
Abstract
A bidirectional communication exists between the brain and the gut, in which the gut microbiota influences cognitive function and vice-versa. Gut dysbiosis has been linked to several diseases, including Alzheimer's disease and related dementias (ADRD). However, the relationship between gut dysbiosis and markers of cerebral small vessel disease (cSVD), a major contributor to ADRD, is unknown. In this cross-sectional study, we examined the connection between the gut microbiome, cognitive, and neuroimaging markers of cSVD in the Framingham Heart Study (FHS). Markers of cSVD included white matter hyperintensities (WMH), peak width of skeletonized mean diffusivity (PSMD), and executive function (EF), estimated as the difference between the trail-making tests B and A. We included 972 FHS participants with MRI scans, neurocognitive measures, and stool samples and quantified the gut microbiota composition using 16S rRNA sequencing. We used multivariable association and differential abundance analyses adjusting for age, sex, BMI, and education level to estimate the association between gut microbiota and WMH, PSMD, and EF measures. Our results suggest an increased abundance of Pseudobutyrivibrio and Ruminococcus genera was associated with lower WMH and PSMD (p values < 0.001), as well as better executive function (p values < 0.01). In addition, in both differential and multivariable analyses, we found that the gram-negative bacterium Barnesiella intestinihominis was strongly associated with markers indicating a higher cSVD burden. Finally, functional analyses using PICRUSt implicated various KEGG pathways, including microbial quorum sensing, AMP/GMP-activated protein kinase, phenylpyruvate, and β-hydroxybutyrate production previously associated with cognitive performance and dementia. Our study provides important insights into the association between the gut microbiome and cSVD, but further studies are needed to replicate the findings.
Similar content being viewed by others

Introduction
With the increase in life expectancy, age-related diseases are anticipated to cause an enormous burden on health care and social systems. Cerebral small vessel disease (cSVD) contributes to cognitive impairment and dementia in the elderly, with characteristic early deficits in information processing and executive function (EF)1,2,3,4,5,6.
Several magnetic resonance imaging (MRI) markers have been established as vital tools for the diagnosis and research on cSVD, including the peak width of skeletonized mean diffusivity (PSMD) and white matter hyperintensity volumes (WMH)1,6,7,8. PSMD is a fully automated diffusion tensor imaging (DTI) marker based on the skeletonization of white matter tracts and histograms analysis of mean diffusivity (MD)9,10. PSMD is highly sensitive to vascular-related white matter lesions and is associated with processing speed in patients with cSVD11. WHM (total and periventricular) are focal and multifocal confluent lesions of white matter injury that reflect axonal loss due to chronic ischemia caused by cSVD5,11,12. Studies have shown that extensive WMH burden is associated with an increased risk of incident stroke and dementia. The initial stage of cSVD is characterized by a decline in EF, commonly observed as a deficit in problem-solving, attention, and decreased information processing speed. Combined with MRI measures of brain structure and neuropsychological assessment, measuring executive function allows for both a structural and functional approach to understanding the etiology of cSVD13,14,15,16,17,18.
Gut microflora imbalances are associated with neurological diseases, including Parkinson's disease, Alzheimer's disease, and stroke19,20,21,22,23,24,25. The gut microbiome modulates the host brain function through the microbiota-gut-brain axis. This bidirectional communication involves various signaling pathways, including the endocrine, enteric nervous systems, immune systems, and tryptophan metabolism19,20,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40. Moreover, it is suggested that the gut microbiota may play a role in vascular cognitive impairment and dementia, as well as related risk factors, such as cSVD. For example, the administration of Clostridium butyricum, a butyrate-producing gram-positive bacterium, to vascular dementia mouse models has been shown to attenuate cognitive dysfunction and protect against cerebral ischemia/reperfusion injury41. Gut dysbiosis has also been associated with cSVD, where patients with Enterotype I were more likely to have cognitive decline and high total cSVD scores42. Others have also shown that gut-derived Metabolite Phenylacetylglutamine levels are associated with WMH burden in patients with acute ischemic stroke43.
Although studies have reported an association between the gut microbiome and cSVD, whether this association can be extended to structural (i.e., WMH and PSMD) MRI markers of cSVD and cognitive performance (i.e., EF) is unknown42. In this study, we used stool, MRI, and neurocognitive data from the Framingham Heart Study to assess the cross-sectional association between the gut microbiome and markers of cSVD.
Material and methods
Sample description
The FHS is an ongoing population-based, longitudinal cohort initiated in 1948, recruiting 5,209 participants from Framingham (MA, USA). The FHS's initial aim was the prospective investigation of the risk factors associated with CVD and has since expanded to other diseases. In 1971, 5,214 children of the Original Cohort (the Offspring Cohort) and their spouses were enrolled and underwent similar examinations as the Original Cohort every four years. The children of the Offspring participants and their spouses have been followed since 2002 as the 3rd Generation cohort, and participants have been examined on several occasions. The current project was limited to participants from the New Offspring Spouse, the 3rd Generation, and the OMNI 2 cohorts who attended the third examination (2016–2019) and provided their stool specimens. All participants provided written informed consent for blood drawn, MRI testing, and cognition assessments at each examination. The IRB of Boston University School of Medicine approved the FHS protocols and participant consent forms.
MRI acquisition and processing
Participants were imaged on a 1.5 or 3T scanner. We used 3D-T1, fluid-attenuated inversion recovery (FLAIR), and Diffusion Tensor Imaging (DTI) sequences to derive the neuroimaging markers. Image analyses were performed at the Imaging of Dementia and Aging (IDeA) laboratory at UC Davis, using established pipelines44. All images were analyzed by operators blinded to all participant characteristics, including cognitive performance on neuropsychological testing. Peak width of skeletonized mean diffusivity (PSMD) is a novel and fully automated MRI biomarker, which has shown clinical relevance in SVD, an essential contributor to VCI45,46. It is based on skeletonization and histogram analysis of diffusion imaging DTI data. PSMD scores have also been shown to be strongly correlated with processing speed47. PSMD is measured as the difference between the 5th and 95th percentiles of the distribution of the voxel MD value across the skeleton of the brain white matter. PSMD and WMH measures were expressed as the percentage of total intracranial volume to correct for head size and were further log-transformed due to skewness before analyses.
