
Abstract
Triphenyltin (TPT) is a widespread synthetic chemical used in many fields and its potential risk to organisms has been comprehensively investigated using different animal models and species. Currently, little is known about the effects of TPT exposure on microbial midgut diversity, therefore we explored these effects in the lepidopterous silkworm model using 16S rDNA sequencing. In total, 5273 and 5065 operational taxonomic units (OTUs) were identified in control and TPT-exposure group samples, ranging from 424 to 728 OTUs/sample. Alpha-diversity analyses revealed that TPT exposure induced the fluctuations of gut microbial diversity and abundance while beta-diversity analyses identified a distinct impact on major gut microbiota components. In our microbiome analyses, 23 phyla and 353 genera were recognized in the control group, while 20 phyla and 358 genera were recognized in the TPT exposure group. At the genus level, midgut microbiota were composed of several predominant bacterial genera, including Muribaculaceae, Lactobacillus, and UCG-010. In the TPT exposure group, o__Bacillales, f__Bacillaceae, and f__Caldicoprobacteraceae abundance was relatively high, while f__Oscillospiraceae, f__Fusobacteriaceae, and f__SC_I_84 abundance was relatively high in the control group. Gene function analyses in silkworm microbiota after TPT exposure showed that biosynthesis of ansamycins, fructose and mannose metabolism, glycerolipid metabolism, type II diabetes mellitus, glycolysis/gluconeogenesis, lipid metabolism, translation proteins, atrazine degradation, DNA repair and recombination proteins, nicotinate and nicotinamide metabolism were significantly increased. Collectively, our silkworm model identified gut microbial diversity risks and the adverse effects from TPT exposure, which were similar to other aquatic animals. Therefore, TPT levels in environmental samples must be monitored to prevent ecological harm.
Similar content being viewed by others


Introduction
Triphenyltin (TPT) is a synthetic chemical used as an antifouling paint in industry and a fungicide in agriculture1. When TPT is exposed to high temperatures and rain, it is released into water bodies2,3. From 2008, TPT-use was prohibited in antifouling paints as TPT concentrations in the environment and aquatic organisms can potentially harm human health4. In coastal areas of Hong Kong, China, it was previously reported that TPT concentrations were up to 3.8–11.7 ng/L in the upper seawater and 71.8–91.7 ng/g (w/w) in the sediment5.
It is accepted that TPT is an endocrine disruptor with high toxicity6,7. Many studies have reported its potential risks in aquatic organisms and other species and concluded that TPT impacts immunity, development, reproduction, and endocrine systems7,8. TPT disrupts lipid metabolism in Marisa cornuarietis at environmental concentrations9. TPT also interferes with retinal axon development in fish at low doses10, induces oxidative stress and apoptosis in zebrafish11, and disrupts glucocorticoid synthesis in the rat adrenal cortex via several mechanisms: 1) it lowers serine/threonine kinase 1 (AKT1) phosphorylation and Sirtuin 1 (SIRT1)/peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) levels, 2) activates AMP-activated protein kinase (AMPK), and 3) possibly induces reactive oxidative species (ROS) production12. TPT also appears to affect reproduction and mortality at decreasing concentrations in temporally overlapping generations13.
As a representative lepidopteran insect vital for agricultural and forestry silk production, the silkworm (Bombyx mori) is an important model organism14. Bombyx mori has relatively weak resistance to stress and disease and is thus a promising biological model system for drug screening and antibiotic resistance and environmental pollutant toxicity studies15,16,17. In our previous study, we evaluated TPT toxicity using this model and reported that TPT exposure hindered ontogeny by affecting carbohydrate, lipid, and amino acid metabolism in the midgut18. Similarly, TPT toxic effects have been comprehensively investigated in multiple species, however, its effects on insect gut microbiota diversity remain obscure. In this study, 16S rDNA sequencing was used to examine the impact of TPT exposure on microbial diversity in the silkworm midgut. Our findings shed new light on how TPT exposure impacts gut microbial homeostasis and highlights TPT ecotoxicity risks in environment and aquatic organisms.
Materials and methods
Chemicals
TPT chloride (CAS 639-58-7 and > 96% purity) was obtained from Sigma Aldrich (St. Louis, MO, USA) and diluted in absolute ethanol to generate a 10 μg/mL stock solution.
