid supercapacitor. Int. J. Hydrog. Energy 41, 9443–9453 (2016).
Zhang, L., Hui, K. N., San Hui, K. & Lee, H. High-performance hybrid supercapacitor with 3D hierarchical porous flower-like layered double hydroxide grown on nickel foam as binder-free electrode. J. Power Sources 318, 76–85 (2016).
Guan, C. et al. Hybrid structure of cobalt monoxide nanowire@nickel hydroxidenitrate nanoflake aligned on nickel foam for high-rate supercapacitor. Energy Environ. Sci. 4, 4496–4499 (2011).
Acknowledgements
This study was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (Grant Numbers: RS-2023-00207763 and NRF-2022R1A2C2010350) and the Science and Technology Project of Shandong Province (Heze University Grant No. 010008002042007) and.
Author information
Authors and Affiliations
Contributions
X.C.: Wrote the manuscript and did the experiments M.S.: Wrote the manuscript and did the experiments F.J., provided extra funding to support the tests and Supervised and revised the manuscript. E.Z.N.: Supervised the project and edited the manuscript. K.S.H.: Supervised the project and edited the manuscript. Z.L.: Wrote the manuscript and did the experiments S.B.: Supervised the project and edited the manuscript. M.D., helped to do XRD/XPS test and characterization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Sun, M., Jaber, F. et al. A flexible wearable self-supporting hybrid supercapacitor device based on hierarchical nickel cobalt sulfide@C electrode. Sci Rep 13, 15555 (2023). https://doi.org/10.1038/s41598-023-42278-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-42278-9
This article is cited by
-
Evaluation and validation of reference genes for RT-qPCR gene expression in Naegleria gruberi
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