Executive function
Clinical neuropsychologists and trained research assistants administered validated neuropsychological tests. We derived the difference between the Trail Making Test B and Trail Making Test A (Trails B-A) as a measure of EF48,49. Values of Trails B-A were natural log-transformed to normalize its distribution before analyses and further inversed such that higher EF scores indicate poor performance. Details of the neuropsychological protocol can be found elsewhere48,49.
Microbiome
Sample handling and DNA extraction
Stool samples were collected in 100% ethanol as previously described50,51,52 and stored at − 80 °C. Briefly, for DNA and RNA co-extraction, the QIAamp 96 PowerFecal Qiacube HT Kit (Qiagen Cat No./ID: 51531) was paired with the Allprep DNA/RNA 96 Kit (Qiagen Cat No./ID: 80311), and IRS solution (Qiagen Cat No./ID: 26000-50-2) for a custom protocol. For initial lysis, 50–200 mg of stool per sample were frozen into individual wells of the PowerBead plate containing 0.1 mm glass beads (Cat No./ID: 27500-4-EP-BP) on a dry ice block. Next, 650 µl of 55 °C heated PW1 buffer and 25 µL of freshly-prepared 1 M DTT were added directly to each sample well before lysis by beating on a TissueLyzer II at 20 Hz for a total of 10 min (in two 5-min intervals with plate rotation in between). Next, samples were pelleted by centrifugation for 6 min at 4500 × g, and supernatants were transferred to a new S block (supplied in PowerFecal Kit), combined with 150 µl of IRS solution, and vortexed briefly before a one-minute incubation. Next, sealed samples were centrifuged again for 6 min at 4500 × g, and up to 450 µl of supernatant was transferred to a new S block, combined with 600 µl of Buffer C4 (PowerFecal Kit), mixed by pipetting ten times and incubated for 1 min. Next, samples were transferred into an AllPrep 96 DNA plate on clean S blocks and centrifuged for 3 min at 4500 × g. This centrifugation step was repeated until the entire sample had been centrifuged. Finally, the AllPrep 96 DNA plate was stored at 4 °C until after DNA extraction.
The Allprep 96 DNA plate was removed from 4 °C and placed on top of a 2 mL waste block for DNA extraction. First, 500 µl AW1 buffer was added to the DNA plate and sealed before centrifuging for 4 min at 4500 × g. The waste block was emptied after each wash step. Next, 500 µl AW2 buffer was added to the DNA plate, sealed with AirPore tape, and centrifuged for 10 min at 4500 × g. Next, the Allprep 96 DNA plate was placed on the elution plate, and 100 µl of 70 °C heated EB Buffer was added to each sample column and incubated for 5 min. The DNA plate was sealed and then centrifuged for 4 min at 4500 × g to elute the DNA, which was stored at -20 °C. All incubation and centrifugation steps were performed at room temperature.
16S rRNA gene sequencing
16S rRNA gene libraries targeting the V4 region were prepared by first using qPCR to normalize template concentrations and determine the optimal cycle number as described in Lavoie et al.52 Briefly, library construction was performed in quadruplicate with the primers 515F (5′-AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGTAA-3′) and unique reverse barcode primers from the Golay primer set53,54. After amplification, sample replicates were pooled and cleaned via the Agencourt AMPure XP-PCR purification system. Prior to final pooling, purified libraries were normalized via qPCR in two 25 µL reactions, 2 × iQ SYBR SUPERMix (Bio-Rad, REF: 1708880) with Read 1 (5′-TATGGTAATT GT GTGYCAGCMGCCGCGGTAA-3′), Read 2 (5′-AGTCAGTCAG CC GGACTACNVGGGTWTCTAAT-3′) primers. Pools were quantified by Qubit (Life Technologies, Inc.) and sequenced on an Illumina MiSeq with 2 × 150 bp reads using custom index 5′-ATTAGAWACCCBDGTAGTCC GG CTGACTGACT-3′ and custom Read 1 and Read 2 primers mentioned above.
Microbiome data analysis
We used QIIME255 for downstream processing of microbiome sequencing data. Briefly, forward and reverse reads were truncated to preserve minimum Phred quality scores of 28 in 75% of reads. Next, high-quality sequencing reads were clustered into operational taxonomic units (OTUs) at a 97% dissimilarity threshold. Then, the clustered OTUs were classified against the Greengenes database to assign taxonomy to the sequences. Lastly, we constructed phylogenetic trees using the MAFFT algorithm in QIIME2. To avoid deviation caused by the effects of different sequencing depths and low representatives, we removed OTUs with fewer than four reads in less than 10% of samples. We also constructed a table of relative abundance counts without rarefaction for multivariate analyses. Finally, we exported the OTUs, taxonomy, phylogeny, and metadata tables to the R platform for further research. Finally, the cross-associations between MRI measures and gut microbial composition were assessed using multivariate linear models, considering MRI measures as continual variables, and differential analyses in which MRI measures were divided into burden groups.
Microbiome multivariable linear regression
Multivariable association analyses were conducted using the R package MaAsLin256 using relative abundance counts without rarefaction and the different diversity indexes. We used the following model
where Taxa is relative abundance counts, Marker is the log-transformed cSVD marker (WMH, PSMD, EF) measure, and \(time\) is the time difference between the stool sample collection and the MRI scan. In addition, we considered the age at which MRI scans were completed, and only EF measures were adjusted for the participant’s education level. Finally, MaAsLin2 analysis was done using the following settings: minimum abundance (relative) cutoff at 10−3, the minimum percent of samples for which a feature is detected at minimum abundance was set a 10%, and we selected the negative binomial as the regression method.