Silkworm feeding
Bombyx mori (Jingsong × Haoyue strain) were fed formula feed18 supplemented with 0 and 2 μg/kg TPT for the entire 5th instar larval stage. In briefly, all larvae were reared under a 12-h light/dark cycle at a constant temperature of 26 °C. The control group (90 larvae) and TPT-exposure group (90 larvae) contained 3 replicates respectively. The formula feed was prepared and cut into 4*4 cm. To avoid the loss of water in artificial feed, and the prepared feed were place in a sterilized plastic box. The control group and TPT-exposure group were as consistent as possible. The midguts of silkworms were collected for further investigation after 4 days of feeding.
DNA extraction, amplification, and 16S rDNA sequencing
Gut content bacterial DNA was extracted using the QIAamp PowerFecal DNA Kit (Qiagen, Hilden, Germany), amplified using polymerase chain reaction targeting the V3–V4 hypervariable region of 16S rDNA, and then sequenced using Illumina NovaSeq6000 (Illumina Inc., San Diego, CA, USA). Raw 16S rDNA sequencing data were submitted to the Sequence Read Archive with the accession number PRJNA963166 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA963166).
Data preprocessing and statistical analysis
Trimmomatic (Version 0.35)19 and FLASH (Version 1.2.11)20 programs were used to remove low-quality reads and assemble paired-end reads, respectively. Clean reads were clustered into operational taxonomic units (OTUs) using Vsearch (Version 2.4.2)21. All valid reads were blasted against the Silva Version 138 database (16s/18s rDNA) using the RDP classifier (Version 2.2)22 and Unite database using BLAST23. The microbial diversity in the midgut content isolated from silkworm larvae was analyzed by the α-diversity, including species richness (Chao 1) and community diversity (Shannon, Simpson and PD whole tree). The Unifrac Principal coordinates analysis (PCoA) between the TPT exposure group and the control group was drawn with the QIIME software (version 1.8.0). To characterize the microbial differences between the different groups, linear discriminant analysis (LDA) effect size (LEfSe) analysis was employed using Galaxy online tools (http://huttenhower.sph.harvard.edu/lefse/).
Through the pathway enrichment analysis of differential metabolites, it is helpful to understand the mechanism of metabolic pathway change in different samples. Pathway enrichment analysis was performed using KEGG ID of differential metabolites, and the enrichment results of metabolic pathways were obtained. Hypergeometric tests were applied to identify pathway entries that were significantly enriched in significantly differentially expressed metabolites compared to the overall background. Its calculation formula is as follows:
N is the total number of metabolites; n is the number of differentially expressed metabolites in N. M is the number of metabolites annotated in a particular pathway; m is the number of differential metabolites in a particular pathway. With p-value ≤ 0.05 as the threshold, the pathway that meets this condition is the pathway that is significantly enriched in differential metabolites. The smaller the p-value, the more significant the difference of the metabolic pathway.
Results
Data acquisition and analysis
In total, 805,966 and 810,383 raw sequences were collected from the control (CK) and TPT exposure groups (TPT), respectively (Table S1). After quality testing, 765,471 and 763,686 high-quality reads were respectively acquired with an average of 76,458 (range 75,112–78,588) clean reads/sample. Rarefaction and rank abundance curve results indicated that nearly all microbial species were recognized (Fig. 1a,b). Using Vsearch (Version 2.4.2) software to classify OTUs based on nucleotide-sequence similarity, 5273 and 5065 OTUs were identified in CK and TPT-exposure group samples, respectively, ranging from 424 to 728 OTUs/sample (Fig. 1c).

TPT exposure effects on microbial species and operational taxonomic units (OTUs). (a) Rarefaction and (b) rank abundance curves. (c) OTUs were identified between the control (CK) and TPT-exposure group (TPT) samples.