Microbiome differential abundance analysis
We used the following procedure to stratify participants into cSVD burden groups according to WMH and PSMD markers, and a similar strategy was used for cognitive function (EF): (i) cSVD markers were log-transformed to ensure normality; (ii) for each marker, we created two groups of interest dichotomized by the upper quintile (20% of participants, “High burden” group) and the bottom four quintiles (80% of participants, “Lower burden” group) of the residual. The differential analysis of microbiome composition analyses was conducted by comparing differences in abundance and alpha-diversity (Observed, Chao1, Shannon, and Simpson index) using a repeated-measures analysis of variance (ANOVA). Beta-diversity was estimated using principal coordinated analysis (PCoA) with Bray–Curtis, weighted and unweighted UniFrac distances. PCoA is an unsupervised dimensionality reduction method that can be used to visualize group separations of compositional data. Finally, we used permutational analysis of variance (PERMANOVA) implemented in the vegan package to test for differences in community composition among the “Lower burden” and “High burden” groups, followed by pairwise tests between groups. The microbiome uniqueness, which reflects how dissimilar individuals are from their neighbors in the population, was computed as in Wilmanski et al.57 Statistically significant differences were reported after adjusting for multiple testing using the Benjamini–Hochberg procedure. We conducted the differential abundance analysis using MaAsLin2, with relative abundance counts, adjusting for relevant covariates (age, age2, sex, BMI, education, and \(time\))56. For taxonomy features significantly different between groups, we performed functional profiling of the bacterial metagenome using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt).
Ethics approval and consent to participate
This study was conducted at the University of Texas Health Science Center at San Antonio, USA, using data collected from the Framingham Heart Study (FHS) participants (Generation 3, New Offspring Spouse, OMNI2 at their third examination cycle) between 2016 and 2019. Participants provided their written informed consent to participate in this study. This study protocol was reviewed and approved by the FHS Executive Committee and the Institutional Review Board at the Boston University Medical Center. All research methods were performed in accordance with relevant guidelines/regulations, and informed consent was obtained from all participants and/or their legal guardians.
Results
Study population
A total of 1,406 participants successfully returned their stool collection kits. Of these, 9 samples failed the quality control process after DNA extraction and sequencing, and 417 participants did not have MRI or neurocognitive measures. Therefore, the present study was limited to the 980 participants who were administered MRI scans in a previous examination or during the visit at which stool samples were requested. In addition, we excluded participants with dementia (3) and incident stroke (5) and, thus, retained 972 participants for further analyses. Participants' characteristics are presented in Table 1. The study flowchart is provided as supplementary material (Supplementary File SF1, Fig. S1). In addition, a correlation plot depicting the relationship between cSVD markers and the different covariates is provided in Supplementary File SF1, Fig. S2.
High cSVD burden is associated with decreased Bacteroidetes abundance
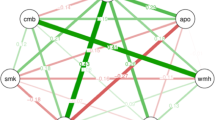
We performed multivariable linear regression analyses to test the associations between the relative abundance of the gut microbiome taxa and cSVD markers (PSMD, WMH, and EF). Statistically significant (adjusted p values < 0.05) associations are summarized in Fig. 1 and Table 2, and detailed association results are provided in Supplementary File SF2, Tables S1–S3. At the phylum level, measures of all cSVD markers were negatively correlated with Bacteroidetes and positively correlated with Proteobacteria, both gram-negative bacteria. Additionally, the gram-positive Firmicutes was positively correlated with PSMD and EF but negatively correlated with measures of WMH volumes. Finally, the gram-negative Synergistetes showed a positive correlation with measures of PSMD and no association with other markers.

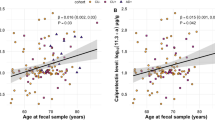
Multivariable association between the gut microbiome and cSVD markers (PSMD, WMH, EF). (A) Volcano plots depicting a significant association (BH-adjusted p value < 0.05) of several genera and PSMD, WMH, and EF (from left to right). (B) Heatmap of common statistically significant genera associated with cSVD markers. (C) Scatter plots of genera Clostridium and Barnesiella relative abundance and normalized PSMD measures.
Barnesiella, a lowly abundant bacterium (< 1% of the human gut), was negatively correlated with all measures of cSVD markers at the genus level. In addition, the abundance of the gram-negative Pseudobutyrivibrio was positively correlated with measures of EF but negatively correlated with MRI markers PSMD and WMH. Ruminococcus was negatively correlated with WMH and EF but positively associated with PSMD. Furthermore, the gram-positive Clostridium was positively correlated with EF and PSMD and negatively correlated with WMH volumes. Other genera were positively correlated with PSMD, including Cloacibacillus and Streptococcus.
At the species level, Xylanivorans, a butyrate-producing bacterium from the rumen, was negatively correlated with PSMD and WMH and positively correlated with EF. The gram-negative Intestinihominis, a species from the Barnesiella genus, was negatively correlated with all three measures of cSVD markers. The gram-positive bolteae was positively correlated with EF and PSMD and negatively correlated with WMH volumes. Additional species correlated positively (Callidus, mitsuokai) and negatively (finegoldii) with PSMD. Finally, we also found a negative correlation between WMH volumes and the abundance of mitsuokai and callidus species.
Altogether, these results suggested an association between cSVD markers' measures and the gut microbiome's relative abundance. Therefore, we next performed differential analyses to assess the difference in microbiome composition between FHS participants stratified by cSVD markers burden groups.
Differential abundance analysis of the gut microbiome and cSVD markers
Study participants were stratified by burden groups (Lower and High burden), as explained in the methods section. Then, we conducted differential abundance analyses to assess microbial composition differences between groups. The results are summarized in Fig. 2 and Table 2, and the complete list of bacteria differentially abundant between Healthy (Lower burden) and Unhealthy (High burden) groups is provided in Supplementary File SF2, Tables S4–S6.

Differential abundance analysis of the gut microbiome and cSVD markers (PSMD, WMH, EF). (Top) Stacked plots at 10% minimum abundance highlight differences at the genus level between Healthy and Unhealthy groups of cSVD. (Down) relative abundance of Barnesiella, Christensenella, and Ruminococcus genera in PSMD, WMH, and EF burden groups, respectively (Healthy = Lower burden, Unhealthy = High burden).
At the phylum level, the Candidate Phylum OD1 bacteria (OD1) had reduced relative abundance (adjusted p value < 0.05) in the Unhealthy groups compared to Healthy (Supplementary File SF1 Figs. S2, S3 and S4) for all three cSVD markers. We also observed a reduced abundance of the gram-positive Firmicutes in Unhealthy groups of WMH and EF. Additionally, Actinobacteria and Bacteroidetes had reduced abundance, and Proteobacteria had increased abundance in the Unhealthy WMH group.
At the genus level, the relative abundance of genera Barnesiella, Ruminococcus, and Methanobrevibacter were significantly reduced in the Unhealthy groups of WMH, PSMD, and EF. In addition, Clostridium, a gram-positive bacterium that includes several human pathogens, had reduced abundance, while Christensenella had increased abundance in Unhealthy PSMD and WHM groups. The genera cc_115 and Oscillospira have increased abundance in PSMD but decreased abundance in WMH Unhealthy groups. Other genera associated with measures of cSVD markers included Victivallis (Decreased abundance in PSMD), Anaerosinus (decreased abundance in PSMD), Alistipes (decreased abundance in EF), and Collinsella (Increased abundance in EF).
At the species level, Intestinihominis and lactaris had decreased abundance in Unhealthy groups, whereas Aldenense has increased abundance in Healthy groups of PSMD, WMH, and EF. In addition, ruminantium was found to have increased abundance in PSMD and EF but decreased abundance in WMH. Furthermore, lavalense, a gram-positive bacterium from the Clostridium genius, has increased abundance in EF but decreased in PSMD. Finally, other species associated with cSVD markers in differential abundance analysis included.
Aerofaciens (increased abundance in EF), vadensis (decreased abundance in PSMD, and Spiroforme, guilliermondii, and variabile with increased abundance in PSMD.
The complete list of differentially abundant taxa in cSVD stratified groups is provided in Supplementary File SF2 Tables S4–S6; the box plots of the significant (adjusted p value < 0.05) taxa are provided in Supplementary File SF1 Figs. S2–S4.
Barnesiella intestinihominis showed a consistent direction of association in both differential and multivariable association analyses
Multivariable association and differential abundance analyses identified different bacteria associated with markers of cSVD. We hypothesized that bacteria showing a consistent direction of association for both methods are more likely to be accurate. At the genus level, Barnesiella was negatively correlated with all cSVD markers and has decreased abundance in Unhealthy individuals, thus supporting the reliability of these associations with cSVD markers. We observed a similar trend at the species level, where the gram-negative intestinihominis has decreased abundance in all cSVD markers. Both observations led us to conclude that Unhealthy measures of cSVD (PSMD, WMH, and EF) markers in the FHS are associated with a reduced abundance of Barnesiella intestinihominis.
cSVD markers do not associate with gut microbiome diversity
Biodiversity analysis: α-diversity and β-diversity
We used multivariable and differential analyses to assess the association between cSVD markers and gut microbiome alpha and beta diversity. Measures of α-diversity included observed species, Chao1, Shannon, and Simpson diversity index. Association analyses were adjusted for sex, age, BMI, education, and the time difference between the visits at which data were collected. For differential analysis, we used a one-way analysis of variance (ANOVA) to compute the statistical difference between burden groups (Lower burden, High burden). We did not observe statistical differences between alpha diversity indexes and measures of cSVD markers in multivariable or differential analysis (Supplementary Figs. S17–S20). To estimate the change in the diversity of OTUs between cSVD markers burden groups, we computed the beta diversity and calculated differences using PCoA, as shown in Supplementary Figs. S21–S22. Overall, the different burden groups of all cSVD markers exhibited similar distribution leading to the conclusion that there are very few species differences between samples of FHS participants. Using multivariable analysis, we found that only the Bray–Curtis minimum dissimilarity index (min_bray) significantly correlated with WMH (Supplementary Fig. S20), whereas min_wnifrac did not associate with cSVD markers.
Predictive functional profiling of microbial communities associated with cSVD
We assessed the potential functional role of the microbial communities associated with cSVD markers using the PICRUSt approach described in the Methods. PICRUSt predictions were based on KEGG orthologs (KO), Enzyme Commission (EC) numbers, and MetaCyc metabolic pathways (MePath) enrichments, comparing Healthy to Unhealthy measures of cSVD markers. The predicted MePath associated with PSMD included L-tyrosine, L-methionine, L-phenylalanine, and teichoic acid biosynthesis. In addition, several KO were associated with PSMD, including adenosine kinase, hemerythrin, and glv operon transcriptional regulator (Supplementary File 2, Tables S7–S9, Supplementary File 1, Fig. S23). Predicted MePath associated with WMH included taurine and adenosine degradation, significant KOs were related to a methyltransferase, motility quorum sensor regulator, and ECs included Nitrate reductase, hydrogenase, and gluconate 2-dehydrogenase. Detailed results of the functional profiling of gut bacteria associated with WMH are provided in Fig. S24 of Supplementary File 1 and Tables S10–S12 of Supplementary Fig. S2. For the EF, we found that KOs terms bacterial CydC and CydD, methyltransferase, and type 1 pantothenate kinase, MePath terms taxadiene biosynthesis, pyruvate dehydrogenase, and reductive TCA cycle-1 were all significantly different between Lower and Higher burden EF groups (Supplementary File 1, Fig. S25; Supplementary File S2, Tables S13–S15). Overall, the functional modules associated with the PICRUSt predicted metagenome included AMP/GMP-activated kinase, xanthine, homogentisate, tyrosine, phenylpyruvate, and dicarboxylate-hydroxybutyrate. A cross-comparison of the enriched terms between all cSVD markers revealed that common KOs (Fig. 3) belong to KEGG modules with genes involved in AMP/GMP-activated protein kinase, phenylpyruvate, and β-hydroxybutyrate production.