Comparative analyses of gut microbial diversity
To further explore the effects of TPT exposure on silkworm midgut microbiota, alpha and beta diversity indices indicating microbial diversity were analyzed. Comparative analyses of diversity indices between two groups showed no significant differences in Chao1, Simpson, Shannon, and PD_whole_tree indices (Fig. 2a–d). Diversity index dilution curve showed that the curve will eventually flatten, indicating that the sequencing depth is large enough and the established libraries can truly and effectively reflect the diversity of bacterial in the samples with research significance and practical value (Fig. S1). Alpha-diversity analyses revealed that TPT exposure could induce the fluctuations of gut microbial diversity and abundance in silkworms.

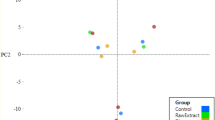
Comparative analyses of gut microbial diversity. Alpha diversity index analyses: (a) Chao1, (b) Simpson, (c) Shannon, and (d) PD_whole_tree indices. (e) Beta-diversity analysis using PCoA.
Beta-diversity was analyzed using principal coordinate analysis (PCoA) (Fig. 2e), which reflected gut microbial similarities and differences between groups, and showed that most of individuals in both groups were separated and minority individuals were clustered together, suggesting that TPT exposure had an obvious impact on major gut microbiota components.
Comparative analysis of microbial taxonomic composition
In total, 23 phyla and 353 genera were recognized in the CK group, while 20 phyla and 358 genera were recognized in the TPT group (Table S2). At the genus level, midgut microbiota were composed of several predominant bacterial genera, including Muribaculaceae, Lactobacillus, UCG-010, Bacteroides, UCG-005, Clostridium_sensu_stricto_1, Rikenellaceae_RC9_gut_group, Escherichia-Shigella, Sphingomonas, Burkholderia-Caballeronia-Paraburkholderia, Lachnospiraceae_NK4A136_group, Alistipes, Prevotella, [Eubacterium]_coprostanoligenes_group, and Christensenellaceae_R-7_group.
Biomarkers reflecting taxonomic levels between groups were analyzed using the linear discriminant analysis coupled with effect size measurements (LEfSe). Analysis results of significantly different bacteria show that f__Coriobacteriaceae, f__Campylobacteraceae, f__Bacillaceae, o__Bacillales, f__Mycoplasmataceae, o__Mycoplasmatales, f__Caldicoprobacteraceae, o__Caldicoprobacterales, f__MBA03, o__MBA03, and f__Pseudomonadaceae were more abundant in the TPT group; whereas o__Gaiellales, c__Thermoleophilia, f__Fibrobacteraceae, o__Fibrobacterales, c__Fibrobacteria, f__Butyricicoccaceae, f__Oscillospiraceae, f__Fusobacteriaceae, f__Rhodobacteraceae, o__Rhodobacterales, f__SC_I_84, and f__Morganellaceae were more abundant in the CK group (Fig. 3a,b, Tables S3, S4).

Comparative analysis of microbial taxonomic composition. (a) Annotation analysis of significantly different bacteria (SDB) based on the linear discriminant analysis coupled with effect size measurements (LEfSe). (b) Score plot of SDB based on LEfSe analysis. Green and red indicates a SDB that is more abundant in the TPT and CK group, respectively.
Predicted functional potential of the altered microbiome
To explore altered genes in silkworm microbiota after TPT exposure, Kyoto Encyclopedia of Genes and Genomes24,25 (KEGG, www.KEGG.jp/KEGG/kegg1.html) orthology level 3 pathways were identified using PICRUSt2, and showed that biosynthesis of ansamycins, fructose and mannose metabolism, glycerolipid metabolism, type II diabetes mellitus, glycolysis/gluconeogenesis, lipid metabolism, translation proteins, atrazine degradation, DNA repair and recombination proteins, nicotinate and nicotinamide metabolism were significantly increased after TPT exposure (Fig. S2).
Discussion
TPT is a toxic chemical used in various industrial and agricultural applications. TPT exposure risk studies have reported harmful effects on human health, including reproductive toxicity, immunotoxicity, neurotoxicity, and endocrine disruption7,8. TPT exposure occurs via inhalation, ingestion, or skin contact with contaminated water, soil, or food26,27. Therefore, proper protective measures should be taken to minimize TPT exposure and limit environmental contamination in occupational settings. In this study, we examined TPT exposure on silkworm microbial diversity and showed that TPT exposure induced the gut microbial diversity and abundance fluctuations (alpha-diversity index analyses). Similarly, beta-diversity analyses showed that TPT exposure had an obvious impact on major gut microbiota components.