Functional profiling of microbial communities associated with markers of cSVD. PICRUSt predictions were based on KEGG Orthologs (KO), Enzyme Commission (EC), and MetaCyc pathways (MePath). The common KOs belong to KEGG modules with genes involved in AMP/GMP-activated protein kinase, phenylpyruvate, and β-hydroxybutyrate production, which have been shown to the associated with cognitive decline and dementia.
Discussion
In this cross-sectional association study, we used stool samples, MRI (PSMD, WMH), and neurocognitive assessment (Trail making tests A and B) data from 972 middle-aged participants of the FHS to assess the association between the gut microbiome and markers of cSVD. We found that several phyla, genera, and species were associated with individual markers of cSVD, but overall, the gram-negative Barnesiella intestinihominis has decreased relative abundance in High burden measures of all cSVD markers.
Our results indicated that measures of PSMD positively correlated with the abundance of Firmicutes and Proteobacteria but negatively correlated with Bacteroidetes. However, in the differential analysis comparing Healthy to Unhealthy participants stratified by PSMD measures, only the Candidate Phylum OD1 bacteria (OD1) was found to have decreased abundance in the Unhealthy group. Conversely, elevated WMH measures, predictive of incident stroke, MCI, and dementia, were associated with increased Proteobacteria and decreased Firmicutes and Bacteroidetes in multivariable and differential abundances analyses. Despite the discrepancy in their direction of association, these phyla were previously implicated in cognitive performance58,59. Also, the gram-negative Bacteroidetes, which excretes pro-inflammatory endotoxin Lipopolysaccharide (LPS), was previously associated with AD in animal and human studies with a contradictory direction of association60,61,62,63,64. Furthermore, the abundance of phylum Firmicutes, Proteobacteria, and Proteobacteria were also observed in patients with mild cognitive impairment without dementia42,65.
We also found several genera were associated with markers of cSVD, including Ruminococcus, Catenibacterium, Clostridium, Cloacibacillus, Alistipes, cc_115, Victivallis, Streptococcus, Pseudobutyrivibrio, Barnesiella, Methanobrevibacter, Oscillospira, Anaerosinus, and Collinsella. Moreover, the genus Barnesiella has a consistent direction of association with all cSVD markers. Our results were consistent with previous studies reporting the association between these genera and normal cognition, mild cognitive impairment (MCI), and AD59,66,67. Also, decreased abundance of Barnesiella has been previously associated with age-related cognitive decline in mice66. Finally, the relative abundance of Barnesiella was shown to correlate with cognitive ability in patients with Parkinson's disease67,68, and differentially abundant in mice after induced traumatic brain injury69.
At the species level, our results indicated an association between cSVD markers and several taxa, including Callidus, intestinihominis, guilliermondii, Lactaris, ruminantium, Xylanivorans, and bolteae. Consistent with the observation at the genus level, we found that intestinihominis has decreased abundance in the Unhealthy group in all cSVD markers.
Our results suggested that the functional modules associated with PICRUSt predicted metagenome included AMP/GMP-activated kinase, xanthine, homogentisate, tyrosine, phenylpyruvate, and dicarboxylate-hydroxybutyrate. These findings align with the known functional roles of the related metabolites in neurodegenerative diseases. Indeed, AMP-activated kinase is a metabolic biosensor with anti-inflammatory activities previously associated with AD, and its activation was proposed to eliminate post-synaptic proteins70,71,72,73,74. Similarly, hydroxybutyrate production inhibited inflammasome activation, attenuated AD pathology in mice, and improved cognition in memory-impaired adults75,76. Altogether, these results suggest that elevated measures of cSVD markers are associated with loss of bacteria with neuroprotective effects and increased neuroinflammation.
This study has several limitations, and the results should be replicated and mechanistically validated before clinical interpretation. First, the stool samples, MRI scans, and neuropsychological batteries were not collected simultaneously. Although we adjusted for the time difference in our analyses, further studies where all relevant data are collected simultaneously are required. Second, it is widely known that medication and diet influence the gut microbiota and cSVD. In our study, we did not adjust for diet or medication intake as they were unavailable for the studied sample. Further, we may have omitted relevant confounders in this analysis, potentially biasing the interpretation of our results. However, these limitations are compensated by the large sample size and the statistical approaches used to assess the link between the gut microbiome and cSVD markers. The statistically significant taxa we reported showed a similar trend in both multivariable association and differential abundance analyses.
Conclusion
We used data from 972 participants of the Framingham Heart Study to assess whether the gut microbiota is associated with MRI (PSMD, WMH) and neurocognitive (EF) markers of cSVD. In multivariable and differential analyses, we found the abundance of several phyla, including Firmicutes, Proteobacteria, and Bacteroidetes, associated with unhealthy measures of cSVD makers. This observation was extended at the genus with Pseudobutyrivibrio, Barnesiella, and at the species level with Intestinihominis, Xylanivorans, and lactaris. Altogether, our results suggest that the gram-negative bacterium Barnesiella intestinihominis is strongly associated with unhealthy measures of cSVD markers. In addition, functional analyses indicated that the differentially abundant bacteria gene contents were involved in AMP/GMP-activated kinase, xanthine, homogentisate, tyrosine, phenylpyruvate, and dicarboxylate-hydroxybutyrate processes, all of which were previously associated with cognitive impairment and dementia. Our study provides important insights into the association between the gut microbiome and cSVD, but further studies are needed to replicate the results.
Data availability
All data supporting the findings of this study are publicly accessible through dbGap (Study ID: phs000007.v32.p13, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000007.v32.p13).
References
Dichgans, M. et al. METACOHORTS for the study of vascular disease and its contribution to cognitive decline and neurodegeneration: An initiative of the Joint Programme for Neurodegenerative Disease Research. Alzheimers Dement 12, 1235–1249. https://doi.org/10.1016/j.jalz.2016.06.004 (2016).
Wallin, A. et al. Biochemical markers in vascular cognitive impairment associated with subcortical small vessel disease—A consensus report. BMC Neurol. 17, 1–12. https://doi.org/10.1186/s12883-017-0877-3 (2017).
Banerjee, G. et al. Total MRI small vessel disease burden correlates with cognitive performance, cortical atrophy, and network measures in a memory clinic population. J. Alzheimer’s Dis. JAD https://doi.org/10.3233/JAD-170943 (2018).
Caunca, M. R., De Leon-Benedetti, A., Latour, L., Leigh, R. & Wright, C. B. Neuroimaging of cerebral small vessel disease and age-related cognitive changes. Front. Aging Neurosci. 11, 145. https://doi.org/10.3389/fnagi.2019.00145 (2019).
Du, J. & Xu, Q. Neuroimaging studies on cognitive impairment due to cerebral small vessel disease. Stroke Vasc. Neurol. https://doi.org/10.1136/svn-2018-000209 (2019).
Fan, Y., Xu, Y., Shen, M., Guo, H. & Zhang, Z. Total cerebral small vessel disease burden on MRI correlates with cognitive impairment in outpatients with amnestic disorders. Front. Neurol. https://doi.org/10.3389/fneur.2021.747115 (2021).
Charidimou, A. et al. Clinical significance of cerebral microbleeds on MRI: A comprehensive meta-analysis of risk of intracerebral hemorrhage, ischemic stroke, mortality, and dementia in cohort studies (v1). Int. J. Stroke 13, 454–468. https://doi.org/10.1177/1747493017751931 (2018).
Huijts, M. et al. Accumulation of MRI markers of cerebral small vessel disease is associated with decreased cognitive function. A study in first-ever lacunar stroke and hypertensive patients. Front. Aging Neurosci. https://doi.org/10.3389/fnagi.2013.00072 (2013).
Vinciguerra, C. et al. Peak width of skeletonized mean diffusivity (PSMD) as marker of widespread white matter tissue damage in multiple sclerosis. Multiple Scler. Relat. Disord. https://doi.org/10.1016/j.msard.2018.11.011 (2019).
Vinciguerra, C. et al. Peak width of skeletonized mean diffusivity (PSMD) and cognitive functions in relapsing-remitting multiple sclerosis. Brain Imaging Behav. https://doi.org/10.1007/s11682-020-00394-4 (2021).
Baykara, E. et al. A novel imaging marker for small vessel disease based on skeletonization of white matter tracts and diffusion histograms. Ann. Neurol. https://doi.org/10.1002/ana.24758 (2016).
Gustavsson, A. M. et al. Cerebral microbleeds and white matter hyperintensities in cognitively healthy elderly: A cross-sectional cohort study evaluating the effect of arterial stiffness. Cerebrovasc. Dis. Extra 5, 41–51. https://doi.org/10.1159/000377710 (2015).
Alber, J. et al. in Alzheimers Dement (N Y) Vol. 5, 107–117 (2019).
Arvanitakis, Z., Capuano, A. W., Leurgans, S. E., Bennett, D. A. & Schneider, J. A. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 15, 934–943. https://doi.org/10.1016/s1474-4422(16)30029-1 (2016).
Persyn, E. et al. Genome-wide association study of MRI markers of cerebral small vessel disease in 42,310 participants. Nat. Commun. 11, 1–12. https://doi.org/10.1038/s41467-020-15932-3 (2020).
Freeze, W. M. et al. White matter hyperintensities potentiate hippocampal volume reduction in non-demented older individuals with abnormal amyloid-beta. J. Alzheimers Dis. 55, 333–342. https://doi.org/10.3233/jad-160474 (2017).
Wardlaw, J. M., Hernández, M. C. V. & Muñoz-Maniega, S. What are white matter hyperintensities made of?. J. Am. Heart Assoc. https://doi.org/10.1161/JAHA.114.001140 (2015).
Prins, N. D. et al. Cerebral small-vessel disease and decline in information processing speed, executive function and memory. Brain J. Neurol. https://doi.org/10.1093/brain/awh553 (2005).
Bullich, C., Keshavarzian, A., Garssen, J., Kraneveld, A. & Perez-Pardo, P. Gut vibes in Parkinson’s disease: The microbiota-gut-brain axis. Mov. Disord. Clin. Pract. 6, 639–651. https://doi.org/10.1002/mdc3.12840 (2019).
Cryan, J. F. et al. The microbiota-gut-brain axis. Physiol. Rev. 99, 1877–2013. https://doi.org/10.1152/physrev.00018.2018 (2019).
Cryan, J. F., O’Riordan, K. J., Sandhu, K., Peterson, V. & Dinan, T. G. The gut microbiome in neurological disorders. Lancet Neurol. https://doi.org/10.1016/S1474-4422(19)30356-4 (2020).
Jiang, C., Li, G., Huang, P., Liu, Z. & Zhao, B. The gut microbiota and Alzheimer’s disease. J. Alzheimers Dis. 58, 1–15. https://doi.org/10.3233/jad-161141 (2017).
Kowalski, K. & Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. https://doi.org/10.5056/jnm18087 (2019).
Lubomski, M. et al. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. https://doi.org/10.1007/s00415-019-09320-1 (2019).