It is reported that TPT exposure exerts a negative impact on microbial diversity, including bacteria, fungi, and algae28,29,30. Exposure alters microbial community structures, thereby reducing bacterial abundance and diversity while promoting pathogen growth. In our study, we found that TPT can upregulate the abundance of pathogen in the midgut, including f__Campylobacteraceae, f__Mycoplasmataceae, and o__Mycoplasmatales (Fig. 3). Members of the family Campylobacteraceae neither ferment nor oxidize carbohydrates, instead they obtain energy from amino acids, or tricarboxylic acid cycle intermediates31. Members of the family Mycoplasmataceae can cause cell damage through different mechanisms, such as obtaining lipids and cholesterol on the cell membrane, causing membrane damage, and releasing neurotoxins, phosphatases, and hydrogen peroxide32,33. Therefore, we infer that TPT can cause damage to midgut cells by upregulating pathogenic bacteria in the intestine.
It was reported that the physiological stress of TPT has a negative impact on many intestinal bacteria, significantly reducing microbial diversity34. Furthermore, TPT could cause lipid metabolism abnormalities by affecting intestinal microbiome34. Our investigation found that TPT induced a significant increase in fructose and mannose metabolism, glycerolipid metabolism, glycolysis/gluconeogenesis, lipid metabolism, and translation proteins in the midgut microbiome of silkworms (Fig. S2). These changes may be related to the abnormality of carbohydrate, lipid, and amino acid metabolism in the midgut of silkworms caused by TPT exposure18. In our study, we used the silkworm model to explore the effects of TPT exposure on gut microbial diversity and observed that adverse effects were similar to other aquatic animals34. Therefore, TPT levels must be monitored in environmental samples to prevent potential ecological harm.
Data availability
The datasets generated and/or analysed during the current study are available in the NCBI repository, accession number PRJNA963166.
References
Li, Z. H. et al. Systematic toxicological analysis of the effect of salinity on the physiological stress induced by triphenyltin in Nile tilapia. Aquat. Toxicol. 257, 106441 (2023).
You, J. et al. Organotins in a food web from the three Gorges Reservoir, China: Trophic enrichment and potential health risk. Sci. Total. Environ. 845, 157276 (2022).
Chen, C., Chen, L., Huang, Q., Chen, Z. & Zhang, W. Organotin contamination in commercial and wild oysters from China: Increasing occurrence of triphenyltin. Sci. Total Environ. 650, 2527–2534 (2019).
Anastasiou, T. I., Chatzinikolaou, E., Mandalakis, M. & Arvanitidis, C. Imposex and organotin compounds in ports of the Mediterranean and the Atlantic: Is the story over?. Sci. Total. Environ. 569–570, 1315–1329 (2016).
Sham, R. C. et al. Occurrence, ecological and human health risks of phenyltin compounds in the marine environment of Hong Kong. Mar. Pollut. Bull. 154, 111093 (2020).
Beg, M. A. et al. Organotin antifouling compounds and sex-steroid nuclear receptor perturbation: Some structural insights. Toxics 11, 25 (2022).
He, S., Li, P. & Li, Z. H. Review on endocrine disrupting toxicity of triphenyltin from the perspective of species evolution: Aquatic, amphibious and mammalian. Chemosphere 269, 128711 (2021).
Yi, A. X., Leung, K. M., Lam, M. H., Lee, J. S. & Giesy, J. P. Review of measured concentrations of triphenyltin compounds in marine ecosystems and meta-analysis of their risks to humans and the environment. Chemosphere 89, 1015–1025 (2012).
Lyssimachou, A., Navarro, J. C., Bachmann, J. & Porte, C. Triphenyltin alters lipid homeostasis in females of the ramshorn snail Marisa cornuarietis. Environ. Pollut. 157, 1714–1720 (2009).
Xiao, Y., Jiang, J., Hu, W., Zhao, Y. & Hu, J. Toxicity of triphenyltin on the development of retinal axons in zebrafish at low dose. Aquat. Toxicol. 189, 9–15 (2017).