Quigley, E. M. M. Microbiota-brain-gut axis and neurodegenerative diseases. Curr. Neurol. Neurosci. Rep. 17, 94. https://doi.org/10.1007/s11910-017-0802-6 (2017).
Long-Smith, C. et al. Microbiota-gut-brain axis: New therapeutic opportunities. (2020). https://doi.org/10.1146/annurev-pharmtox-010919-023628.
Caputi, V. & Giron, M. C. Microbiome-gut-brain axis and toll-like receptors in Parkinson’s disease. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19061689 (2018).
Chernikova, M. A. et al. The Brain-gut-microbiome system: Pathways and implications for autism spectrum disorder. Nutrients https://doi.org/10.3390/nu13124497 (2021).
De la Fuente, M. The role of the microbiota-gut-brain axis in the health and illness condition: A focus on Alzheimer’s disease. J. Alzheimer’s Dis. JAD https://doi.org/10.3233/JAD-201587 (2021).
Dinan, T. G. & Cryan, J. F. The microbiome-gut-brain axis in health and disease. Gastroenterol. Clin. N. Am. 46, 77–89. https://doi.org/10.1016/j.gtc.2016.09.007 (2017).
Doifode, T. et al. The impact of the microbiota-gut-brain axis on Alzheimer’s disease pathophysiology. Pharmacol. Res. https://doi.org/10.1016/j.phrs.2020.105314 (2021).
Ghezzi, L., Cantoni, C., Rotondo, E. & Galimberti, D. The gut microbiome-brain crosstalk in neurodegenerative diseases. Biomedicines https://doi.org/10.3390/biomedicines10071486 (2022).
Isaiah, S. et al. Overview of brain-to-gut axis exposed to chronic CNS bacterial infection(s) and a predictive urinary metabolic profile of a brain infected by Mycobacterium tuberculosis. Front. Neurosci. https://doi.org/10.3389/fnins.2020.00296 (2020).
Kelly, J. R., Minuto, C., Cryan, J. F., Clarke, G. & Dinan, T. G. Cross talk: The microbiota and neurodevelopmental disorders. Front. Neurosci. 11, 490. https://doi.org/10.3389/fnins.2017.00490 (2017).
Khlevner, J., Park, Y. & Margolis, K. G. Brain-gut axis: Clinical implications. Gastroenterol. Clin. N. Am. https://doi.org/10.1016/j.gtc.2018.07.002 (2018).
Kowalski, K. & Mulak, A. Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil. 25, 48–60. https://doi.org/10.5056/jnm18087 (2019).
EA, M., K, T. & A, G. Gut/brain axis and the microbiota. The Journal of clinical investigation 125 (2015). https://doi.org:https://doi.org/10.1172/JCI76304
LH, M., HL, S. & SK, M. The gut microbiota-brain axis in behaviour and brain disorders. Nature reviews. Microbiology 19 (2021). https://doi.org:https://doi.org/10.1038/s41579-020-00460-0
Vogt, N. M. et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 7, 13537. https://doi.org/10.1038/s41598-017-13601-y (2017).
Cattaneo, A. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68. https://doi.org/10.1016/j.neurobiolaging.2016.08.019 (2017).
Liu, J. et al. Neuroprotective effects of Clostridium butyricum against vascular dementia in mice via metabolic butyrate. Biomed. Res. Int. 2015, 412946. https://doi.org/10.1155/2015/412946 (2015).
Saji, N. et al. The association between cerebral small vessel disease and the gut microbiome: A cross-sectional analysis. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105568 (2021).
Yu, F. et al. Gut-derived metabolite phenylacetylglutamine and white matter hyperintensities in patients with acute ischemic stroke. Front. Aging Neurosci. https://doi.org/10.3389/fnagi.2021.675158 (2021).
Maillard, P. et al. Instrumental validation of free water, peak-width of skeletonized mean diffusivity, and white matter hyperintensities: MarkVCID neuroimaging kits. Alzheimers Dement (Amst.) 14, e12261. https://doi.org/10.1002/dad2.12261 (2022).
Wei, N. et al. A neuroimaging marker based on diffusion tensor imaging and cognitive impairment due to cerebral white matter lesions. Front. Neurol. 10, 81. https://doi.org/10.3389/fneur.2019.00081 (2019).
Shi, Y. & Wardlaw, J. M. Update on cerebral small vessel disease: A dynamic whole-brain disease. Stroke Vasc. Neurol. 1, 83–92. https://doi.org/10.1136/svn-2016-000035 (2016).
Baykara, E. et al. A novel imaging marker for small vessel disease based on skeletonization of white matter tracts and diffusion histograms. Ann. Neurol. 80, 581–592. https://doi.org/10.1002/ana.24758 (2016).
Au, R. et al. New norms for a new generation: Cognitive performance in the framingham offspring cohort. Exp. Aging Res. https://doi.org/10.1080/03610730490484380 (2004).
Pase, M. P. et al. Association of aortic stiffness with cognition and brain aging in young and middle-aged adults: The Framingham third generation cohort study. Hypertension https://doi.org/10.1161/HYPERTENSIONAHA.115.06610 (2016).
Lloyd-Price, J., Abu-Ali, G. & Huttenhower, C. The healthy human microbiome. Genome Med. 8, 51. https://doi.org/10.1186/s13073-016-0307-y (2016).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662. https://doi.org/10.1038/s41586-019-1237-9 (2019).
Lavoie, S. et al. The Crohn’s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. eLife https://doi.org/10.7554/eLife.39982 (2019).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. https://doi.org/10.1073/pnas.1000080107 (2011).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. https://doi.org/10.1038/ismej.2012.8 (2012).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. https://doi.org/10.1038/s41587-019-0209-9 (2019).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1009442 (2021).
Wilmanski, T. et al. Gut microbiome pattern reflects healthy ageing and predicts survival in humans. Nat. Metabol. https://doi.org/10.1038/s42255-021-00348-0 (2021).
Manderino, L. et al. Preliminary evidence for an association between the composition of the gut microbiome and cognitive function in neurologically healthy older adults. J. Int. Neuropsychol. Soc. JINS https://doi.org/10.1017/S1355617717000492 (2017).
Meyer, K. et al. Association of the gut microbiota with cognitive function in midlife. JAMA Netw. Open https://doi.org/10.1001/jamanetworkopen.2021.43941 (2022).
Brandscheid, C. et al. Altered gut microbiome composition and tryptic activity of the 5xFAD Alzheimer’s mouse model. J. Alzheimer’s Dis. JAD https://doi.org/10.3233/JAD-160926 (2017).
Sun, B. L. et al. Gut microbiota alteration and its time course in a tauopathy mouse model. J. Alzheimer’s Dis. JAD https://doi.org/10.3233/JAD-181220 (2019).
Bäuerl, C., Collado, M. C., Diaz Cuevas, A., Viña, J. & Pérez, Martínez G. Shifts in gut microbiota composition in an APP/PSS1 transgenic mouse model of Alzheimer’s disease during lifespan. Lett. Appl. Microbiol. https://doi.org/10.1111/lam.12882 (2018).
Chen, D. et al. Prebiotic effect of fructooligosaccharides from Morinda officinalis on Alzheimer’s disease in rodent models by targeting the microbiota-gut-brain axis. Front. Aging Neurosci. https://doi.org/10.3389/fnagi.2017.00403 (2017).
Abraham, D. et al. Exercise and probiotics attenuate the development of Alzheimer’s disease in transgenic mice: Role of microbiome. Exp. Gerontol. https://doi.org/10.1016/j.exger.2018.12.005 (2019).
Saji, N. et al. The relationship between the gut microbiome and mild cognitive impairment in patients without dementia: A cross-sectional study conducted in Japan. Sci. Rep. https://doi.org/10.1038/s41598-019-55851-y (2019).
Wu, M. L., Yang, X. Q., Xue, L., Duan, W. & Du, J. R. Age-related cognitive decline is associated with microbiota-gut-brain axis disorders and neuroinflammation in mice. Behav. Brain Res. https://doi.org/10.1016/j.bbr.2021.113125 (2021).
Ren, T. et al. Gut microbiota altered in mild cognitive impairment compared with normal cognition in sporadic Parkinson’s disease. Front. Neurol. 11, 137. https://doi.org/10.3389/fneur.2020.00137 (2020).
Sun, X., Xue, L., Wang, Z. & Xie, A. Update to the treatment of Parkinson’s disease based on the gut-brain axis mechanism. Front. Neurosci. https://doi.org/10.3389/fnins.2022.878239 (2022).
Treangen, T. J., Wagner, J., Burns, M. P. & Villapol, S. Traumatic brain injury in mice induces acute bacterial dysbiosis within the fecal microbiome. Front. Immunol. https://doi.org/10.3389/fimmu.2018.02757 (2018).
Cai, Z., Yan, L. J., Li, K., Quazi, S. H. & Zhao, B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromol. Med. https://doi.org/10.1007/s12017-012-8173-2 (2012).
Barone, E., Di Domenico, F., Perluigi, M. & Butterfield, D. A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. https://doi.org/10.1016/j.freeradbiomed.2021.09.006 (2021).
Chen, M. et al. AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. https://doi.org/10.1016/j.bbr.2020.113043 (2021).
Salminen, A., Kaarniranta, K., Haapasalo, A., Soininen, H. & Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. https://doi.org/10.1111/j.1471-4159.2011.07331.x (2011).
Domise, M. et al. Neuronal AMP-activated protein kinase hyper-activation induces synaptic loss by an autophagy-mediated process. Cell Death Dis. https://doi.org/10.1038/s41419-019-1464-x (2019).
Reger, M. A. et al. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging https://doi.org/10.1016/S0197-4580(03)00087-3 (2004).
Shippy, D. C., Wilhelm, C., Viharkumar, P. A., Raife, T. J. & Ulland, T. K. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. https://doi.org/10.1186/s12974-020-01948-5 (2020).
Acknowledgements
The Authors thank Dr. Ramnik Xavier from the Broad Institute of MIT and Harvard, and the Center for Microbiome Informatics and Therapeutics (Massachusetts Institute of Technology, Cambridge, MA, USA) for providing access to the FHS microbiome data.
Funding
This study was funded in part by the UT Health San Antonio Center for Biomedical Neuroscience (CBN) and Grants from the NIA (AG059421, AG054076, AG049607, AG033090, AG066524, P30 AG066546, 5P30AG059305-03, RF1 AG061729A1, 5U01AG052409-04) and NINDS (NS017950, UF1NS125513, K01NS126489). In addition, Drs. Fongang, Kautz, Seshadri, Satizabal, Maestre, Cavazos, and Himali are partially supported by the South Texas Alzheimer’s Disease Research Center (P30AG066546). Drs. Seshadri and Himali receive support from The Bill and Rebecca Reed Endowment for Precision Therapies and Palliative Care. Dr. Himali is supported by an endowment from the William Castella family as William Castella Distinguished University Chair for Alzheimer’s Disease Research, and Dr. Seshadri by an endowment from the Barker Foundation as the Robert R Barker Distinguished University Professor of Neurology, Psychiatry and Cellular and Integrative Physiology.
Author information
Authors and Affiliations
Contributions
B.F., C.S., and S.S. conceived the study. C.S., J.H., and A.B. prepared the data. B.F. performed all statistical analyses. B.F. wrote the manuscript. All authors discussed the results, provided feedback during the writing process, and commented on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fongang, B., Satizabal, C., Kautz, T.F. et al. Cerebral small vessel disease burden is associated with decreased abundance of gut Barnesiella intestinihominis bacterium in the Framingham Heart Study. Sci Rep 13, 13622 (2023). https://doi.org/10.1038/s41598-023-40872-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-40872-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