Zhang, C., Jiang, D., Wang, J. & Qi, Q. The effects of TPT and dietary quercetin on growth, hepatic oxidative damage and apoptosis in zebrafish. Ecotoxicol. Environ. Saf. 224, 112697 (2021).
Wu, K. et al. Long-term triphenyltin exposure disrupts adrenal function in adult male rats. Chemosphere 243, 125149 (2020).
Horie, Y. et al. Effects of triphenyltin on reproduction in Japanese medaka (Oryzias latipes) across two generations. Aquat. Toxicol. 192, 16–23 (2017).
Meng, X., Zhu, F. & Chen, K. Silkworm: A promising model organism in life science. J. Insect Sci. 17, 97 (2017).
Bian, D. et al. Evaluation of tolerance to λ-cyhalothrin and response of detoxification enzymes in silkworms reared on artificial diet. Ecotoxicol. Environ. Saf. 232, 113232 (2022).
Muhammad, A. et al. Toxic effects of acute exposure to polystyrene microplastics and nanoplastics on the model insect, silkworm Bombyx mori. Environ. Pollut. 285, 117255 (2021).
Chen, B. et al. Gut bacteria of the silkworm Bombyx mori facilitate host resistance against the toxic effects of organophosphate insecticides. Environ. Int. 143, 105886 (2020).
Chen, X. D. et al. Toxic effects of triphenyltin on the silkworm Bombyx mori as a lepidopterous insect model. Ecotoxicol. Environ. Saf. 247, 114245 (2022).
Bolger, A. M. et al. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Reyon, D. et al. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 30, 460–465 (2012).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Mount, D. W. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007, pdb.top17 (2007).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Hou, Y. et al. Triphenyltin exposure induced abnormal morphological colouration in adult male guppies (Poecilia reticulata). Ecotoxicol. Environ. Saf. 242, 113912 (2022).
Sham, R. C. et al. Occurrence and trophic magnification profile of triphenyltin compounds in marine mammals and their corresponding food webs. Environ. Int. 137, 105567 (2020).
Cao, Z. et al. Physiological responses of marine Chlorella sp. exposed to environmental levels of triphenyltin. Environ. Sci. Pollut. Res. Int. 30, 26387–26396 (2023).
Shapiro-Ilan, D. I., Reilly, C. C. & Hotchkiss, M. W. Comparative impact of artificial selection for fungicide resistance on Beauveria bassiana and Metarhizium brunneum. Environ. Entomol. 40, 59–65 (2011).
Boopathy, R. & Daniels, L. Pattern of organotin inhibition of methanogenic bacteria. Appl. Environ. Microbiol. 57, 1189–1193 (1991).
Fitzgerald, C. Campylobacter. Clin. Lab. Med. 35, 289–298 (2015).
Vizarraga, D. et al. The sialoglycan binding adhesins of Mycoplasma genitalium and Mycoplasma pneumoniae. Trends Microbiol. 29, 477–481 (2021).
Razin, S. et al. Phospholipid and cholesterol uptake by Mycoplasma cells and membranes. Biochim. Biophys. Acta. 598, 628–640 (1980).
He, S. et al. Triphenyltin exposure causes changes in health-associated gut microbiome and metabolites in marine medaka. Environ. Pollut. 288, 117751 (2021).
Acknowledgements
This research was funded by the Key Scientific and Technological Grant of Zhejiang for Breeding New Agricultural Varieties (2021C02072-1); the earmarked fund for CARS-18; Zhejiang Provincial Key R&D Project (2019C02044); the Zhejiang Provincial Silkworm and Bee Resources Utilization and Innovation Research Key Laboratory (2020E10025).
Author information
Authors and Affiliations
Contributions
W.Z., X.Z., and X.H. designed the research and analyzed the data. W.Z., X.C., X.W., A.Y., J.C., and X.H. conducted the research. W.Z., X.Z., and X.H. wrote the article. W.Z. had primary responsibility for the final content, and all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, W., Zhang, X., Chen, X. et al. Short-term triphenyltin exposure alters microbial homeostasis in the silkworm (Bombyx mori) midgut. Sci Rep 13, 15183 (2023). https://doi.org/10.1038/s41598-023-41948-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-41948-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
